
Development of Pelubiprofen Tromethamine with Improved Gastrointestinal Safety and Absorption

 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of the PEL-T Powder
2.3. Physico-Chemical Properties of PEL and PEL-T Powder
2.4. HPLC Determination of PEL
2.5. Solubility, Dissolution, and Stress Stability Test
2.6. Permeability Test in Caco-2 Cells
2.7. Gastric Mucosal Injury Test in Rats
2.8. PK Studies in Rats
2.9. LC-MS/MS Method and PK Studies
2.10. Statistical Analysis
3. Results and Discussion
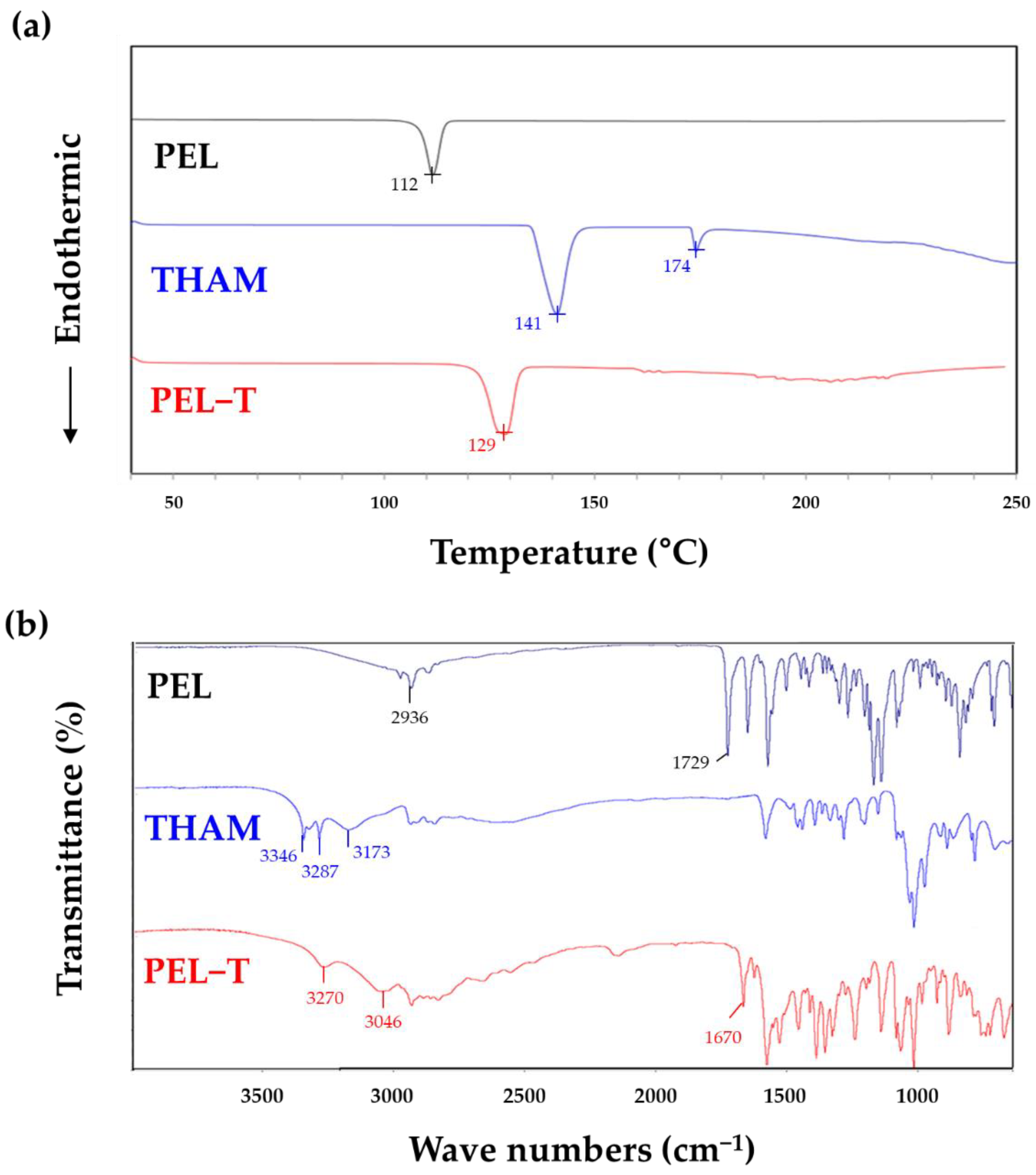
3.1. Physicochemical Characteristics of PEL-T
