
Analytical Techniques for Structural Characterization of Proteins in Solid Pharmaceutical Forms: An Overview


Abstract
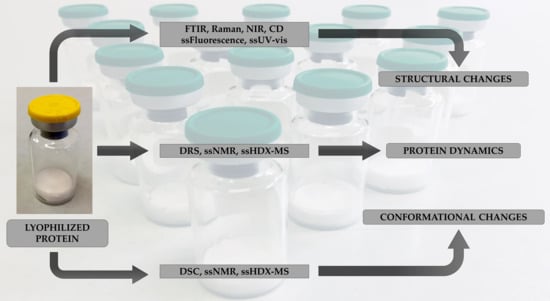
:
1. Introduction
2. Methods for Structural Characterization
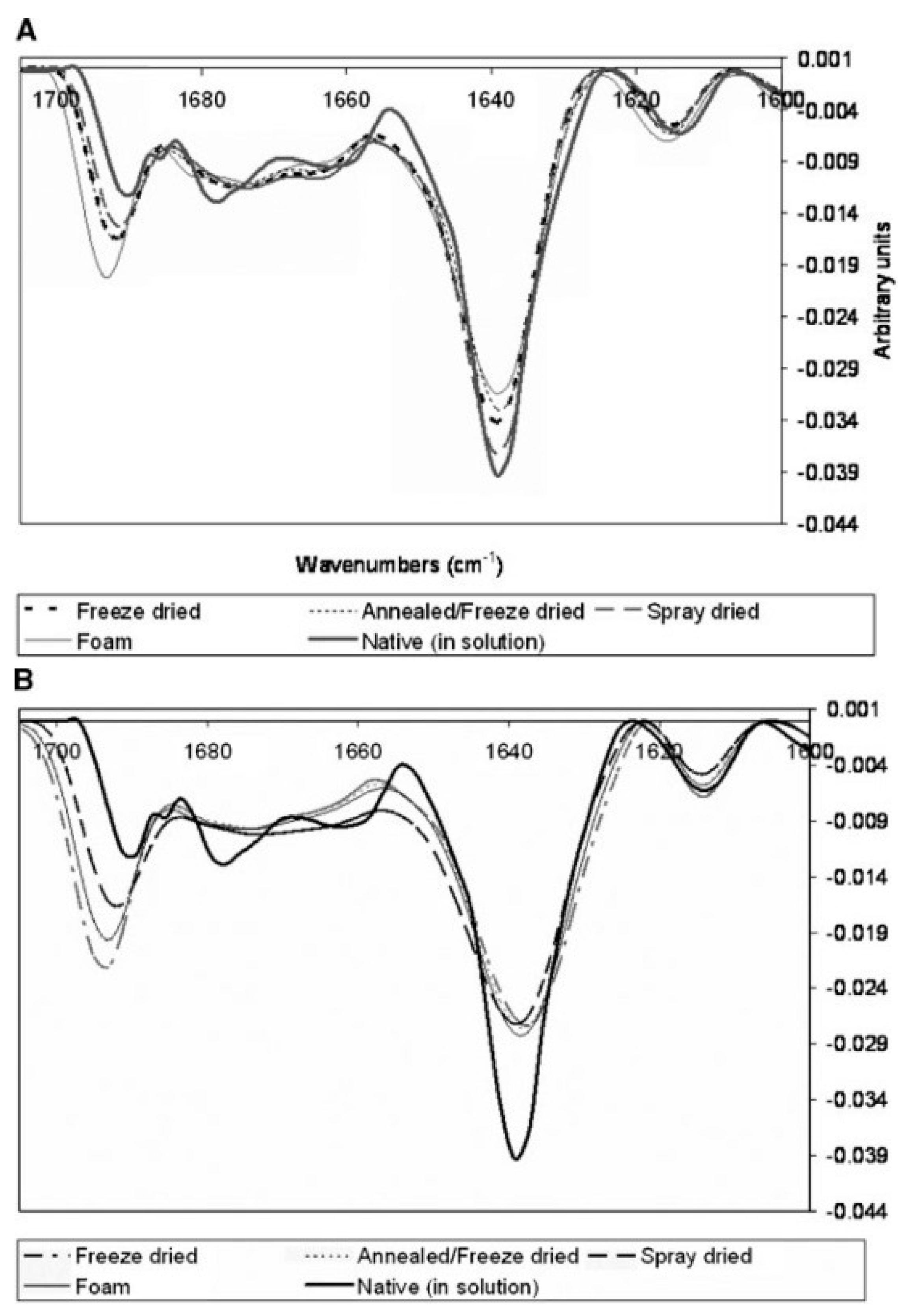
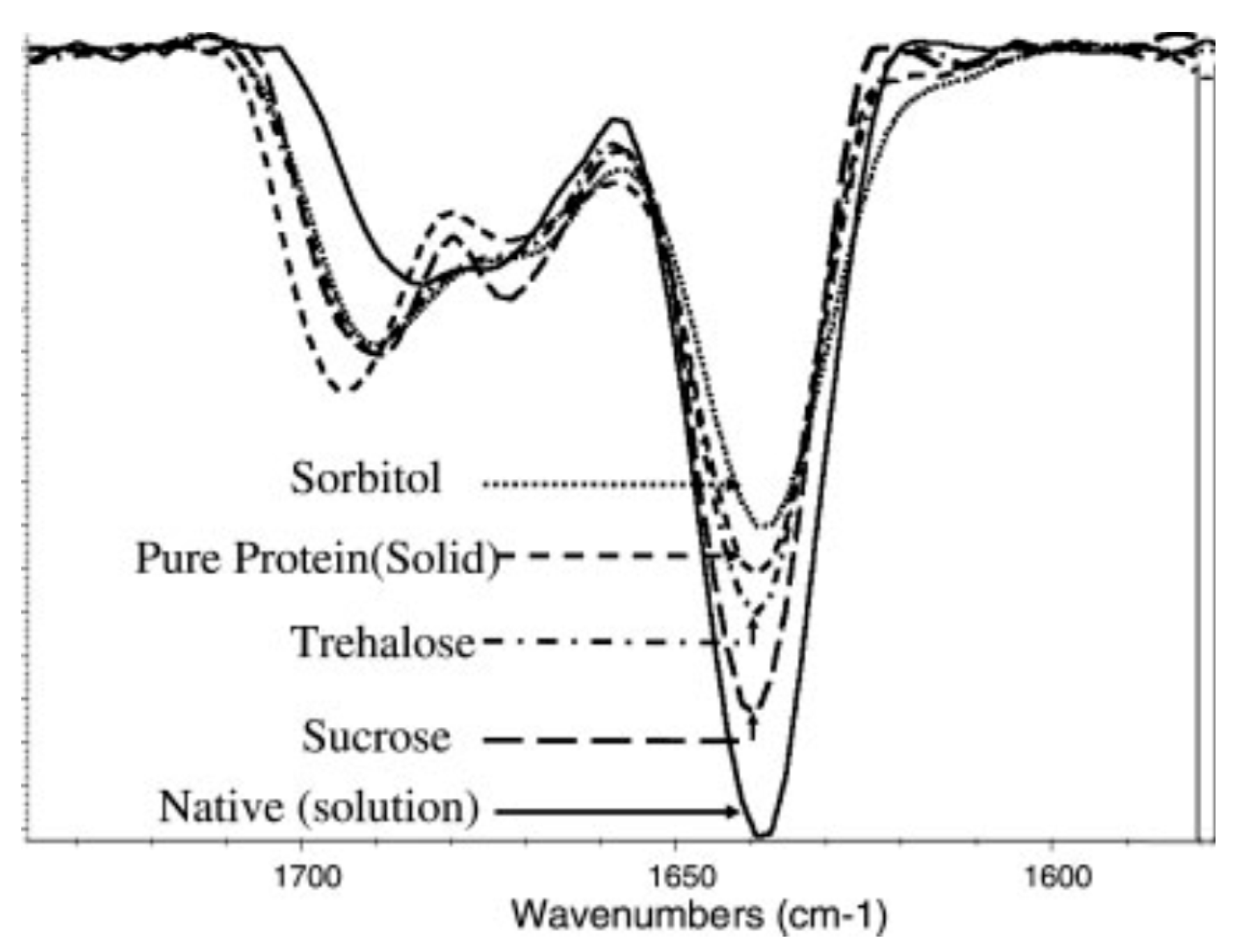
2.1. Fourier-Transform Infrared Spectroscopy (FTIR)
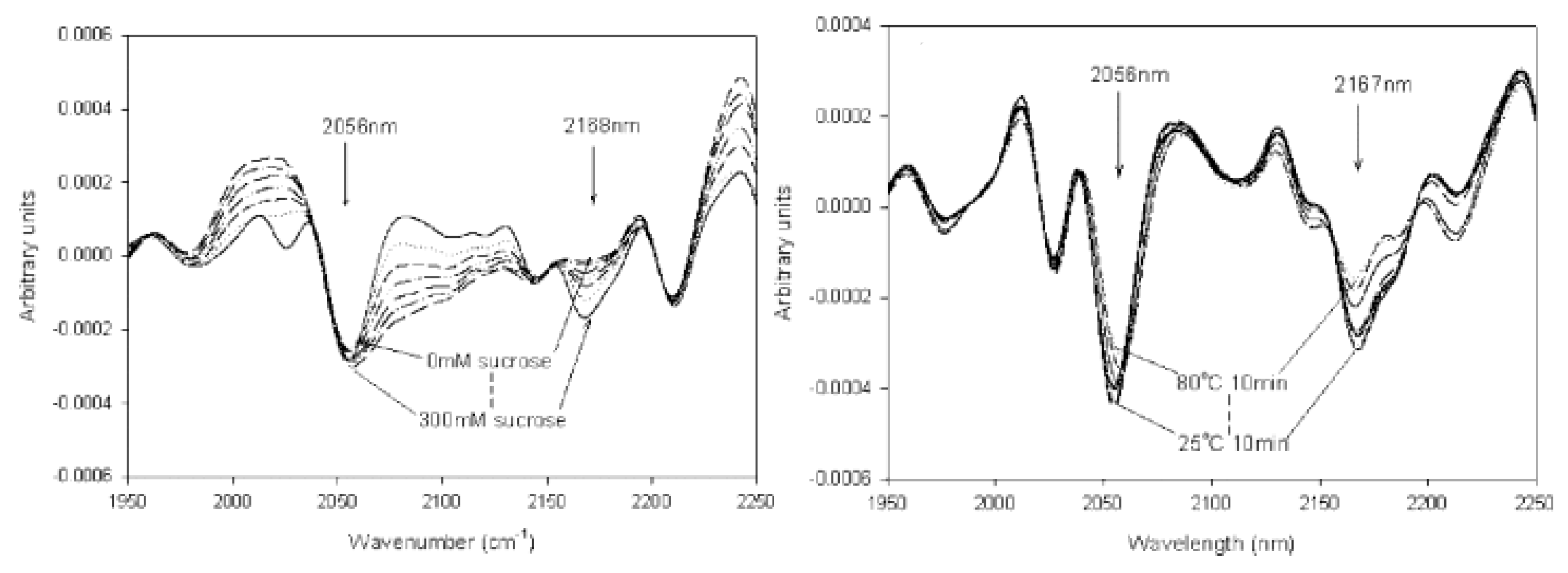
2.2. Near-Infrared Spectroscopy (NIR)
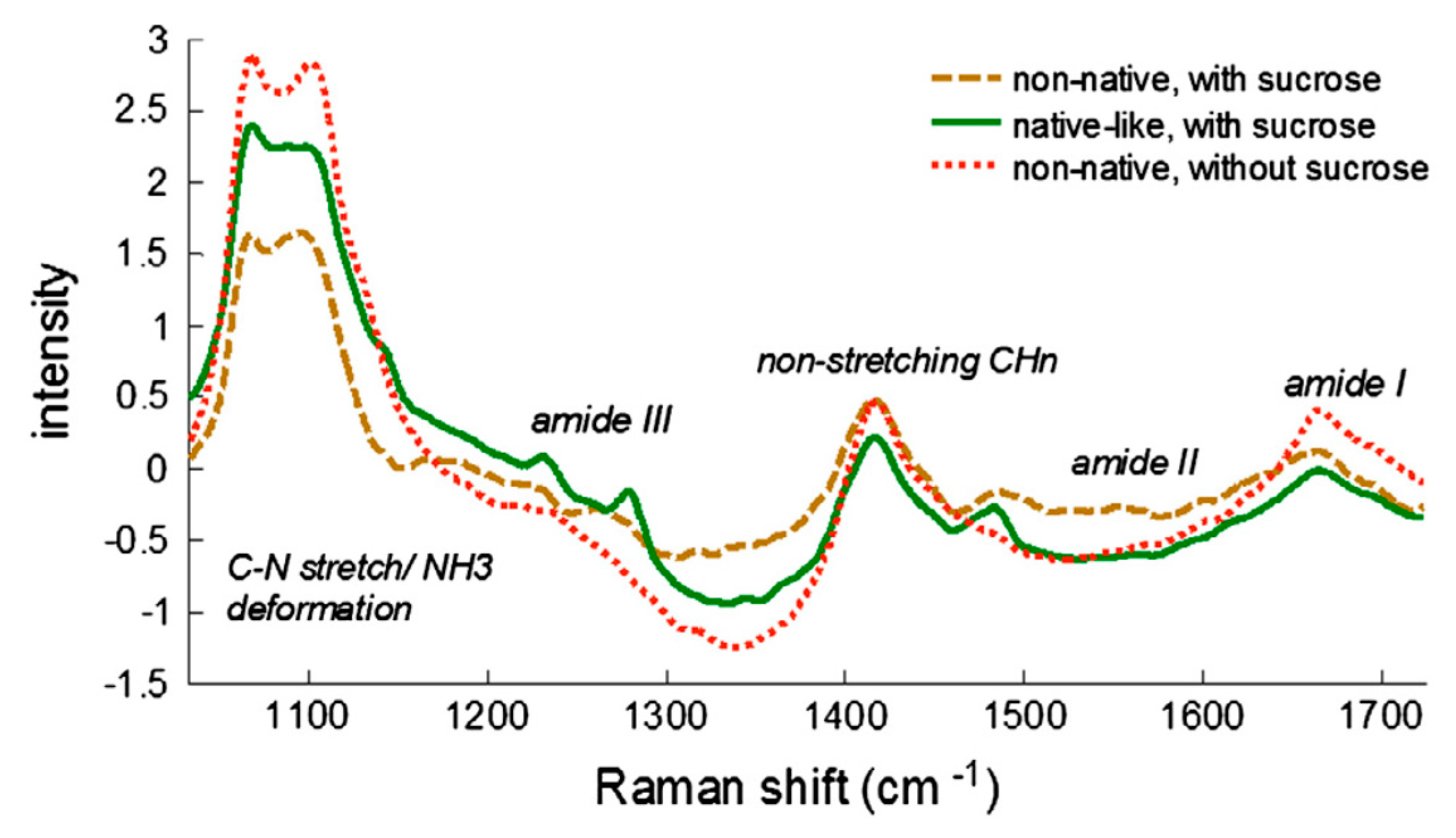
2.3. Raman Spectroscopy
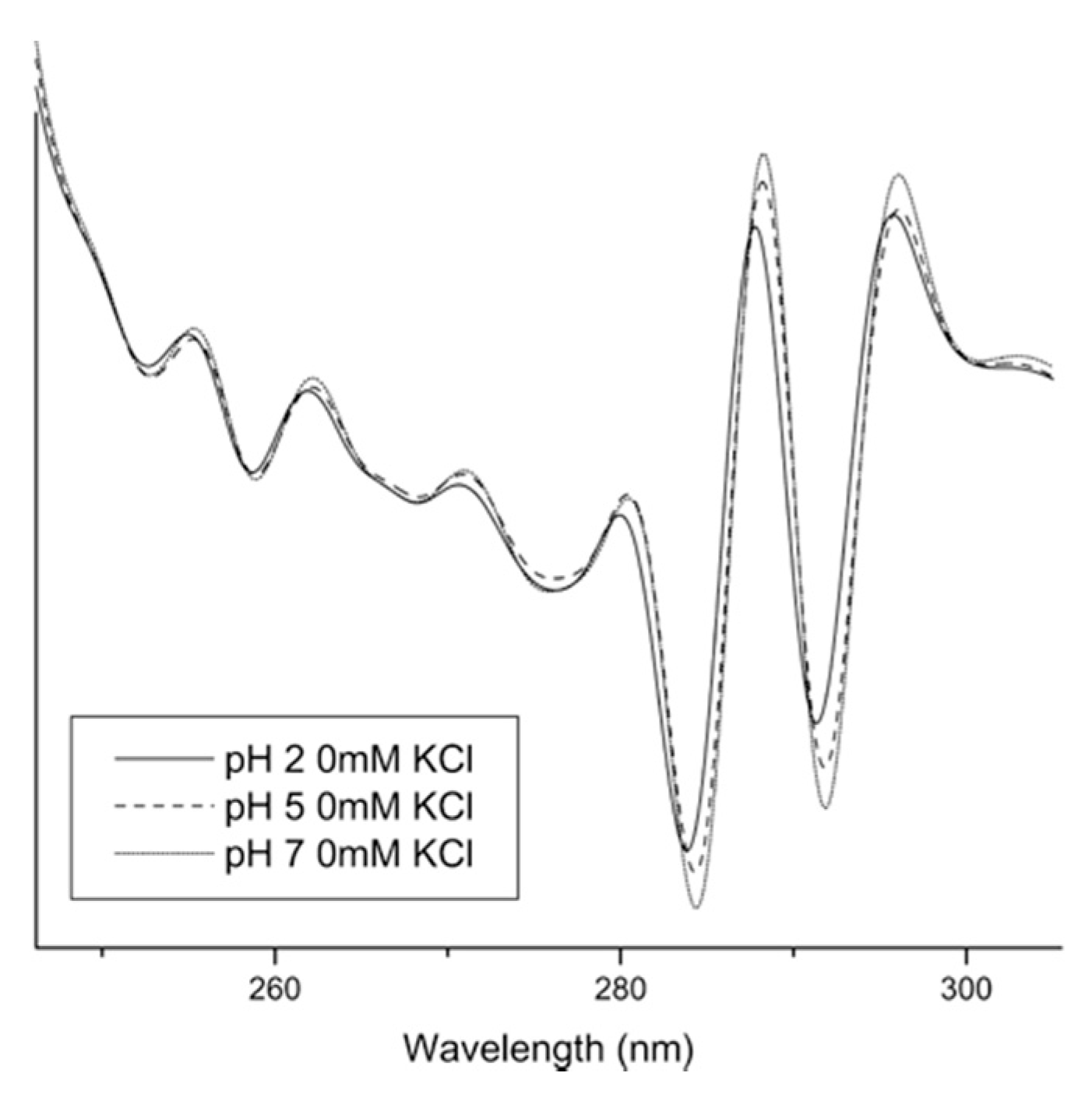
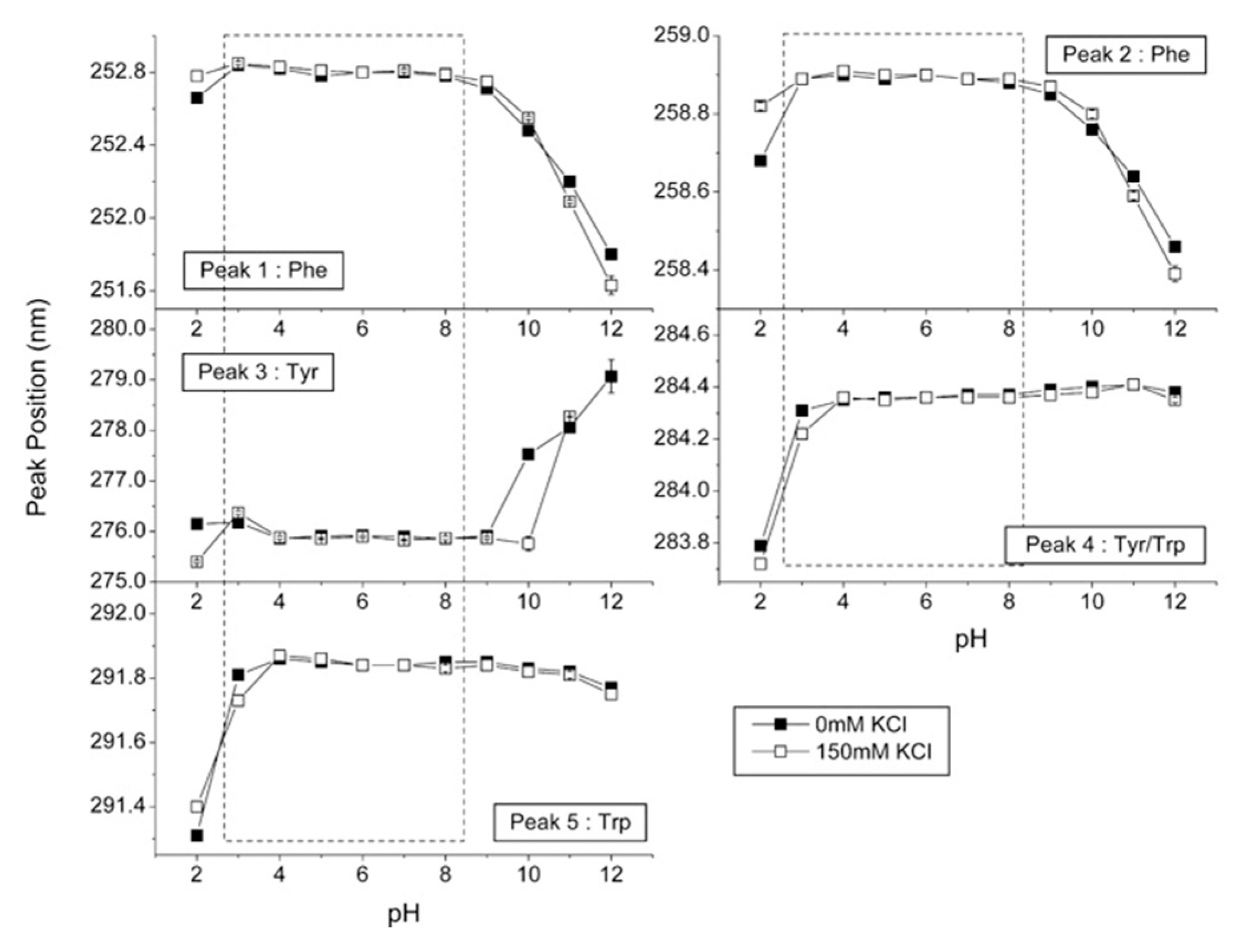
2.4. Solid-State UV–Vis Spectroscopy
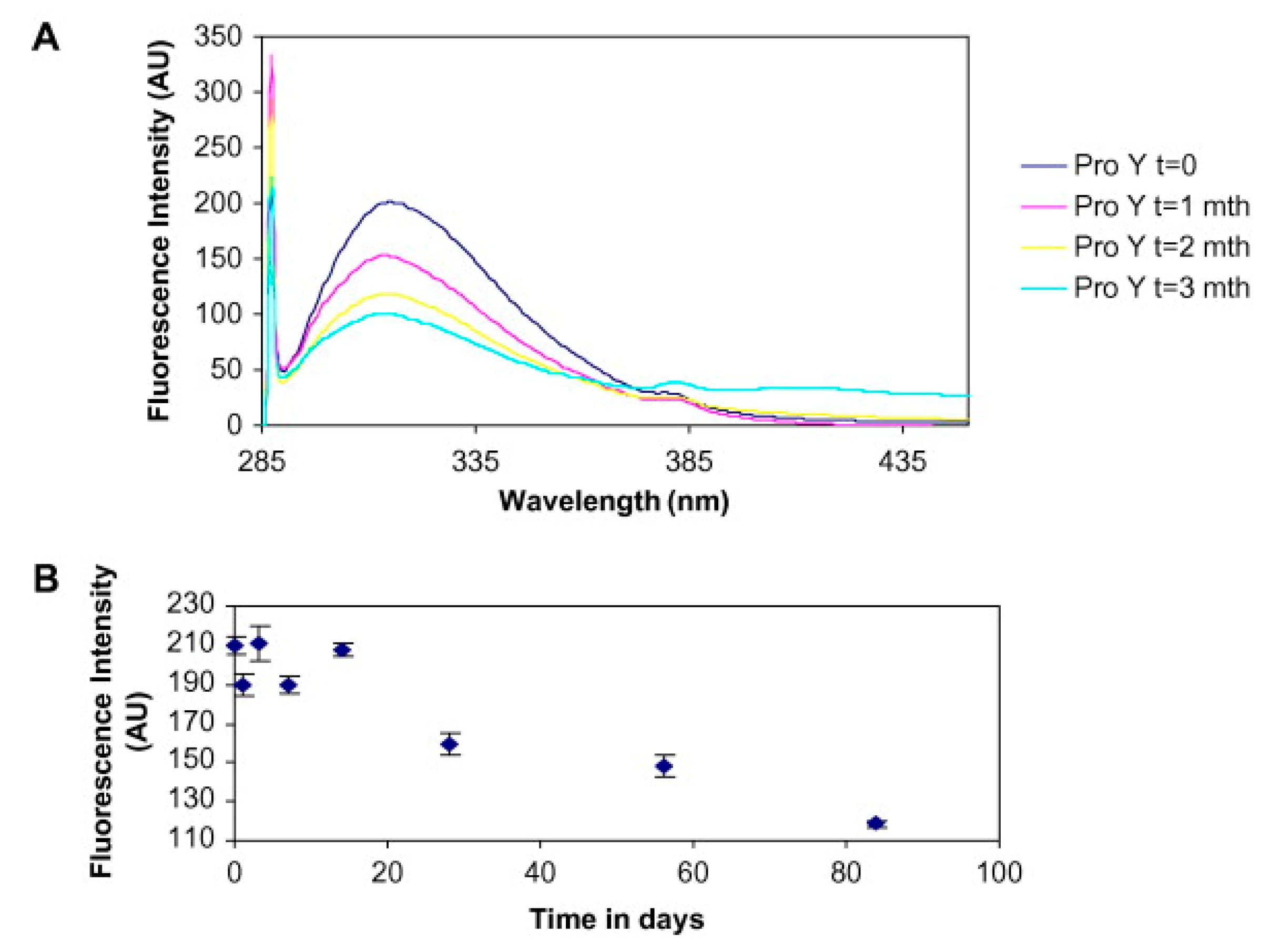
2.5. Solid-State Fluorescence Spectroscopy
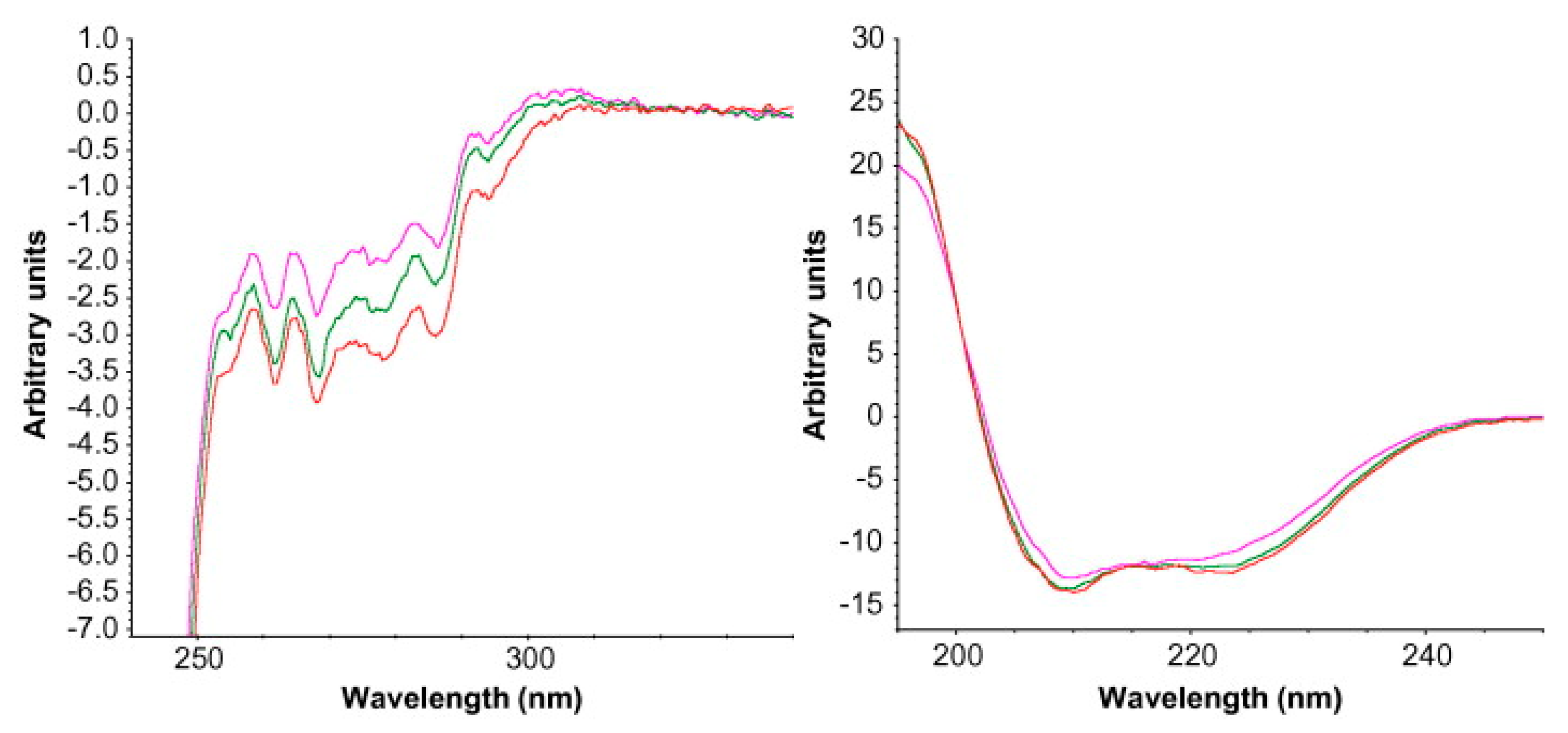
2.6. Circular Dichroism (CD)
2.7. Solid-State Nuclear Magnetic Resonance Spectroscopy (ssNMR)
2.8. Differential Scanning Calorimetry (DSC)
2.9. Dielectric Relaxation Spectroscopy (DRS)
2.10. X-ray Diffraction (XRD)
2.11. Solid-State Hydrogen-Deuterium Exchange Mass Spectrometry (ssHDX-MS)
3. Methods for Aggregation Studies
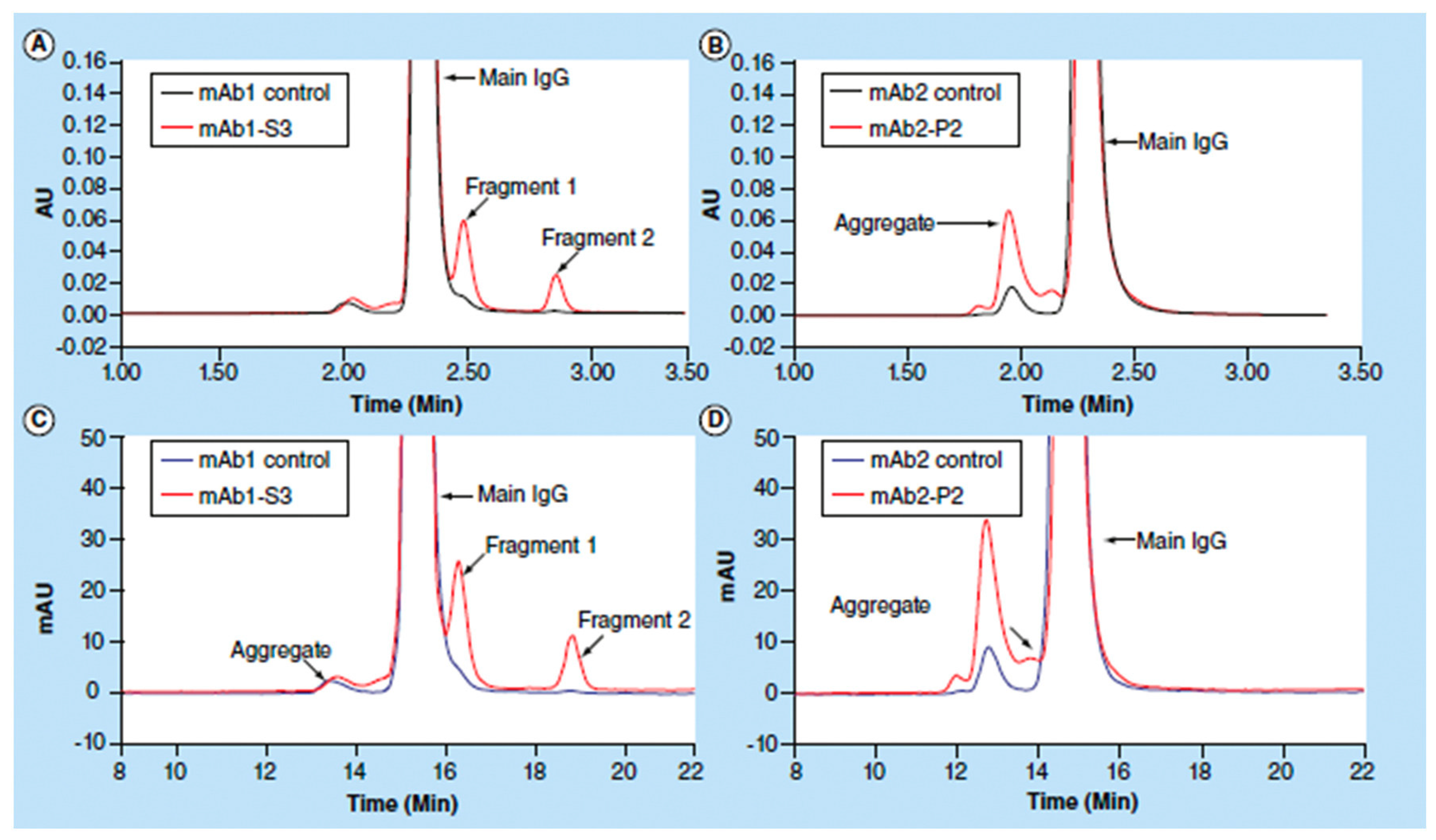
3.1. Size-Exclusion Chromatography (SEC)
3.2. Dynamic Light Scattering (DLS)
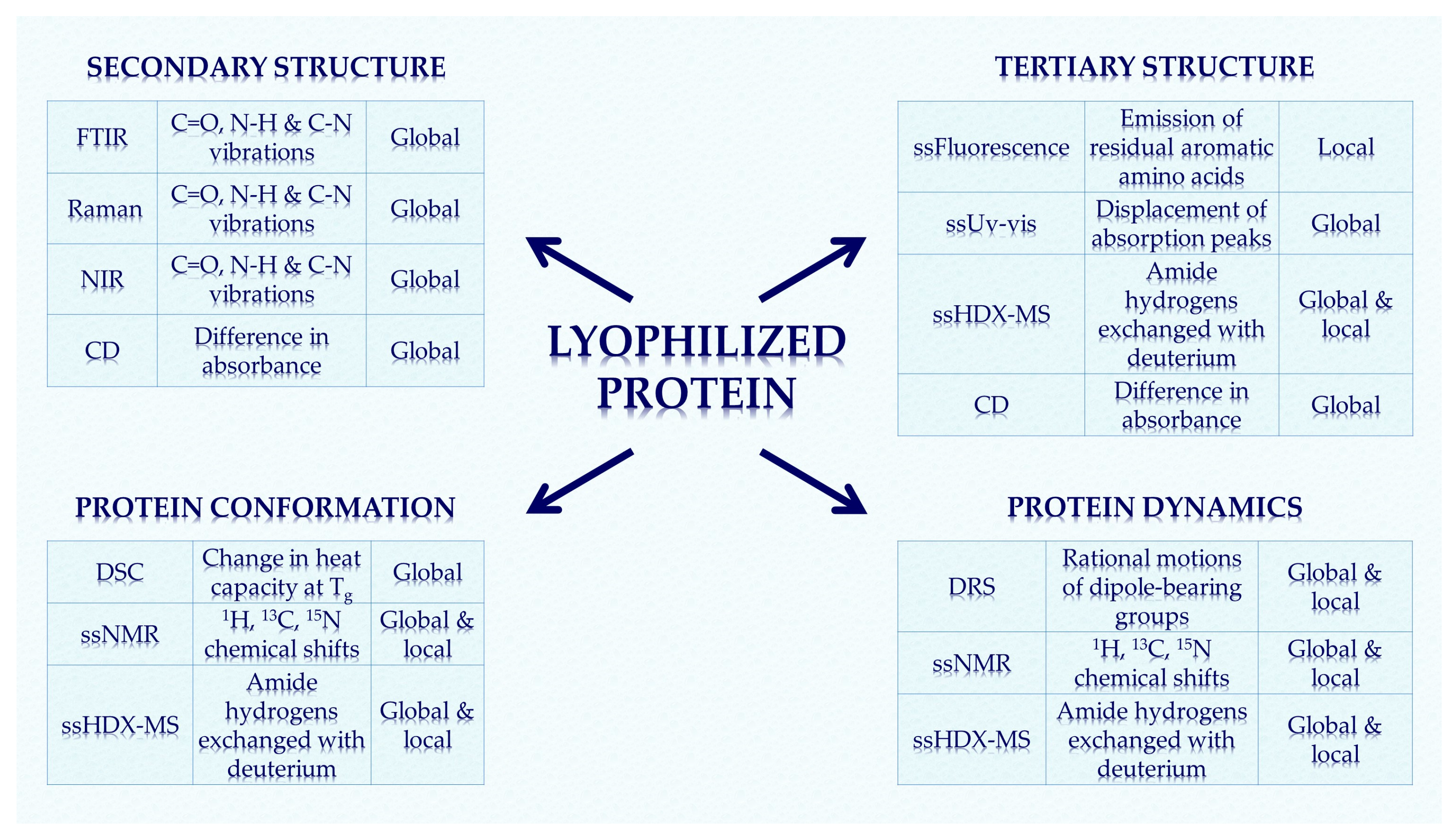
4. Method Summary
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, W.; Ohtake, S. Science and art of protein formulation development. Int. J. Pharm. 2019, 568, 118–505. [Google Scholar] [CrossRef]
- Abdul-Fattah, A.M.; Kalonia, D.S.; Pikal, M.J. The Challenge of Drying Method Selection for Protein Pharmaceuticals: Product Quality Implications. J. Pharm. Sci. 2007, 96, 1886–1916. [Google Scholar] [CrossRef] [PubMed]
- Dong, A.; Prestrelski, S.J.; Allison, S.D.; Carpenter, J.F. Infrared spectroscopic studies of lyophilization- and temperature-induced protein aggregation. J. Pharm. Sci. 1995, 84, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Prestrelski, S.J.; Tedeschi, N.; Arakawa, T.; Carpenter, J.F. Dehydration-induced conformational transitions in proteins and their inhibition by stabilizers. Biophys. J. 1993, 65, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Prestrelski, S.J.; Pikal, K.A.; Arakawa, T. Optimization of Lyophilization Conditions for Recombinant Human Interleukin-2 by Dried-State Conformational Analysis Using Fourier-Transform Infrared Spectroscopy. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1995, 12, 1250–1259. [Google Scholar] [CrossRef]