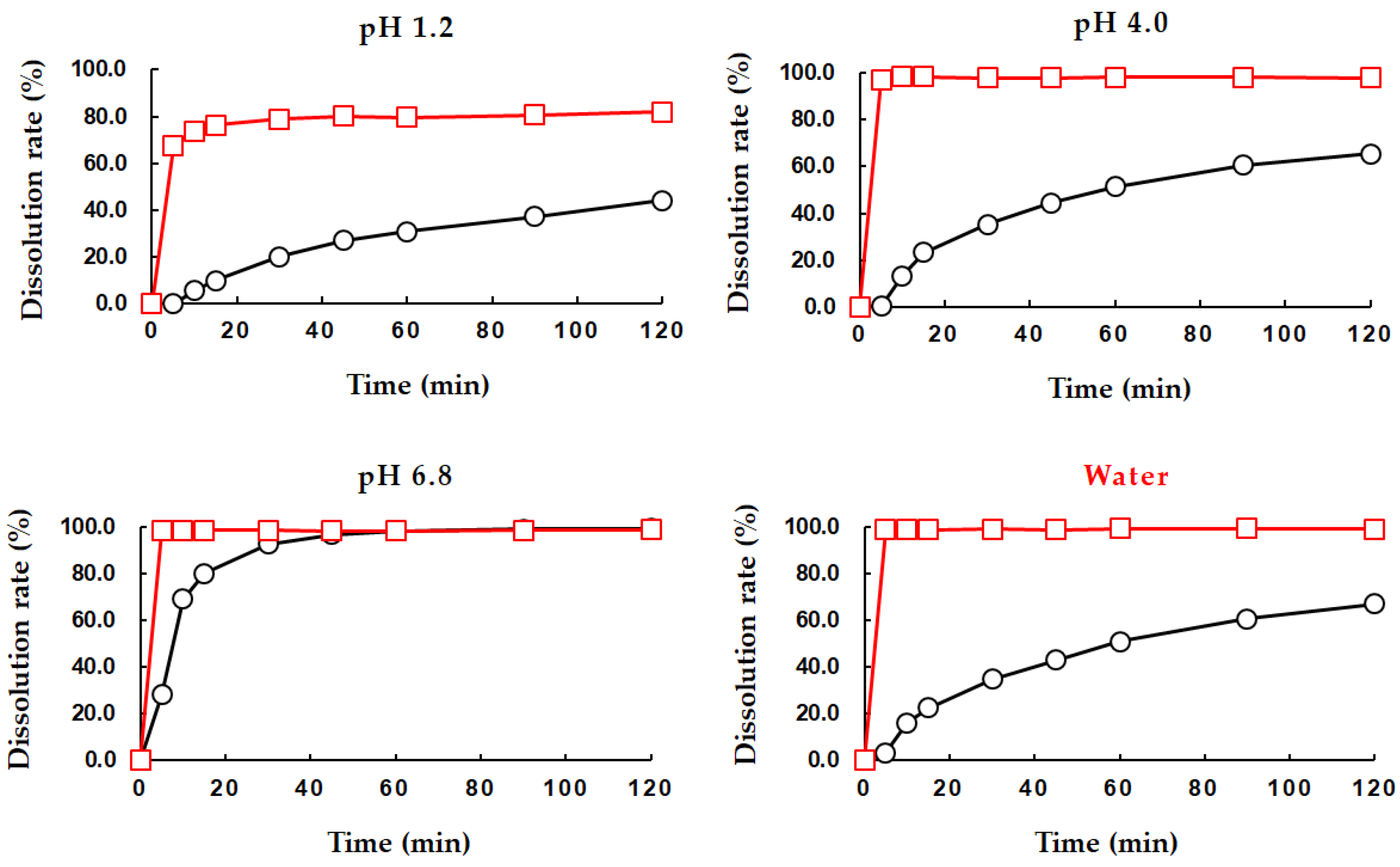
3.2. Solubility and Dissolution Behavior of PEL-T
3.3. Permeability Test of PEL-T in Caco-2 Cells
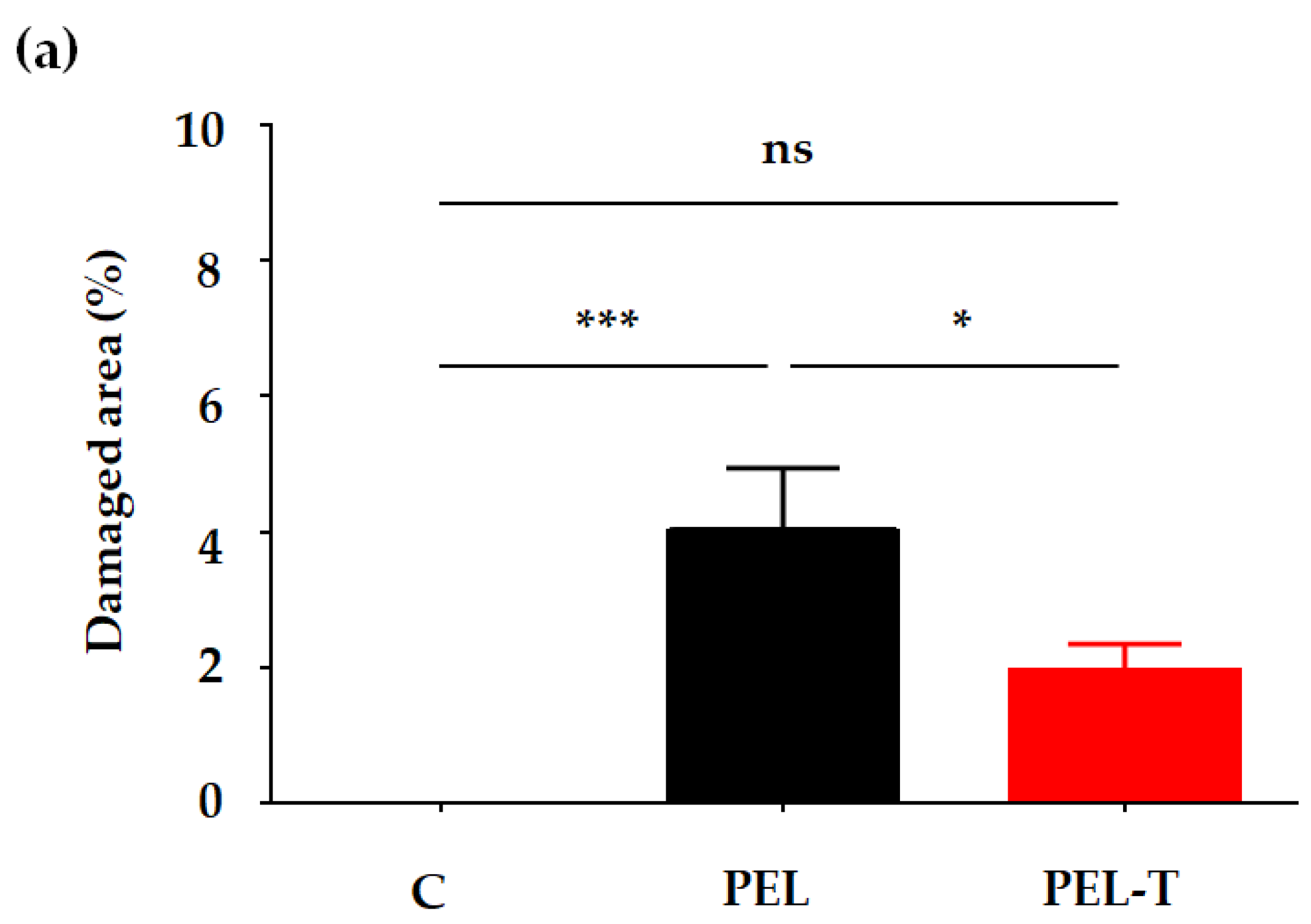
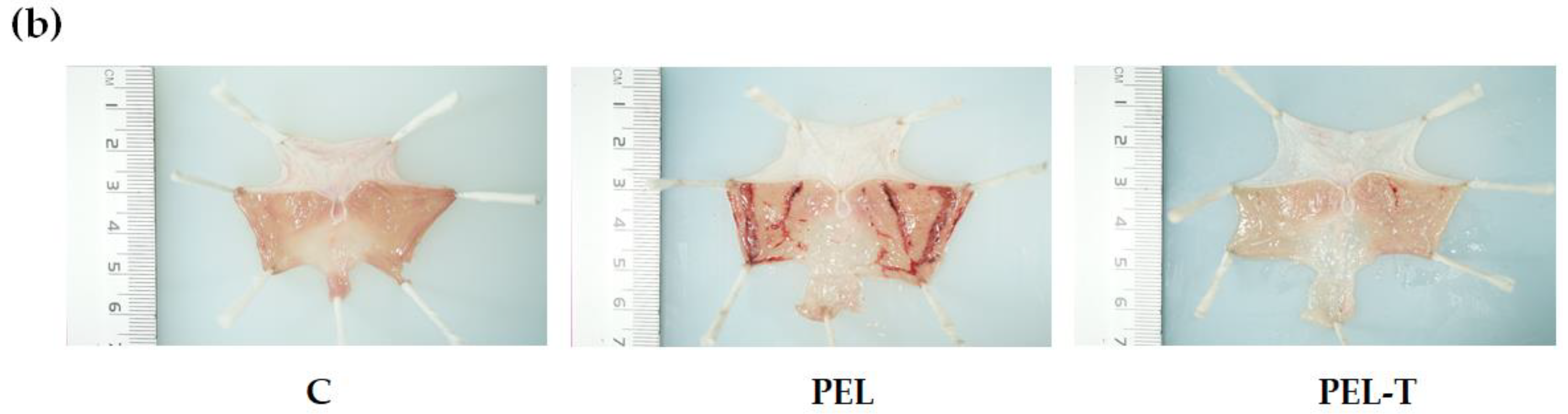
3.4. PEL-T Reduced Gastric Mucosal Injury in Rats
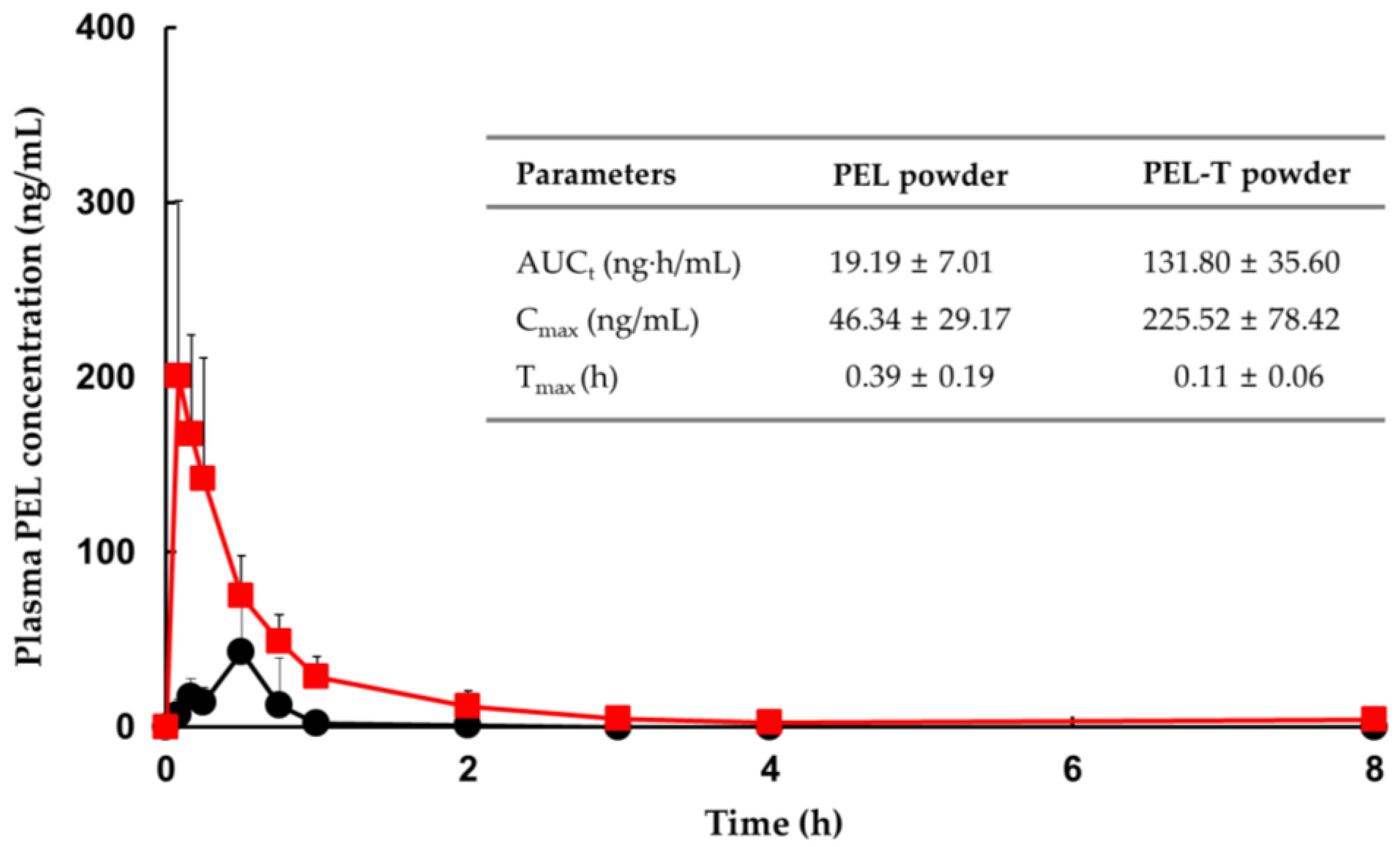
3.5. Comparative PK in Rats
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- NEDRUG. Pelubi® (Pelubiprofen) [Korean Prescribing Information]. Available online: https://nedrug.mfds.go.kr (accessed on 17 May 2021).