- Chang, B.S.; Beauvais, R.M.; Dong, A.; Carpenter, J.F. Physical Factors Affecting the Storage Stability of Freeze-Dried Interleukin-1 Receptor Antagonist: Glass Transition and Protein Conformation. Arch. Biochem. Biophys. 1996, 331, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, J.F.; Prestrelski, S.J.; Dong, A. Application of infrared spectroscopy to development of stable lyophilized protein formulations. Eur. J. Pharm. Biopharm. 1998, 45, 231–238. [Google Scholar] [CrossRef]
- Ken-ichi, I.; Sumie, Y.; Tadao, T. Stabilization of β-galactosidase by amphiphilic additives during freeze-drying. Int. J. Pharm. 1993, 90, 187–194. [Google Scholar] [CrossRef]
- Cicerone, M.T.; Pikal, M.J.; Qian, K.K. Stabilization of proteins in solid form. Adv. Drug Deliv. Rev. 2015, 93, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Lam, X.M.; Costantino, H.R.; Overcashier, D.E.; Nguyen, T.H.; Hsu, C.C. Replacing succinate with glycolate buffer improves the stability of lyophilized interferon-. Int. J. Pharm. 1996, 142, 85–95. [Google Scholar] [CrossRef]
- Carpenter, J.F.; Crowe, J.H. The mechanism of cryoprotection of proteins by solutes. Cryobiology 1988, 25, 244–255. [Google Scholar] [CrossRef]
- Chang, B.S.; Kendrick, B.S.; Carpenter, J.F. Surface-induced denaturation of proteins during freezing and its inhibition by surfactants. J. Pharm. Sci. 1996, 85, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Crowe, J.H.; Carpenter, J.F.; Crowe, L.M.; Anchordoguy, T.J. Are freezing and dehydration similar stress vectors? A comparison of modes of interaction of stabilizing solutes with biomolecules. Cryobiology 1990, 27, 219–231. [Google Scholar] [CrossRef]
- Prestrelski, S.J.; Arakawa, T.; Carpenter, J.F. Separation of freezing- and drying-induced denaturation of lyophilized proteins using stress-specific stabilization: II. Structural studies using infrared spectroscopy. Arch. Biochem. Biophys. 1993, 303, 465–473. [Google Scholar] [CrossRef]
- Carpenter, J.F.; Prestrelski, S.J.; Arakawa, T. Separation of Freezing- and Drying-Induced Denaturation of Lyophilized Proteins Using Stress-Specific Stabilization: I. Enzyme Activity and Calorimetric Studies. Arch. Biochem. Biophys. 1993, 303, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Crowe, L.M.; Reid, D.S.; Crowe, J.H. Is trehalose special for preserving dry biomaterials? Biophys. J. 1996, 71, 2087–2093. [Google Scholar] [CrossRef] [Green Version]
- Izutsu, K.I.; Yoshioka, S.; Kojima, S. Increased Stabilizing Effects of Amphiphilic Excipients on Freeze-Drying of Lactate Dehydrogenase (LDH) by Dispersion into Sugar Matrices. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1995, 12, 838–843. [Google Scholar] [CrossRef]
- Ramos, A.; Raven, N.D.H.; Sharp, R.J.; Bartolucci, S.; Rossi, M.; Cannio, R.; Lebbink, J.; Van Der Oost, J.; De Vos, W.M.; Santos, H. Stabilization of Enzymes against Thermal Stress and Freeze-Drying by Mannosylglycerate. Appl. Environ. Microbiol. 1997, 63, 4020–4025. [Google Scholar] [CrossRef] [Green Version]
- Wahl, V.; Khinast, J.; Paudel, A. Lyophilized protein powders: A review of analytical tools for root cause analysis of lot-to-lot variability. TrAC Trends Anal. Chem. 2016, 82, 468–491. [Google Scholar] [CrossRef]
- Moorthy, B.; Iyer, L.; Topp, E. Characterizing Protein Structure, Dynamics and Conformation in Lyophilized Solids. Curr. Pharm. Des. 2015, 21, 5845–5853. [Google Scholar] [CrossRef] [Green Version]
- Wang, W. Lyophilization and development of solid protein pharmaceuticals. Int. J. Pharm. 2000, 203, 1–60. [Google Scholar] [CrossRef]
- Duddu, S.P.; Zhang, G.; Dal Monte, P.R. The relationship between protein aggregation and molecular mobility below the glass transition temperature of lyophilized formulations containing a monoclonal antibody. Pharm. Res. 1997, 14, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Protein aggregation and its inhibition in biopharmaceutics. Int. J. Pharm. 2005, 289, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Pieters, S.; Vander Heyden, Y.; Roger, J.M.; D’Hondt, M.; Hansen, L.; Palagos, B.; De Spiegeleer, B.; Remon, J.P.; Vervaet, C.; De Beer, T. Raman spectroscopy and multivariate analysis for the rapid discrimination between native-like and non-native states in freeze-dried protein formulations. Eur. J. Pharm. Biopharm. 2013, 85, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Bandekar, J. Amide modes and protein conformation. Biochim. Biophys. Acta (BBA)/Protein Struct. Mol. 1992, 1120, 123–143. [Google Scholar] [CrossRef]
- Cooper, E.A.; Knutson, K. Fourier transform infrared spectroscopy investigations of protein structure. Pharm. Biotechnol. 1995, 7, 101–143. [Google Scholar] [CrossRef]
- Jiskoot, W.; Crommelin, D. Methods for Structural Analysis of Protein Pharmaceuticals. Available online: https://books.google.com/books?id=CyeilBm5az8C&pgis=1 (accessed on 8 January 2021).
- Barth, A.; Zscherp, C. What vibrations tell us about proteins. Q. Rev. Biophys. 2002, 35, 369–430. [Google Scholar] [CrossRef]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [Green Version]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Tian, F.; Middaugh, C.R.; Offerdahl, T.; Munson, E.; Sane, S.; Rytting, J.H. Spectroscopic evaluation of the stabilization of humanized monoclonal antibodies in amino acid formulations. Int. J. Pharm. 2007, 335, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Andya, J.D.; Hsu, C.C.; Shire, S.J. Mechanisms of aggregate formation and carbohydrate excipient stabilization of lyophilized humanized monoclonal antibody formulations. AAPS J. 2003, 5, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Fattah, A.M.; Truong-Le, V.; Yee, L.; Nguyen, L.; Kalonia, D.S.; Cicerone, M.T.; Pikal, M.J. Drying-induced variations in physico-chemical properties of amorphous pharmaceuticals and their impact on stability (I): Stability of a monoclonal antibody. J. Pharm. Sci. 2007, 96, 1983–2008. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Carpenter, J.F.; Cicerone, M.T.; Randolph, T.W. Contributions of local mobility and degree of retention of native secondary structure to the stability of recombinant human growth hormone (rhGH) in glassy lyophilized formulations. Soft Matter 2013, 9, 7855–7865. [Google Scholar] [CrossRef]
- Allison, S.D.; Chang, B.; Randolph, T.W.; Carpenter, J.F. Hydrogen bonding between sugar and protein is responsible for inhibition of dehydration-induced protein unfolding. Arch. Biochem. Biophys. 1999, 365, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Griebenow, K.; Klibanov, A.M. Lyophilization-induced reversible changes in the secondary structure of proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 10969–10976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüle, S.; Frieß, W.; Bechtold-Peters, K.; Garidel, P. Conformational analysis of protein secondary structure during spray-drying of antibody/mannitol formulations. Eur. J. Pharm. Biopharm. 2007, 65, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.L.; Shepherd, D.; Sun, J.; Tang, X.C.; Pikal, M.J. Effect of sorbitol and residual moisture on the stability of lyophilized antibodies: Implications for the mechanism of protein stabilization in the solid state. J. Pharm. Sci. 2005, 94, 1445–1455. [Google Scholar] [CrossRef]
- Van De Weert, M.; Haris, P.I.; Hennink, W.E.; Crommelin, D.J.A. Fourier transform infrared spectrometric analysis of protein conformation: Effect of sampling method and stress factors. Anal. Biochem. 2001, 297, 160–169. [Google Scholar] [CrossRef]
- Costantino, H.R.; Carrasquillo, K.G.; Cordero, R.A.; Mumenthaler, M.; Hsu, C.C.; Griebenow, K. Effect of excipients on the stability and structure of lyophilized recombinant human growth hormone. J. Pharm. Sci. 1998, 87, 1412–1420. [Google Scholar] [CrossRef]
- Chang, L.; Shepherd, D.; Sun, J.; Ouellette, D.; Grant, K.L.; Tang, X.; Pikal, M.J. Mechanism of protein stabilization by sugars during freeze-drying and storage: Native structure preservation, specific interaction, and/or immobilization in a glassy matrix? J. Pharm. Sci. 2005, 94, 1427–1444. [Google Scholar] [CrossRef]
- Wang, B.; Tchessalov, S.; Warne, N.W.; Pikal, M.J. Impact of sucrose level on storage stability of proteins in freeze-dried solids: I. Correlation of protein-sugar interaction with native structure preservation. J. Pharm. Sci. 2009, 98, 3131–3144. [Google Scholar] [CrossRef]
- Moorthy, B.S.; Schultz, S.G.; Kim, S.G.; Topp, E.M. Predicting protein aggregation during storage in lyophilized solids using solid state amide hydrogen/deuterium exchange with mass spectrometric analysis (ssHDX-MS). Mol. Pharm. 2014, 11, 1869–1879. [Google Scholar] [CrossRef]
- Blanco, M.; Coello, J.; Iturriaga, H.; Maspoch, S.; De La Pezuela, C. Near-infrared spectroscopy in the pharmaceutical industry. Analyst 1998, 123, 135R–150R. [Google Scholar] [CrossRef]
- Ciurczak, E.W. Uses of near-infrared spectroscopy in pharmaceutical analysis. Appl. Spectrosc. Rev. 1987, 23, 147–163. [Google Scholar] [CrossRef]
- Morisseau, K.M.; Rhodes, C.T. Pharmaceutical uses of near-infrared spectroscopy. Drug Dev. Ind. Pharm. 1995, 21, 1071–1090. [Google Scholar] [CrossRef]
- Miyazawa, M.; Sonoyama, M. Studies on the Structural Characterisation of Proteins M Second derivative near infrared studies on the structural characterisation of proteins. J. Near Infrared Spectrosc. 1998, 6, 253–257. [Google Scholar] [CrossRef]
- Chan, H.K.; Ongpipattanakul, B.; Au-Yeung, J. Aggregation of rhDNase occurred during the compression of KBr pellets used for FTIR spectroscopy. Pharm. Res. 1996, 13, 238–242. [Google Scholar] [CrossRef]
- Luner, P.E.; Majuru, S.; Seyer, J.J.; Kemper, M.S. Quantifying crystalline form composition in binary powder mixtures using near-infrared reflectance spectroscopy. Pharm. Dev. Technol. 2000, 5, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Watanabe, T.; Tsuchikawa, S. The effect of path length, light intensity and co-added time on the detection limit associated with NIR spectroscopy of potassium hydrogen phthalate in aqueous solution. PLoS ONE 2017, 12, e0176920. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Nayar, R.; Carpenter, J.F.; Manning, M.C. Noninvasive determination of protein conformation in the solid state using near infrared (NIR) spectroscopy. J. Pharm. Sci. 2005, 94, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Derksen, M.W.J.; Van De Oetelaar, P.J.M.; Maris, F.A. The use of near-infrared spectroscopy in the efficient prediction of a specification for the residual moisture content of a freeze-dried product. J. Pharm. Biomed. Anal. 1998, 17, 473–480. [Google Scholar] [CrossRef]
- Jones, J.A.; Last, I.R.; MacDonald, B.F.; Prebble, K.A. Development and transferability of near-infrared methods for determination of moisture in a freeze-dried injection product. J. Pharm. Biomed. Anal. 1993, 11, 1227–1231. [Google Scholar] [CrossRef]
- Last, I.R.; Prebble, K.A. Suitability of near-infrared methods for the determination of moisture in a freeze-dried injection product containing different amounts of the active ingredient. J. Pharm. Biomed. Anal. 1993, 11, 1071–1076. [Google Scholar] [CrossRef]
- Sadler, A.J.; Horsch, J.G.; Lawson, E.Q.; Harmatz, D.; Brandau, D.T.; Middaugh, C.R. Near-infrared photoacoustic spectroscopy of proteins. Anal. Biochem. 1984, 138, 44–51. [Google Scholar] [CrossRef]
- Dufour, E.; Devaux, M.F.; Mouhous, N.; Robert, P. Monitoring the Secondary Structure of Proteins by Near-Infrared Spectroscopy. Appl. Spectrosc. 1999, 53, 226–232. [Google Scholar]
- Sakuri, K.; Cho, R.-K.; Miura, T.; Liu, Y.; Ozaki, Y. Studies on Spectra/Structure Correlations in Near-Infrared Spectra of Proteins and Polypeptides. Part I: A Marker Band for Hydrogen Bonds. Appl. Spectrosc. 1994, 48, 1249–1254. [Google Scholar]
- Katayama, D.S.; Carpenter, J.F.; Menard, K.P.; Manning, M.C.; Randolph, T.W. Mixing Properties of Lyophilized Protein Systems: A Spectroscopic and Calorimetric Study. J. Pharm. Sci. 2009, 98, 2954–2969. [Google Scholar] [CrossRef] [PubMed]
- Izutsu, K.; Fujimaki, Y.; Kuwabara, A.; Hiyama, Y.; Yomota, C.; Aoyagi, N. Near-infrared analysis of protein secondary structure in aqueous solutions and freeze-dried solids. J. Pharm. Sci. 2006, 95, 781–789. [Google Scholar] [CrossRef]
- Pieters, S.; De Beer, T.; Vander Heyden, Y. Near-Infrared and Raman Spectroscopy: Potential Tools for Monitoring of Protein Conformational Instability during Freeze-Drying Processes. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/38367-Near-infrared-and-Raman-Spectroscopy-Potential-Tools-for-Monitoring-of-Protein-Conformational-Instability-during-Freeze-drying-Processes/ (accessed on 10 April 2021).
- Wen, Z. Raman spectroscopy of protein pharmaceuticals. J. Pharm. Sci. 2007, 96, 2861–2878. [Google Scholar] [CrossRef]
- Paudel, A.; Raijada, D.; Rantanen, J. Raman spectroscopy in pharmaceutical product design. Adv. Drug Deliv. Rev. 2015, 89, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Bunaciu, A.A.; Aboul-Enein, H.Y.; Hoang, V.D. Raman spectroscopy for protein analysis. Appl. Spectrosc. Rev. 2015, 50, 377–386. [Google Scholar] [CrossRef]
- De Beer, T.R.M.; Vercruysse, P.; Burggraeve, A.; Quinten, T.; Ouyang, J.; Zhang, X.; Vervaet, C.; Remon, J.P.; Baeyens, W.R.G. In-line and real-time process monitoring of a freeze drying process using Raman and NIR spectroscopy as complementary process analytical technology (PAT) tools. J. Pharm. Sci. 2009, 98, 3430–3446. [Google Scholar] [CrossRef] [PubMed]
- Roessl, U.; Leitgeb, S.; Pieters, S.; De Beer, T.; Nidetzky, B. In situ protein secondary structure determination in ice: Raman spectroscopy-based process analytical tool for frozen storage of biopharmaceuticals. J. Pharm. Sci. 2014, 103, 2287–2295. [Google Scholar] [CrossRef] [PubMed]
- Hedoux, A.; Paccou, L.; Achir, S.; Guinet, Y. Mechanism of protein stabilization by trehalose during freeze-drying analyzed by in situ micro-raman spectroscopy. J. Pharm. Sci. 2013, 102, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Hédoux, A.; Paccou, L.; Achir, S.; Guinet, Y. In Situ Monitoring of Proteins during Lyophilization using Micro-Raman Spectroscopy: A Description of Structural Changes induced by Dehydration. J. Pharm. Sci. 2012, 101, 2316–2326. [Google Scholar] [CrossRef]
- Kauppinen, A.; Toiviainen, M.; Aaltonen, J.; Korhonen, O.; Järvinen, K.; Juuti, M.; Pellinen, R.; Ketolainen, J. Microscale freeze-drying with Raman spectroscopy as a tool for process development. Anal. Chem. 2013, 85, 2109–2116. [Google Scholar] [CrossRef]
- Elkordy, A.A.; Forbes, R.T.; Barry, B.W. Study of protein conformational stability and integrity using calorimetry and FT-Raman spectroscopy correlated with enzymatic activity. Eur. J. Pharm. Sci. 2008, 33, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Sane, S.U.; Wong, R.; Hsu, C.C. Raman Spectroscopic Characterization of Drying-Induced Structural Changes in a Therapeutic Antibody: Correlating Structural Changes with Long-Term Stability. J. Pharm. Sci. 2004, 93, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Tattini, V.; Parra, D.F.; Polakiewicz, B.; Pitombo, R.N.M. Effect of lyophilization on the structure and phase changes of PEGylated-bovine serum albumin. Int. J. Pharm. 2005, 304, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Terada, H. Estimation of state and amount of phenylalanine residues in proteins by second derivative spectrophotometry. BBA Protein Struct. 1979, 580, 120–128. [Google Scholar] [CrossRef]
- Kueltzo, L.A.; Ersoy, B.; Ralston, J.P.; Middaugh, C.R. Derivative absorbance spectroscopy and protein phase diagrams as tools for comprehensive protein characterization: A bGCSF case study. J. Pharm. Sci. 2003, 92, 1805–1820. [Google Scholar] [CrossRef] [PubMed]
- Kuś, S.; Marczenko, Z.; Obarski, N. Derivative UV-VIS spectrophotometry in analytical chemistry. Chem. Anal. 1996, 41, 899–929. [Google Scholar]
- Ichikawa, T.; Terada, H. Second derivative spectrophotometry as an effective tool for examining phenylalanine residues in proteins. BBA Protein Struct. 1977, 494, 267–270. [Google Scholar] [CrossRef]
- Kueltzo, L.A.; Wang, W.; Randolph, T.W.; Carpenter, J.F. Effects of Solution Conditions, Processing Parameters, and Container Materials on Aggregation of a Monoclonal Antibody during Freeze-Thawing. J. Pharm. Sci. 2008, 97, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Ragone, R.; Colonna, G.; Balestrieri, C.; Servillo, L.; Irace, G. Determination of Tyrosine Exposure in Proteins by Second-Derivative Spectroscopy. Biochemistry 1984, 23, 1871–1875. [Google Scholar] [CrossRef]
- Donovan, J.W. [21] Ultraviolet difference spectroscopy—New techniques and applications. Methods Enzymol. 1973, 27, 497–525. [Google Scholar] [CrossRef] [PubMed]
- Mach, H.; Middaugh, C.R. Simultaneous Monitoring of the Environment of Tryptophan, Tyrosine, and Phenylalanine Residues in Proteins by Near-Ultraviolet Second-Derivative Spectroscopy. Anal. Biochem. 1994, 222, 323–331. [Google Scholar] [CrossRef]
- Balestrieri, C.; Colonna, G.; Giovane, A.; Irace, G.; Servillo, L. Second-Derivative Spectroscopy of Proteins: A Method for the Quantitiative Determination of Aromatic Amino Acids in Proteins. Eur. J. Biochem. 1978, 90, 433–440. [Google Scholar] [CrossRef]
- Kueltzo, L.A.; Normand, N.; O’Hare, P.; Middaugh, C.R. Conformational lability of herpesvirus protein VP22. J. Biol. Chem. 2000, 275, 33213–33221. [Google Scholar] [CrossRef] [Green Version]
- Mach, H.; Thomson, J.A.; Middaugh, C.R.; Lewis, R.V. Examination of phenylalanine microenvironments in proteins by second-derivative absorption spectroscopy. Arch. Biochem. Biophys. 1991, 287, 33–40. [Google Scholar] [CrossRef]
- Lange, R.; Balny, C. UV-visible derivative spectroscopy under high pressure. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2002, 1595, 80–93. [Google Scholar] [CrossRef]
- Kueltzo, L.A.; Middaugh, C.R. Structural characterization of bovine granulocyte colony stimulating factor: Effect of temperature and pH. J. Pharm. Sci. 2003, 92, 1793–1804. [Google Scholar] [CrossRef] [PubMed]
- Ramachander, R.; Jiang, Y.; Li, C.; Eris, T.; Young, M.; Dimitrova, M.; Narhi, L. Solid state fluorescence of lyophilized proteins. Anal. Biochem. 2008, 376, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Kalonia, D.S. Steady-state tryptophan fluorescence spectroscopy study to probe tertiary structure of proteins in solid powders. J. Pharm. Sci. 2003, 92, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Schersch, K.; Betz, O.; Garidel, P.; Muehlau, S.; Bassarab, S.; Winter, G. Systematic investigation of the effect of lyophilizate collapse on pharmaceutically relevant proteins I: Stability after freeze-drying. J. Pharm. Sci. 2010, 99, 2256–2278. [Google Scholar] [CrossRef]
- Schersch, K.; Betz, O.; Garidel, P.; Muehlau, S.; Bassarab, S.; Winter, G. Systematic investigation of the effect of lyophilizate collapse on pharmaceutically relevant proteins, part 2: Stability during storage at elevated temperatures. J. Pharm. Sci. 2012, 101, 2288–2306. [Google Scholar] [CrossRef]
- Park, J.; Nagapudi, K.; Vergara, C.; Ramachander, R.; Laurence, J.S.; Krishnan, S. Effect of pH and excipients on structure, dynamics, and long-term stability of a model IgG1 monoclonal antibody upon freeze-drying. Pharm. Res. 2013, 30, 968–984. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2006; ISBN 0387312781. [Google Scholar]
- Royer, C.A. Probing protein folding and conformational transitions with fluorescence. Chem. Rev. 2006, 106, 1769–1784. [Google Scholar] [CrossRef]
- Demeule, B.; Gurny, R.; Arvinte, T. Detection and characterization of protein aggregates by fluorescence microscopy. Int. J. Pharm. 2007, 329, 37–45. [Google Scholar] [CrossRef]
- Gabellieri, E.; Strambini, G.B. Perturbation of Protein Tertiary Structure in Frozen Solutions Revealed by 1-Anilino-8-Naphthalene Sulfonate Fluorescence. Biophys. J. 2003, 85, 3214–3220. [Google Scholar] [CrossRef] [Green Version]
- Kelly, S.; Price, N. The Use of Circular Dichroism in the Investigation of Protein Structure and Function. Curr. Protein Pept. Sci. 2005, 1, 349–384. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.K.; Kalonia, D.S. Temperature- and pH-Induced Multiple Partially Unfolded States of Recombinant Human Interferon-α2a: Possible Implications in Protein Stability. Pharm. Res. 2003, 20, 1721–1729. [Google Scholar] [CrossRef]
- Tsai, A.M.; van Zanten, J.H.; Betenbaugh, M.J. I. Study of protein aggregation due to heat denaturation: A structural approach using circular dichroism spectroscopy, nuclear magnetic resonance, and static light scattering. Biotechnol. Bioeng. 1998, 59, 273–280. [Google Scholar] [CrossRef]
- Knubovets, T.; Osterhout, J.J.; Klibanov, A.M. Structure of lysozyme dissolved in neat organic solvents as assessed by NMR and CD spectroscopies. Biotechnol. Bioeng. 1999, 63, 242–248. [Google Scholar] [CrossRef]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta Proteins Proteom. 2005, 1751, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ren, D.; Huang, H.; Dankberg, J.; Rosenfeld, R.; Cocco, M.J.; Li, L.; Brems, D.N.; Remmele, R.L. Structure and stability changes of human IgG1 Fc as a consequence of methionine oxidation. Biochemistry 2008, 47, 5088–5100. [Google Scholar] [CrossRef] [PubMed]
- Harn, N.; Allan, C.; Oliver, C.; Middaugh, C.R. Highly concentrated monoclonal antibody solutions: Direct analysis of physical structure and thermal stability. J. Pharm. Sci. 2007, 96, 532–546. [Google Scholar] [CrossRef] [PubMed]
- Joshi, V.; Shivach, T.; Yadav, N.; Rathore, A.S. Circular dichroism spectroscopy as a tool for monitoring aggregation in monoclonal antibody therapeutics. Anal. Chem. 2014, 86, 11606–11613. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, A.W.P.; Norde, W. The thermal stability of immunoglobulin: Unfolding and aggregation of a multi-domain protein. Biophys. J. 2000, 78, 394–404. [Google Scholar] [CrossRef] [Green Version]
- Harada, T.; Moriyama, H. Solid-State Circular Dichroism Spectroscopy. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Castiglioni, E.; Biscarini, P.; Abbate, S. Experimental aspects of solid state circular dichroism. Chirality 2009, 21, E28–E36. [Google Scholar] [CrossRef]
- Kawamura, I.; Sato, H. Solid-state vibrational circular dichroism studies of L- and D-serine. Anal. Biochem. 2019, 580, 14–20. [Google Scholar] [CrossRef]
- Bak, K.H.; Thulstrup, P.W.; Orlien, V. Spectroscopic studies on the effect of high pressure treatment on the soluble protein fraction of porcine longissimus dorsi. Food Chem. 2014. [Google Scholar] [CrossRef]
- Paudel, A.; Geppi, M.; Mooter, G. Van den Structural and Dynamic Properties of Amorphous Solid Dispersions: The Role of Solid-State Nuclear Magnetic Resonance Spectroscopy and Relaxometry. J. Pharm. Sci. 2014, 103, 2635–2662. [Google Scholar] [CrossRef]
- Storey, R.A.; Ymén, I. Solid State Characterization of Pharmaceuticals. Solid State Charact. Pharm. 2011. [Google Scholar] [CrossRef]
- Yoshioka, S.; Aso, Y. Correlations between molecular mobility and chemical stability during storage of amorphous pharmaceuticals. J. Pharm. Sci. 2007, 96, 960–981. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, S.; Forney, K.M.; Aso, Y.; Pikal, M.J. Effect of sugars on the molecular motion of freeze-dried protein formulations reflected by NMR relaxation times. Pharm. Res. 2011, 28, 3237–3247. [Google Scholar] [CrossRef]
- Luthra, S.A.; Hodge, I.M.; Utz, M.; Pikal, M.J. Correlation of annealing with chemical stability in lyophilized pharmaceutical glasses. J. Pharm. Sci. 2008, 97, 5240–5251. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, S.; Aso, Y.; Kojima, S. The effect of excipients on the molecular mobility of lyophilized formulations, as measured by glass transition temperature and NMR relaxation- based critical mobility temperature. Pharm. Res. 1999, 16, 135–140. [Google Scholar] [CrossRef]
- Yoshioka, S.; Aso, Y.; Kojima, S.; Sakurai, S.; Fujiwara, T.; Akutsu, H. Molecular mobility of protein in lyophilized formulations linked to the molecular mobility of polymer excipients, as determined by high resolution 13C solid-state NMR. Pharm. Res. 1999, 16, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, S.; Miyazaki, T.; Aso, Y.; Kawanishi, T. Significance of local mobility in aggregation of β-galactosidase lyophilized with trehalose, sucrose or stachyose. Pharm. Res. 2007, 24, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Suihko, E.J.; Forbes, R.T.; Apperley, D.C. A solid-state NMR study of molecular mobility and phase separation in co-spray-dried protein–sugar particles. Eur. J. Pharm. Sci. 2005, 25, 105–112. [Google Scholar] [CrossRef]
- Lam, Y.H.; Bustami, R.; Phan, T.; Chan, H.K.; Separovic, F. A solid-state NMR study of protein mobility in lyophilized protein-sugar powders. J. Pharm. Sci. 2002, 91, 943–951. [Google Scholar] [CrossRef]
- Separovic, F.; Lam, Y.H.; Ke, X.; Chan, H.K. A solid-state NMR study of protein hydration and stability. Pharm. Res. 1998, 15, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, S. Molecular Mobility of freeze-dried formulations as determined by NMR relaxation, and its effect on storage stability. In Freeze-Drying/Lyophilization of Bharmaceutical and Biological Products; Taylor & Francis Group: London, UK, 2004; ISBN 9780203021323. [Google Scholar]
- Yoshioka, S.; Aso, Y.; Nakai, Y.; Kojima, S. Effect of high molecular mobility of poly(vinyl alcohol) on protein stability of lyophilized γ-globulin formulations. J. Pharm. Sci. 1998, 87, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, S.; Aso, Y.; Kojima, S. Softening temperature of lyophilized bovine serum albumin and γ- globulin as measured by spin-spin relaxation time of protein protons. J. Pharm. Sci. 1997, 86, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Moghadam, T.T.; Ranjbar, B. Differential scanning calorimetry techniques: Applications in biology and nanoscience. J. Biomol. Tech. 2010, 21, 167–193. [Google Scholar]
- Pikal, M.J.; Rigsbee, D.; Roy, M.L. Solid state stability of proteins III: Calorimetric (DSC) and spectroscopic (FTIR) characterization of thermal denaturation in freeze dried human growth hormone (hGH). J. Pharm. Sci. 2008, 97, 5122–5131. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, J. Thermal Analysis of Drugs and Drug Products. Encycl. Pharm. Technol. 2019, 304–329. [Google Scholar] [CrossRef]
- Matheus, S.; Friess, W.; Mahler, H.-C. FTIR and nDSC as Analytical Tools for High-Concentration Protein Formulations. Pharm. Res. 2006, 23, 1350–1363. [Google Scholar] [CrossRef]
- Han, Y.; Jin, B.S.; Lee, S.B.; Sohn, Y.; Joung, J.W.; Lee, J.H. Effects of sugar additives on protein stability of recombinant human serum albumin during lyophilization and storage. Arch. Pharm. Res. 2007, 30, 1124–1131. [Google Scholar] [CrossRef]
- Ihnat, P.M.; Vellekamp, G.; Obenauer-Kutner, L.J.; Duan, J.; Han, M.A.; Witchey-Lakshmanan, L.C.; Grace, M.J. Comparative thermal stabilities of recombinant adenoviruses and hexon protein. Biochim. Biophys. Acta Gen. Subj. 2005, 1726, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Davidson, P.; Sun, W.Q. Effect of sucrose/raffinose mass ratios on the stability of co-lyophilized protein during storage above the Tg. Pharm. Res. 2001, 18, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Strickley, R.G.; Anderson, B.D. Solid-state stability of human insulin II. Effect of water on reactive intermediate partitioning in lyophiles from pH 2-5 solutions: Stabilization against covalent dimer formation. J. Pharm. Sci. 1997, 86, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Darrington, R.T.; Anderson, B.D. The Role of Intramolecular Nucleophilic Catalysis and the Effects of Self-Association on the Deamidation of Human Insulin at Low pH. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1994, 11, 784–793. [Google Scholar] [CrossRef]
- Craig, D.Q.M.; Royall, P.G.; Kett, V.L.; Hopton, M.L. The relevance of the amorphous state to pharmaceutical dosage forms: Glassy drugs and freeze dried systems. Int. J. Pharm. 1999, 179, 179–207. [Google Scholar] [CrossRef] [Green Version]
- Höhne, G.W.H.; Hemminger, W.F.; Flammersheim, H.-J. Differential Scanning Calorimetry; Springer: Berlin/Heidelberg, Germany, 2003; ISBN 978-3-642-05593-5. [Google Scholar]
- Breen, E.D.; Curley, J.G.; Overcashier, D.E.; Hsu, C.C.; Shire, S.J. Effect of moisture on the stability of a lyophilized humanized monoclonal antibody formulation. Pharm. Res. 2001, 18, 1345–1353. [Google Scholar] [CrossRef]
- Duru, C.; Swann, C.; Dunleavy, U.; Mulloy, B.; Matejtschuk, P. The importance of formulation in the successful lyophilization of influenza reference materials. Biologicals 2015, 43, 110–116. [Google Scholar] [CrossRef] [Green Version]
- El Moznine, R.; Smith, G.; Polygalov, E.; Suherman, P.M.; Broadhead, J. Dielectric properties of residual water in amorphous lyophilized mixtures of sugar and drug. J. Phys. D Appl. Phys. 2003, 36, 330–335. [Google Scholar] [CrossRef]
- Searles, J.A.; Carpenter, J.F.; Randolph, T.W. Annealing to optimize the primary drying rate, reduce freezing-induced drying rate heterogeneity, and determine T′g pharmaceutical lyophilization. J. Pharm. Sci. 2001, 90, 872–887. [Google Scholar] [CrossRef]
- Chang, L.L.; Milton, N.; Rigsbee, D.; Mishra, D.S.; Tang, X.C.; Thomas, L.C.; Pikal, M.J. Using modulated DSC to investigate the origin of multiple thermal transitions in frozen 10% sucrose solutions. Thermochim. Acta 2006, 444, 141–147. [Google Scholar] [CrossRef]
- Moussa, E.M.; Singh, S.K.; Kimmel, M.; Nema, S.; Topp, E.M. Probing the Conformation of an IgG1 Monoclonal Antibody in Lyophilized Solids Using Solid-State Hydrogen-Deuterium Exchange with Mass Spectrometric Analysis (ssHDX-MS). Mol. Pharm. 2018, 15, 356–368. [Google Scholar] [CrossRef]
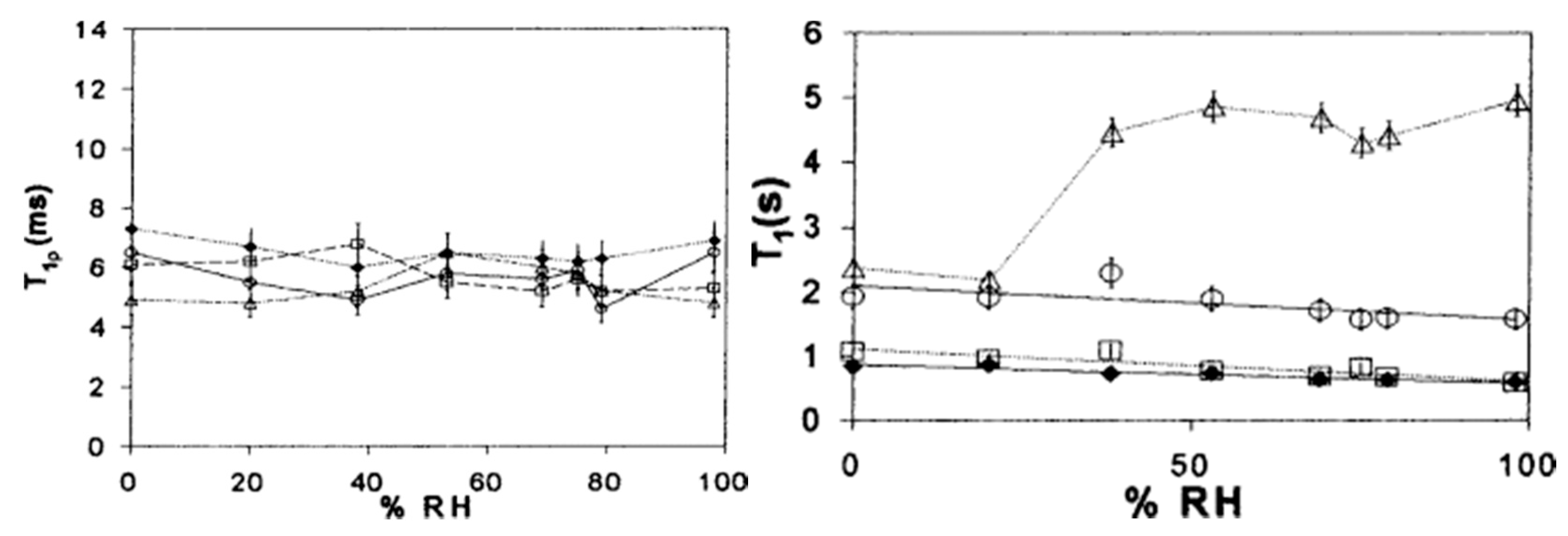
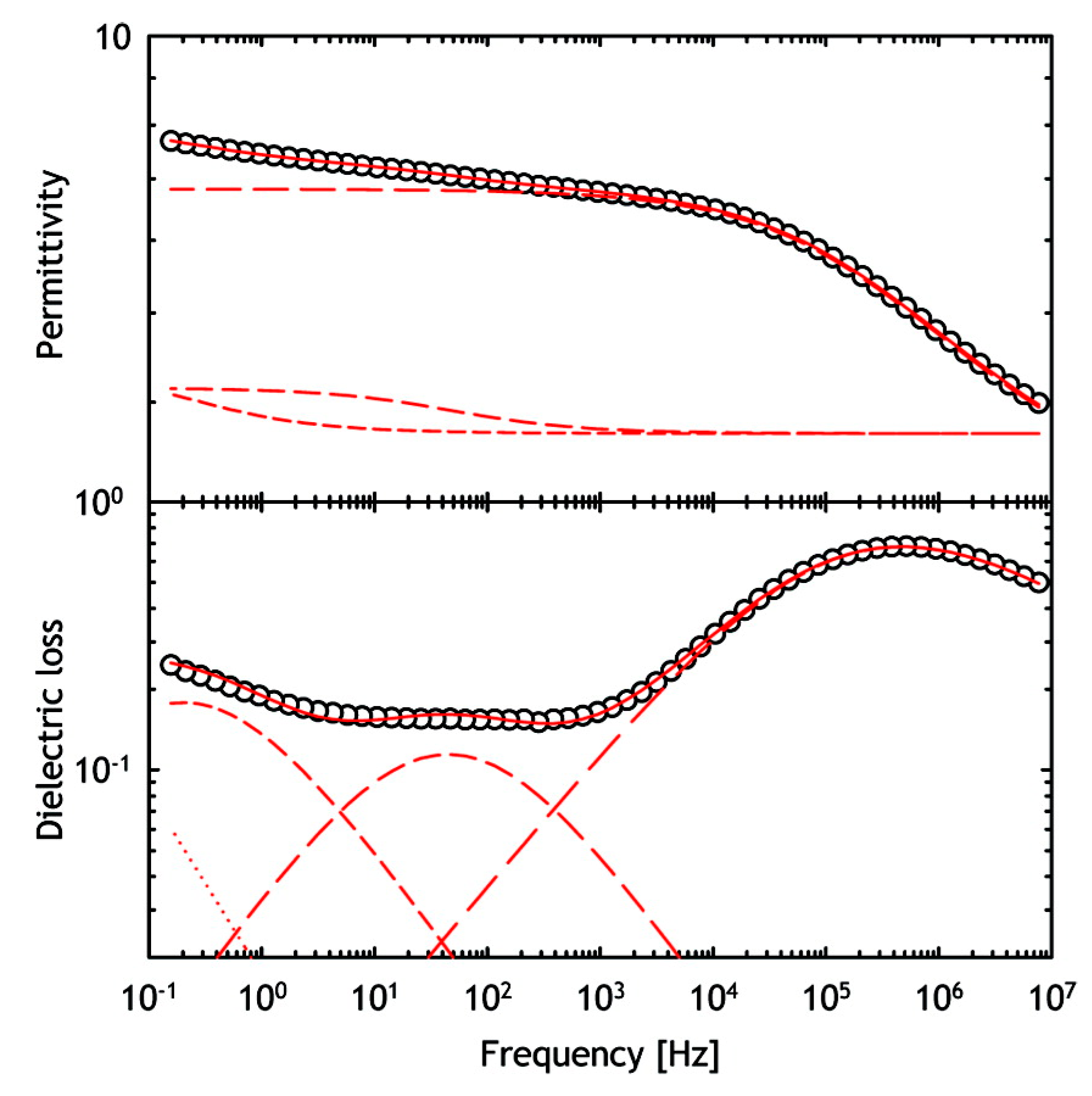
- Pearson, D.S.; Smith, G. Dielectric analysis as a tool for investigating the lyophilization of proteins. Pharm. Sci. Technol. Today 1998, 1, 108–117. [Google Scholar] [CrossRef]
- Duddu, S.P.; Sokoloski, T.D. Dielectric analysis in the characterization of amorphous pharmaceutical solids. 1. Molecular mobility in poly(vinylpyrrolidone)–water systems in the glassy state. J. Pharm. Sci. 1995, 84, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Schirò, G.; Cupane, A.; Vitrano, E.; Bruni, F. Dielectric relaxations in confined hydrated myoglobin. J. Phys. Chem. B 2009, 113, 9606–9613. [Google Scholar] [CrossRef] [PubMed]
- Jansson, H.; Swenson, J. The protein glass transition as measured by dielectric spectroscopy and differential scanning calorimetry. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, G.; Duffy, A.P.; Shen, J.; Olliff, C.J. Dielectric relaxation spectroscopy and some applications in the pharmaceutical sciences. J. Pharm. Sci. 1995, 84, 1029–1044. [Google Scholar] [CrossRef] [PubMed]
- Khodadadi, S.; Pawlus, S.; Sokolov, A.P. Influence of hydration on protein dynamics: Combining dielectric and neutron scattering spectroscopy data. J. Phys. Chem. B 2008, 112, 14273–14280. [Google Scholar] [CrossRef] [PubMed]
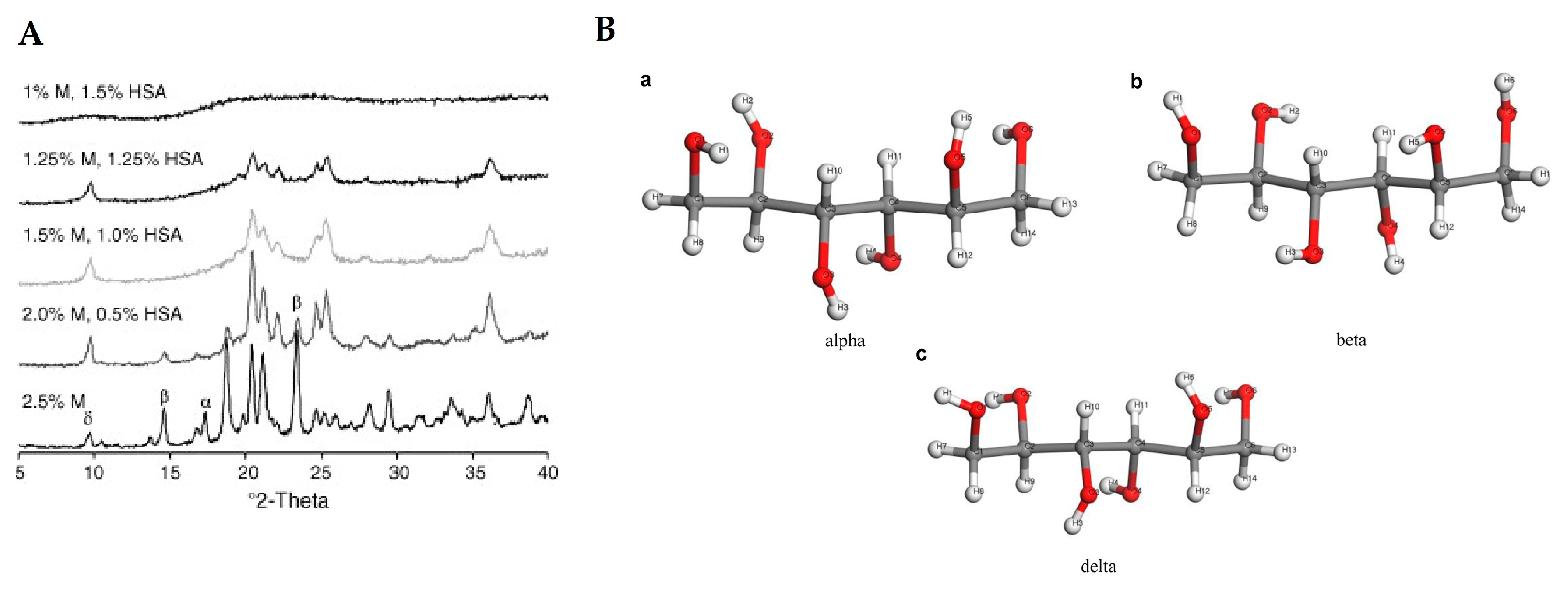
- Saleki-Gerhardt, A.; Ahlneck, C.; Zografi, G. Assessment of disorder in crystalline solids. Int. J. Pharm. 1994, 101, 237–247. [Google Scholar] [CrossRef]
- Shah, B.; Kakumanu, V.K.; Bansal, A.K. Analytical techniques for quantification of amorphous/crystalline phases in pharmaceutical solids. J. Pharm. Sci. 2006, 95, 1641–1665. [Google Scholar] [CrossRef] [PubMed]
- Young, A.L. Powder X-ray diffraction and its application to biotherapeutic formulation development. Am. Pharm. Rev. 2012, 15, 1–10. [Google Scholar]
- Shalaev, E.Y. The impact of buffer on processing and stability of freeze-dried dosage forms, part 1: Solution freezing behavior. Am. Pharm. Rev. 2005, 8, 80–87. [Google Scholar]
- Izutsu, K.I.; Kadoya, S.; Yomota, C.; Kawanishi, T.; Yonemochi, E.; Terada, K. Stabilization of protein structure in freeze-dried amorphous organic acid buffer salts. Chem. Pharm. Bull. 2009, 57, 1231–1236. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.E.; Kirchhoff, C.F.; Gaud, H.T. Mannitol-sucrose mixtures—Versatile formulations for protein lyophilization. J. Pharm. Sci. 2002, 91, 914–922. [Google Scholar] [CrossRef]
- Nunes, C.; Suryanarayanan, R.; Botez, C.E.; Stephens, P.W. Characterization and crystal structure of D-mannitol hemihydrate. J. Pharm. Sci. 2004, 93, 2800–2809. [Google Scholar] [CrossRef]
- Pitkänen, I.; Perkkalainen, P.; Rautiainen, H. Thermoanalytical studies on phases of D-mannitol. Thermochim. Acta 1993, 214, 157–162. [Google Scholar] [CrossRef]
- Telang, C.; Suryanarayanan, R.; Yu, L. Crystallization of D-Mannitol in Binary Mixtures with NaCl: Phase Diagram and Polymorphism. Pharm. Res. 2003, 20, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Hawe, A.; Frieß, W. Physico-chemical lyophilization behavior of mannitol, human serum albumin formulations. Eur. J. Pharm. Sci. 2006, 28, 224–232. [Google Scholar] [CrossRef]
- Su, W.; Zhang, Y.; Liu, J.; Ma, M.; Guo, P.; Liu, X.; Wang, H.; Li, C. Molecular Dynamic Simulation of D-Mannitol Polymorphs in Solid State and in Solution Relating With Spontaneous Nucleation. J. Pharm. Sci. 2020, 109, 1537–1546. [Google Scholar] [CrossRef]
- Norrman, M.; Hubálek, F.; Schluckebier, G. Structural characterization of insulin NPH formulations. Eur. J. Pharm. Sci. 2007, 30, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Smith, D.L. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 1993, 2, 522–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, L.; Pan, J.; Liu, Y.H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 2011, 40, 1224–1234. [Google Scholar] [CrossRef]
- Tsutsui, Y.; Wintrode, P. Hydrogen/Deuterium Exchange-Mass Spectrometry: A Powerful Tool for Probing Protein Structure, Dynamics and Interactions. Curr. Med. Chem. 2007, 14, 2344–2358. [Google Scholar] [CrossRef] [PubMed]
- Engen, J.R. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal. Chem. 2009, 81, 7870–7875. [Google Scholar] [CrossRef] [Green Version]
- Kaltashov, I.A.; Bobst, C.E.; Abzalimov, R.R. H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: Is there a need for a top-down approach? Anal. Chem. 2009, 81, 7892–7899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, M.M.G.; Hoang, L.; Lin, Y.; Englander, S.W. Hydrogen exchange methods to study protein folding. Methods 2004, 34, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Wales, T.E.; Engen, J.R. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006, 25, 158–170. [Google Scholar] [CrossRef]
- Rosa, J.J.; Richards, F.M. An experimental procedure for increasing the structural resolution of chemical hydrogen-exchange measurements on proteins: Application to ribonuclease S peptide. J. Mol. Biol. 1979, 133, 399–416. [Google Scholar] [CrossRef]
- Woods, V.L.; Hamuro, Y. High resolution, high-throughput amide deuterium exchange-mass spectrometry (DXMS) determination of protein binding site structure and dynamics: Utility in pharmaceutical design. J. Cell. Biochem. 2001, 84, 89–98. [Google Scholar] [CrossRef] [PubMed]
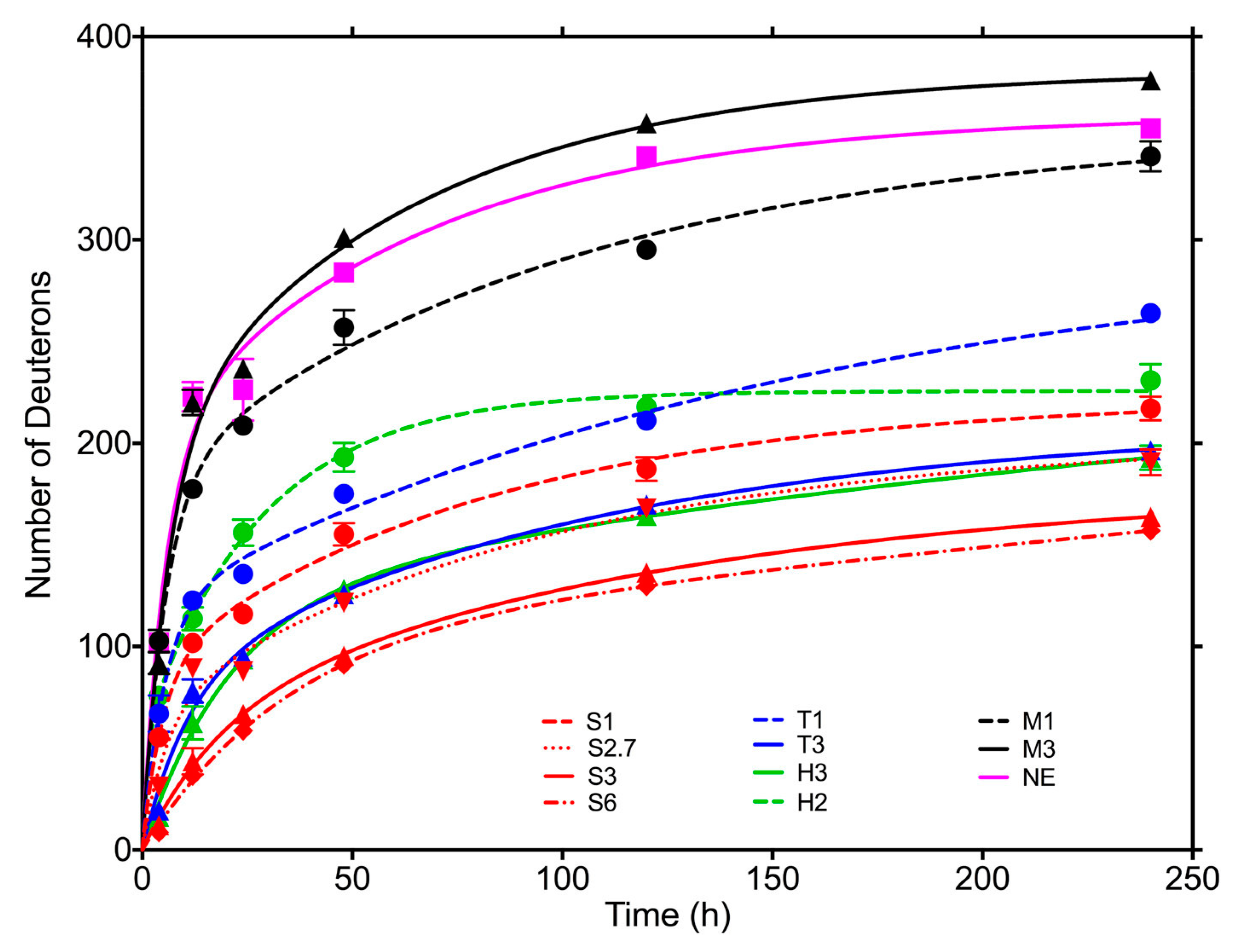
- Moorthy, B.S.; Zarraga, I.E.; Kumar, L.; Walters, B.T.; Goldbach, P.; Topp, E.M.; Allmendinger, A. Solid-State Hydrogen-Deuterium Exchange Mass Spectrometry: Correlation of Deuterium Uptake and Long-Term Stability of Lyophilized Monoclonal Antibody Formulations. Mol. Pharm. 2018, 15, 1–11. [Google Scholar] [CrossRef]
- Li, Y.; Williams, T.D.; Schowen, R.L.; Topp, E.M. Trehalose and calcium exert site-specific effects on calmodulin conformation in Amorphous solids. Biotechnol. Bioeng. 2007, 97, 1650–1653. [Google Scholar] [CrossRef]
- Li, Y.; Williams, T.D.; Schowen, R.L.; Topp, E.M. Characterizing protein structure in amorphous solids using hydrogen/deuterium exchange with mass spectrometry. Anal. Biochem. 2007, 366, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Li, Y.; Williams, T.D.; Topp, E.M. Protein conformation in amorphous solids by FTIR and by hydrogen/deuterium exchange with mass spectrometry. Biophys. J. 2008, 95, 5951–5961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moorthy, B.S.; Iyer, L.K.; Topp, E.M. Mass Spectrometric Approaches to Study Protein Structure and Interactions in Lyophilized Powders. J. Vis. Exp. 2015, 98, 52503. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, R.; Middaugh, C.R.; Weis, D.D.; Volkin, D.B. Hydrogen-Deuterium Exchange Mass Spectrometry as an Emerging Analytical Tool for Stabilization and Formulation Development of Therapeutic Monoclonal Antibodies. J. Pharm. Sci. 2015, 104, 327–345. [Google Scholar] [CrossRef]
- French, D.L.; Arakawa, T.; Li, T. Fourier Transformed Infrared Spectroscopic Investigation of Protein Conformation in Spray-Dried Protein/Trehalose Powders. Biopolymers 2004, 73, 524–531. [Google Scholar] [CrossRef]
- Desai, U.R.; Klibanoy, A.M.; Osterhout, J.J. Protein Structure in the Lyophilized State: A Hydrogen Isotope Exchange/NMR Study with Bovine Pancreatic Trypsin Inhibitor. J. Am. Chem. Soc. 1994, 116, 9420–9422. [Google Scholar] [CrossRef]
- Sophocleous, A.M.; Zhang, J.; Topp, E.M. Localized Hydration in Lyophilized Myoglobin by Hydrogen–Deuterium Exchange Mass Spectrometry. 1. Exchange Mapping. Mol. Pharm. 2012, 9, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Sophocleous, A.M.; Topp, E.M. Localized hydration in lyophilized myoglobin by hydrogen-deuterium exchange mass spectrometry. 2. Exchange kinetics. Mol. Pharm. 2012, 9, 727–733. [Google Scholar] [CrossRef] [Green Version]
- Wilson, N.E.; Topp, E.M.; Zhou, Q.T. Effects of drying method and excipient on structure and stability of protein solids using solid-state hydrogen/deuterium exchange mass spectrometry (ssHDX-MS). Int. J. Pharm. 2019, 567. [Google Scholar] [CrossRef]
- Beck, A.; Wagner-Rousset, E.; Ayoub, D.; Van Dorsselaer, A.; Sanglier-Cianférani, S. Characterization of therapeutic antibodies and related products. Anal. Chem. 2013, 85, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.C.; Patel, K.; Borchardt, R.T. Stability of Protein Pharmaceuticals. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1989, 6, 903–918. [Google Scholar] [CrossRef]
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of protein pharmaceuticals: An update. Pharm. Res. 2010, 27, 544–575. [Google Scholar] [CrossRef] [PubMed]
- Mahler, H.C.; Müller, R.; Frieß, W.; Delille, A.; Matheus, S. Induction and analysis of aggregates in a liquid IgG1-antibody formulation. Eur. J. Pharm. Biopharm. 2005, 59, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Hong, P.; Koza, S.; Bouvier, E.S.P. A review size-exclusion chromatography for the analysis of protein biotherapeutics and their aggregates. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 2923–2950. [Google Scholar] [CrossRef] [Green Version]
- Den Engelsman, J.; Garidel, P.; Smulders, R.; Koll, H.; Smith, B.; Bassarab, S.; Seidl, A.; Hainzl, O.; Jiskoot, W. Strategies for the assessment of protein aggregates in pharmaceutical biotech product development. Pharm. Res. 2011, 28, 920–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Liu, D.; Liu, H.; Motchnik, P. Characterization of monoclonal antibody size variants containing extra light chains. MAbs 2013, 5, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Tang, Q.; Cronin, B.; Markovich, R.; Rustum, A. Development of a high performance size exclusion chromatography method to determine the stability of Human Serum Albumin in a lyophilized formulation of Interferon alfa-2b. J. Chromatogr. A 2008, 1194, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Ejima, D.; Yumioka, R.; Arakawa, T.; Tsumoto, K. Arginine as an effective additive in gel permeation chromatography. J. Chromatogr. A 2005, 1094, 49–55. [Google Scholar] [CrossRef]
- Goyon, A.; Fekete, S.; Beck, A.; Veuthey, J.L.; Guillarme, D. Unraveling the mysteries of modern size exclusion chromatography—The way to achieve confident characterization of therapeutic proteins. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1092, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tchessalov, S.; Cicerone, M.T.; Warne, N.W.; Pikal, M.J. Impact of sucrose level on storage stability of proteins in freeze-dried solids: II. Correlation of aggregation rate with protein structure and molecular mobility. J. Pharm. Sci. 2009, 98, 3145–3166. [Google Scholar] [CrossRef]
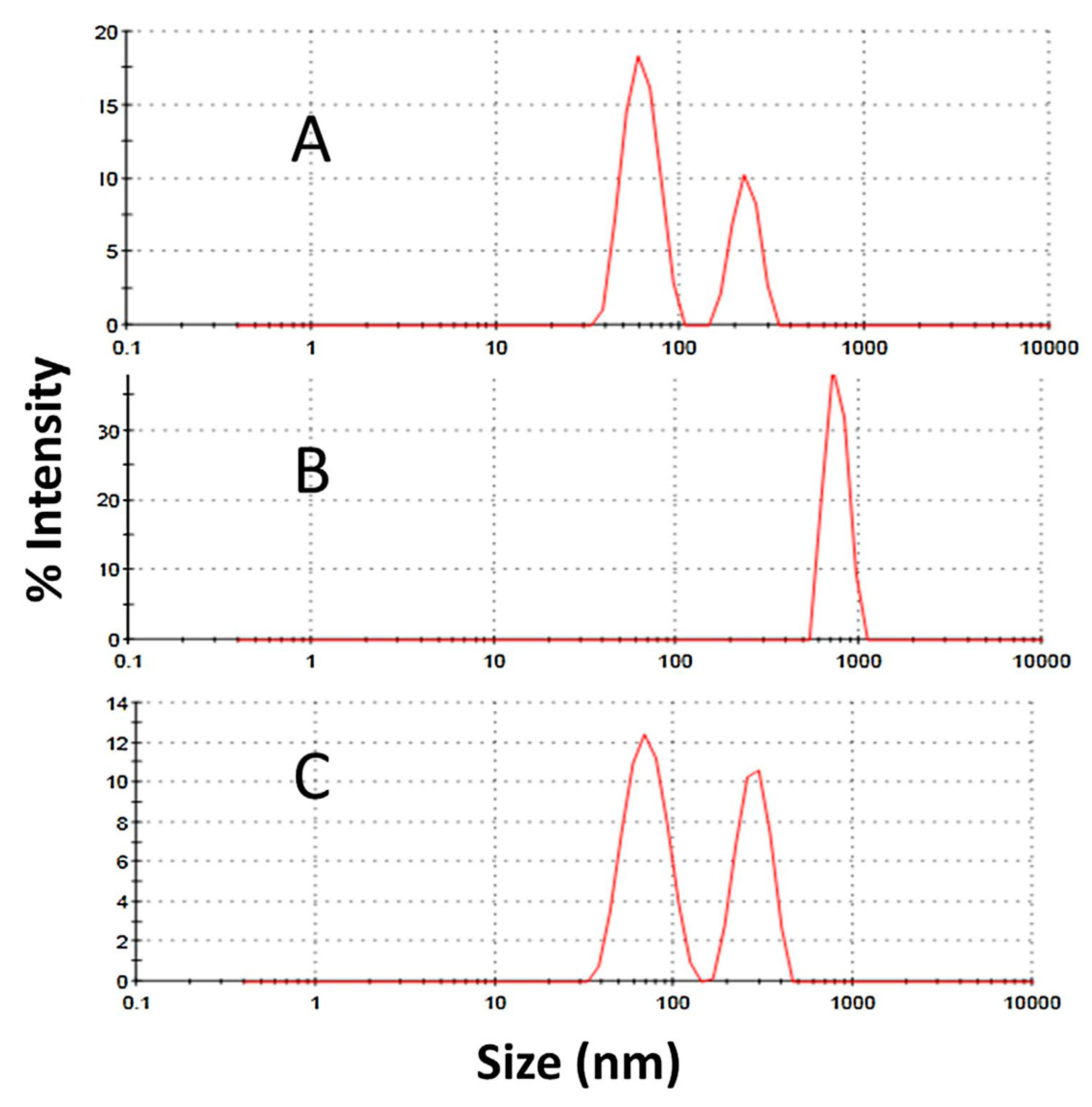
- Al-Ghobashy, M.A.; Mostafa, M.M.; Abed, H.S.; Fathalla, F.A.; Salem, M.Y. Correlation between Dynamic Light Scattering and Size Exclusion High Performance Liquid Chromatography for Monitoring the Effect of pH on Stability of Biopharmaceuticals. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1060, 1–9. [Google Scholar] [CrossRef]
- Carpenter, D.K. Dynamic Light Scattering with Applications to Chemistry, Biology, and Physics (Berne, Bruce J.; Pecora, Robert). J. Chem. Educ. 1977, 54, A430. [Google Scholar] [CrossRef] [Green Version]
- Nobbmann, U.; Connah, M.; Fish, B.; Varley, P.; Gee, C.; Mulot, S.; Chen, J.; Zhou, L.; Lu, Y.; Sheng, F.; et al. Dynamic light scattering as a relative tool for assessing the molecular integrity and stability of monoclonal antibodies. Biotechnol. Genet. Eng. Rev. 2007, 24, 117–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Roberts, C.J. Protein aggregation—Mechanisms, detection, and control. Int. J. Pharm. 2018, 550, 251–268. [Google Scholar] [CrossRef]
- Arkawa, T.; Philo, J.S.; Ejima, D.; Tsumoto, K.; Arisaka, F. Aggregation analysis of therapeutic proteins, part 2. Bioprocess Int. 2007, 5, 36–47. [Google Scholar]
- Muthukutty, P.; Kato, T.; Park, E.Y. Production of human papillomavirus 6b L1 virus-like particles incorporated with enhanced green fluorescent whole protein in silkworm larvae. Biotechnol. Bioprocess Eng. 2013, 18, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Cohan, R.A.; Madadkar-Sobhani, A.; Khanahmad, H.; Roohvand, F.; Aghasadeghi, M.R.; Hedayati, M.H.; Barghi, Z.; Ardestani, M.S.; Inanlou, D.N.; Norouzian, D. Design, modeling, expression, and chemoselective PEGylation of a new nanosize cysteine analog of erythropoietin. Int. J. Nanomed. 2011, 6, 1217. [Google Scholar] [CrossRef] [Green Version]
- Cleland, J.L.; Wang, D.I.C. Transient Association of the First Intermediate during the Refolding of Bovine Carbonic Anhydrase B. Biotechnol. Prog. 1992, 8, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Hammarström, P.; Persson, M.; Freskgård, P.O.; Mårtensson, L.G.; Andersson, D.; Jonsson, B.H.; Carlsson, U. Structural mapping of an aggregation nucleation site in a molten globule intermediate. J. Biol. Chem. 1999, 274, 32897–32903. [Google Scholar] [CrossRef] [Green Version]
- Tomski, S.J.; Murphy, R.M. Kinetics of aggregation of synthetic β-amyloid peptide. Arch. Biochem. Biophys. 1992, 294, 630–638. [Google Scholar] [CrossRef]
- Lomakin, A.; Teplow, D.B.; Kirschner, D.A.; Benedeki, G.B. Kinetic theory of fibrillogenesis of amyloid β-protein. Proc. Natl. Acad. Sci. USA 1997, 94, 7942–7947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawe, A.; Friess, W. Development of HSA-free formulations for a hydrophobic cytokine with improved stability. Eur. J. Pharm. Biopharm. 2008, 68, 169–182. [Google Scholar] [CrossRef]
- Ahrer, K.; Buchacher, A.; Iberer, G.; Jungbauer, A. Thermodynamic stability and formation of aggregates of human immunoglobulin G characterised by differential scanning calorimetry and dynamic light scattering. J. Biochem. Biophys. Methods 2006, 66, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.A.; An, I.B.; Lim, D.G.; Lim, J.Y.; Lee, S.Y.; Shim, W.S.; Kang, N.-G.; Jeong, S.H. Effects of pH and Buffer Concentration on the Thermal Stability of Etanercept Using DSC and DLS. Biol. Pharm. Bull. 2014, 37, 808–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumar, M.; Doyle, B.L.; Combs, J.L.; Pekar, A.H. Opalescent appearance of an IgG1 antibody at high concentrations and its relationship to noncovalent association. Pharm. Res. 2004, 21, 1087–1093. [Google Scholar] [CrossRef] [PubMed]










 —trehalose;
—trehalose;  —80% trehalose, 20% lysozyme; □—20% trehalose, 80% lysozyme;
—80% trehalose, 20% lysozyme; □—20% trehalose, 80% lysozyme;  —lysozyme. Reproduced with permission from [116], Elsevier, 2002.
—trehalose; —80% trehalose, 20% lysozyme; □—20% trehalose, 80% lysozyme; —lysozyme. Reproduced with permission from [116], Elsevier, 2002.
—lysozyme. Reproduced with permission from [116], Elsevier, 2002.
—trehalose; —80% trehalose, 20% lysozyme; □—20% trehalose, 80% lysozyme; —lysozyme. Reproduced with permission from [116], Elsevier, 2002.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
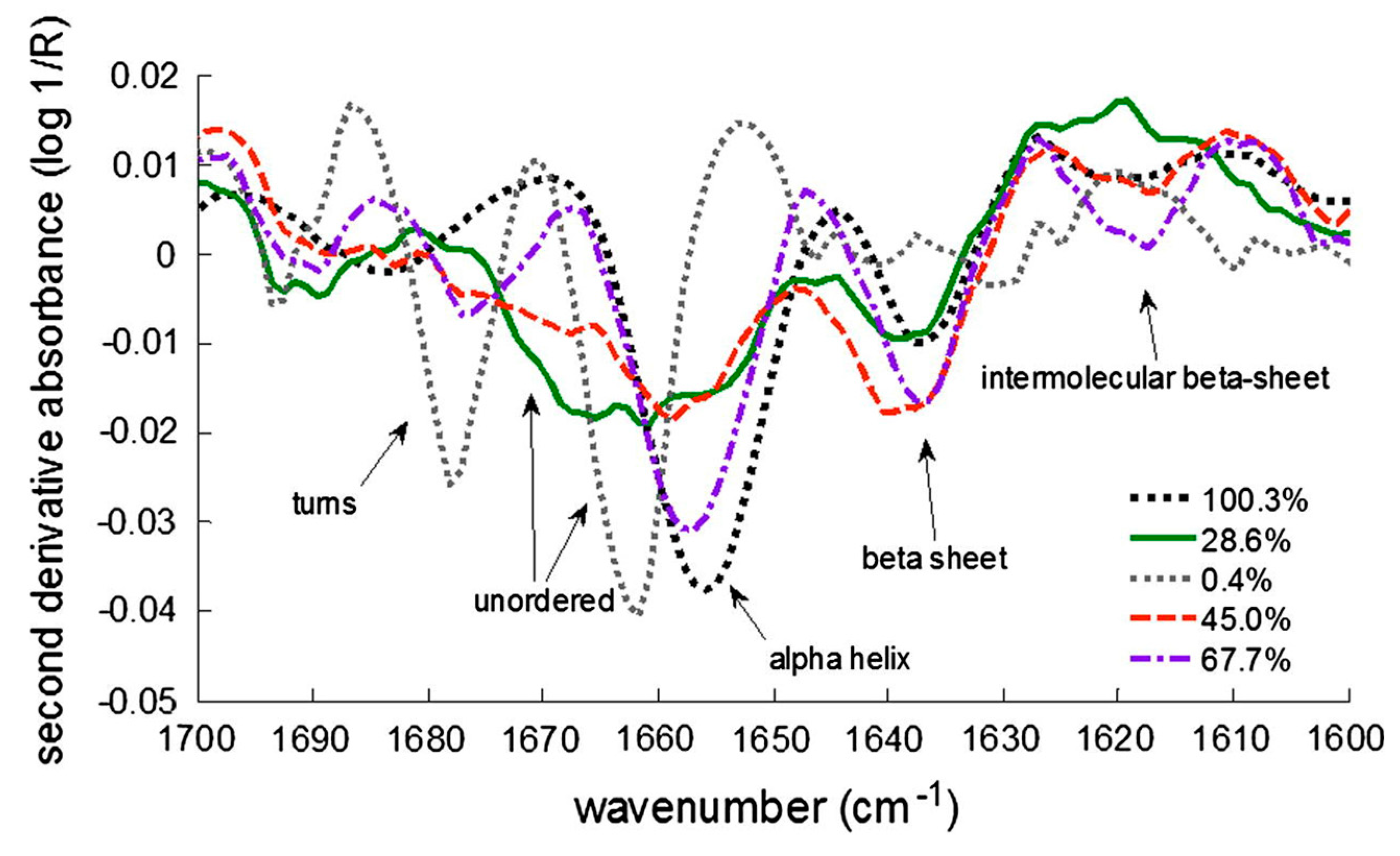
| Frequency of the Band (cm−1) | Amide Region | Vibrations | Type of 2nd Structure |
|---|---|---|---|
| 1680 1670–1680 1650–1655 1640 | Amide I | H-bonded C=O stretch | β-Turn |
| β-Sheet and β-barrel | |||
| α-Helix | |||
| Loose β-sheet | |||
| 1300–1340 | Amide III | N–H and C–H bend | α-Helix |
| 1260 | Disordered | ||
| 1235–1250 | β-Sheet | ||
| 930–950 | Backbone | N–Cα–C stretch | α-Helix |
| Formulations | pH | Sucrose/mAb Ratio (w/w) | Histidine/mAb Ratio (w/w) | Moisture Content | Tg |
|---|---|---|---|---|---|
| 5H2 | 5 | 2:1 | 2.82 ± 1.31 | 94 | |
| 5S1 | 5 | 1:1 | 2.72 ± 0.24 | ND | |
| 5S1H0.5 | 5 | 1:1 | 0.5:1 | 1.67 ± 0.77 | 90 |
| 5S1H1 | 5 | 1:1 | 1:1 | 2.57 ± 0.01 | 90 |
| 6H2 | 6 | 2:1 | 2.79 ± 0.67 | 106 | |
| 6S1 | 6 | 1:1 | 2.01 ± 0.22 | 93 | |
| 6S1H0.5 | 6 | 1:1 | 0.5:1 | 1.60 ± 0.39 | 95 |
| 6S1H1 | 6 | 1:1 | 1:1 | 1.95 ± 0.38 | 101 |
| H2 | 6.8 | 2:1 | 0.99 ± 0.34 | 105 | |
| S1 | 6.8 | 1:1 | 1.10 ± 0.03 | ND | |
| S1H0.5 | 6.8 | 1:1 | 0.5:1 | 2.55 ± 0.13 | 94 |
| S1H1 | 6.8 | 1:1 | 1:1 | 2.12 ± 0.76 | 98 |
| Method | Physical Principles | Sensitivity | Potential Damages | Pros | Cons |
|---|---|---|---|---|---|
| FTIR | C=O, N–H and C–N vibrations are measured as absorption or emission due to infrared light | Low to medium (on a global level) | Protein can be damage if too much pressure is applied when analyzing in ATR mode | Fast measurement, easy setup, non-expensive equipment, no hazardous chemicals are used, with ATR mode the sample can be recovered, small amount of sample needed | Only secondary structure can be evaluated, if KBr pellet mode is used, the sample cannot be recover, only analysis on a global level can be done and cannot provide site-specific information on specific portions of the protein sequence and their interactions with excipients, often unable to detect subtle structural differences |
| NIR | C=O, N–H and C–N vibrations are measured in the near-infrared region | Low to medium (on a global level) | No damages | Fast analysis, small amount of sample needed, no inert gas purging, easy setup and non-expensive equipment | Only secondary structure on a global level can be evaluated, water can interfere within protein spectra in some cases, cannot provide site-specific information on specific portions of the protein sequence and their interactions with excipients, often unable to detect subtle structural differences |
| Raman | C=O, N–H and C–N vibrations are measured as inelastic scattering after light excitation | Medium to high (on a global level) | Samples are usually damaged due to laser light irradiation | Very small amount of sample needed, | Samples cannot be recovered, longer time needed for measurements, more difficult equipment setup, only global level analysis |
| UV–Vis | Displacement of absorption (of UV or visible light) peaks is measured | Low to medium (on a global level) | No damages | Easy and non-expensive equipment setup, samples can be recovered, fast analysis | Only global level analysis of tertiary structure, cannot provide site-specific information on specific portions of the protein sequence and their interactions with excipients, often unable to detect subtle structural differences |
| Fluorescence | Emission of residual aromatic amino acids is measured after absorption of light or electromagnetic radiation | Medium (on a local level) | No damages | Tertiary structure on a local level, intensity and peak maxima position can be measured | Higher amount of sample needed, sample preparation for measurement is crucial |
| CD | Difference in absorbance is measured, involving circularly polarized light (left- and right-handed light) | Medium (on a global level) | No damage (except when temperature dependence experiment is applied) | Secondary and tertiary structure can be analyzed, small amount of sample needed | Only global level analysis, if temperature dependent experiment is applied the sample cannot be recovered, nitrogen gas purging needed |
| ssNMR | 1H, 13C, 15N chemical shifts are measured after magnetic field excitation of the nuclei sample | High (on a global and local level) | No damage | Conformation and dynamics can be measured on a global and local level, different nuclei can be analyzed (proton, carbon, nitrogen) | Expensive equipment, long measurement time, higher amounts of sample are needed, only in some cases the samples can be recovered |
| DSC | Change in heat capacity at Tg is measured (the difference in the amount of heat required to increase the temperature of a sample and reference is measured as a function of temperature) | High to medium (on a global level) | Sample is damaged and cannot be recovered | Conformational changes and crystallinity of the sample can be evaluated | Sample cannot be recovered, only global level analysis, necessary to have well-characterized drug compounds |
| DRS | Rational motions of dipole-bearing groups are measured (as a function of frequency) | High to medium (on a global and local level) | Sample can be damaged | Global and local analysis | Sample usually cannot be recovered, higher amounts of sample are needed, difficult sample preparation, water may interfere with the sample analysis |
| XRD | Crystal structure is evaluated by irradiating the sample material with X-rays and measuring the intensities and scattering angles that leave the material | High (on a global level) | Samples can be damaged due to X-ray irradiation | Conformation and crystallinity of samples can be evaluated | Only global level analysis, sample usually cannot be recovered, expensive equipment |
| ssHDX-MS | Amide hydrogen exchange with deuterium in solid (by exposure to D2O) is measured with LC–MS | High (on a global and local level) | Samples are damaged due to deuteration and MS analysis | Very good correlation with aggregates formation and physical stability on storage, when peptide digestion is employed the samples can be analyzed on a local level providing also site-specific information on interactions between the protein and excipients | Samples cannot be recovered, difficult setup and expensive equipment |
| SEC | Chromatographic method in which molecules are separated by size, and in some cases molecular weight | High (on a global level) | Can be damage in some cases by mobile phase or column | Standard method for aggregation studies, small amount of sample is needed, fast method if automation is employed, also degradation products can be detected | Expensive equipment, some particles may not be detected or separated |
| DLS | Measures the Brownian motion of macromolecules in solution that arises due to bombardment from solvent molecules, and relates this motion to the size of particles to determine their size distribution | High (on a global level) | If temperature dependence or zeta-potential is measured, samples are damaged (aggregated) | Can analyze particles that may not be seen with SEC, easy setup and non-expensive equipment, fast analysis | Cannot differentiate molecules that are closely related (monomer and dimer) since it is a low- resolution method, it must be used on highly dilute solutions, restricted to transparent samples, very sensitive to temperature and solvent viscosity |
| Method | Pros | Cons | Information | Comparison with Other Methods |
|---|---|---|---|---|
| FTIR | Secondary structure determination, samples can be recovered, small amount of sample needed. | Structure determination only on a global level, poor correlation with other methods and especially SEC. | Amide I region was analyzed for each formulation and used to compare protein secondary structure. Little difference was observed with either changes in excipient or processing conditions, with the exception of β-lactoglobulin, where for spray-dried samples an increase in the heterogeneity can be deducted. | In comparison to ssHDX-MS, the results were relatively inconsistent and poor correlation was observed with results from SEC analysis. On the other hand is the only method in this study to characterize proteins secondary structures in solid. Further, is a fast and routinely analysis. |
| Fluorescence | Tertiary structure determination, samples can be recovered. | Poorer correlation with stability studies than with the ssHXD-MS. Measurements are not possible with lower concentrations. | Changes in tertiary structure correspond to shifts in the peak. Fluorescence spectra showed process related differences for BSA, they may be attributed to hydration differences, since spray-dried samples have lower moisture content. Lysozyme samples showed significant differences in peak position that is depended on formulation and processing conditions. Mannitol-containing formulations displayed red shifts, whereas sucrose samples displayed the blue ones. No difference was observed with trehalose samples. | In comparison to ssHDX-MS has weak correlation with long-term storage stability. On the other hand is quicker and has an easy equipment setup. Similar to FTIR is the only method for tertiary structure characterization, which is relatively fast and routinely. |
| XRD | Only method with DSC for sugars crystallization analysis. | Information only on global level with no sample recovery. | Formulations containing sucrose or trehalose were all completely amorphous, whereas mannitol samples showed minor peaks on XRD, indicating the presence of crystalline mannitol. | Mannitol samples showed some crystallization, which was observed also with DSC. The mannitol crystallization might be reflected in poorer storage stability, which was confirmed with ssHDX-MS and SEC. |
| DSC | Together with XRD analyzes and confirms samples crystallinity, as well as measures samples Tg, which can be compared. | Samples cannot be recovered, the information only on a global level only. | Tg values were determined for sucrose and trehalose formulations, whereas Tm was determined for mannitol formulations. Sucrose samples had lower Tg, whereas trehalose samples showed process-dependent differences in Tg, with higher values for spray-dried samples. For samples with mannitol, the Tm confirmed crystallinity. | The results do not correlate good with storage stability measured by SEC, except for mannitol samples, which have shown to be crystalline and therefore less stable on long-term. Mannitol crystallization was observed also in XRD analysis. |
| ssHDX-MS | Good correlation with storage stability, analysis on global and local level (if peptide digestion is employed). | Expensive and rather complex equipment. Samples cannot be recovered after analysis. Longer times require for the experiments. | Mannitol formulations showed greater deuterium uptake and, hence, decreased storage stability (probably due to phase separation caused by the crystallization of the excipient), which correlates great with highest aggregate content measured by SEC. Similar results were obtained with either the deconvoluted peak area or the maximum deuterium incorporation. | In contrast to FTIR and fluorescence, ssHXD-MS gives a very good and consistent correlation with aggregation studies with SEC. On the other hand, the method requires much longer times for the analyses and it is not yet a routinely measurement. |
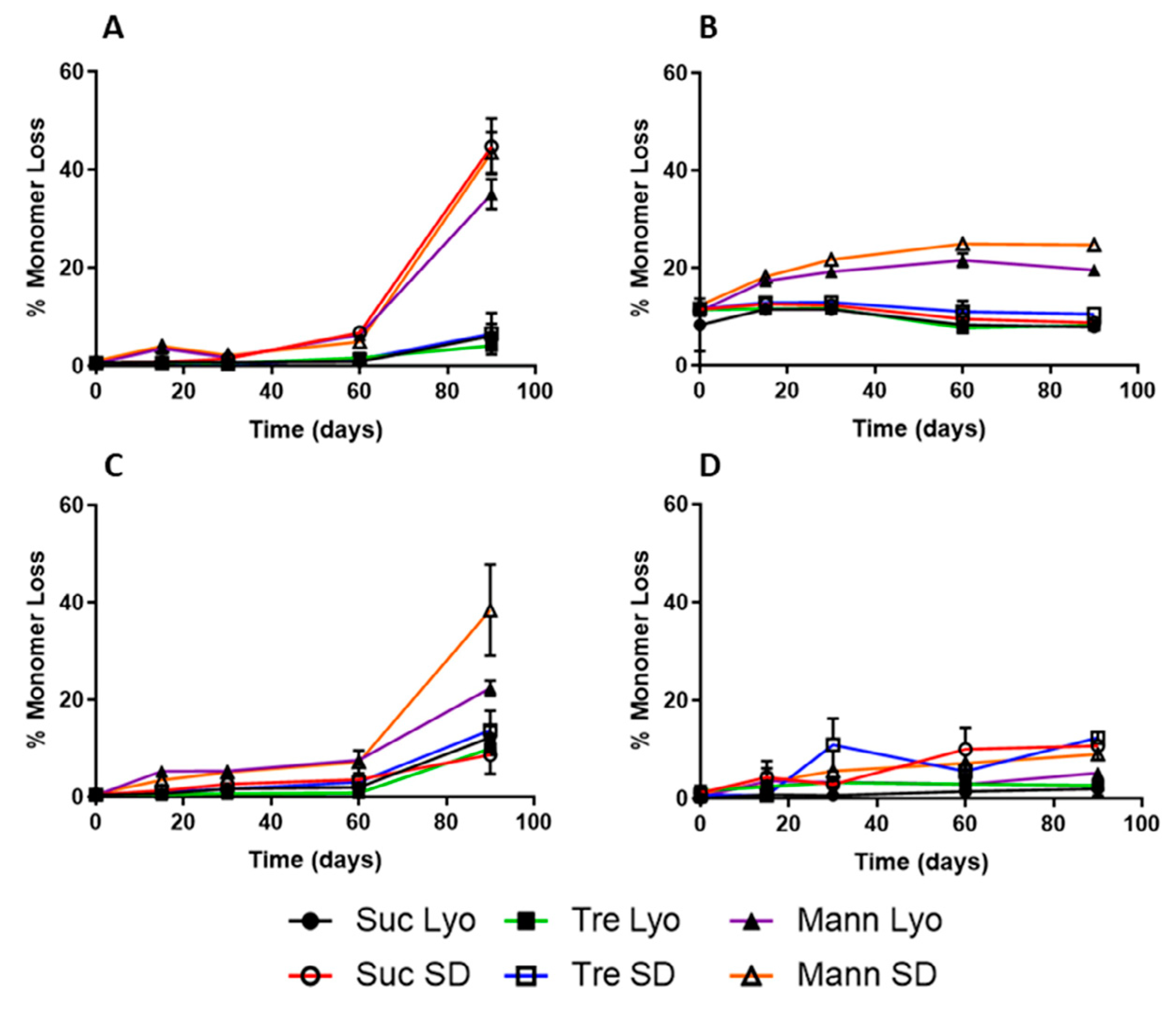
| SEC | Very good and reliable (standard) method for aggregation analysis and storage stability studies. | Measurements are done in solution—conditions only after the reconstitution of proteins; more expensive equipment. | The percentage of aggregates was greatest in mannitol formulations containing mannitol, with the exception of myoglobin spray-dried with sucrose and all formulations of lysozyme (spray-dried samples had greater aggregate content). | Provides information on long-term stability as measures the loss of monomer (aggregates formation). Despite ssHDX-MS, that can predict aggregation to certain degree due to good correlation with storage stability, SEC is the only method to really measure the extent of aggregation that occurred within the samples. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolje, A.; Gobec, S. Analytical Techniques for Structural Characterization of Proteins in Solid Pharmaceutical Forms: An Overview. Pharmaceutics 2021, 13, 534. https://doi.org/10.3390/pharmaceutics13040534
Bolje A, Gobec S. Analytical Techniques for Structural Characterization of Proteins in Solid Pharmaceutical Forms: An Overview. Pharmaceutics. 2021; 13(4):534. https://doi.org/10.3390/pharmaceutics13040534
Chicago/Turabian StyleBolje, Aljoša, and Stanislav Gobec. 2021. "Analytical Techniques for Structural Characterization of Proteins in Solid Pharmaceutical Forms: An Overview" Pharmaceutics 13, no. 4: 534. https://doi.org/10.3390/pharmaceutics13040534
APA StyleBolje, A., & Gobec, S. (2021). Analytical Techniques for Structural Characterization of Proteins in Solid Pharmaceutical Forms: An Overview. Pharmaceutics, 13(4), 534. https://doi.org/10.3390/pharmaceutics13040534