- Shin, J.S.; Baek, S.R.; Sohn, S.I.; Cho, Y.W.; Lee, K.-T. Anti-inflammatory effect of pelubiprofen, 2-[4-(oxocyclohexylidenemethyl)-phenyl]propionic acid, mediated by dual suppression of COX activity and LPS-induced inflammatory gene expression via NF-κB inactivation. J. Cell Biochem. 2011, 112, 3594–3603. [Google Scholar] [CrossRef] [PubMed]
- Song, S.H.; Chae, B.R.; Sohn, S.I.; Yeom, D.W.; Son, H.Y.; Kim, J.H.; Kim, S.R.; Lee, S.G.; Choi, Y.W. Formulation of controlled-release pelubiprofen tablet using Kollidon(®) SR. Int. J. Pharm. 2016, 511, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Asami, M.; Yamamura, M.; Nakajima, E.; Takasaki, W.; Tanaka, Y.; Ohtsuki, T.; Takaichi, M.; Mitsui, T.; Yokoshima, T. Metabolic Studies on CS-670, A New 2-Arylpropionic Acid Nonsteroidal Anti-inflammatory Drug (1): Absorption, Metabolism and Excretion of 14C-CS-670 in Mice, Rats and Dogs. Drug Metab. Pharmacokinet. 1995, 10, 637–650. [Google Scholar] [CrossRef]
- Itoh, K.; Yamamoto, K.; Adachi, M.; Kosaka, T.; Tanaka, Y. Leukotriene B4 12-hydroxydehydrogenase/15-ketoprostaglandin Delta 13-reductase (LTB4 12-HD/PGR) responsible for the reduction of a double-bond of the alpha,beta-unsaturated ketone of an aryl propionic acid non-steroidal anti-inflammatory agent CS-670. Xenobiotica 2008, 38, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.M.; Lichtenstein, D.R.; Singh, G. BCS toxicity of nonsteroidal antiinflammatory drugs. N. Engl. J. Med. 1999, 340, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, D.; Florin, T.; Eagle, C.; Marschner, I.; Singh, G.; Grobler, M.; Fenn, C.; Schou, M.; Curnow, K.M. Risk of serious NSAID-related gastrointestinal events during long-term exposure: A systematic review. Med. J. Aust. 2006, 185, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Correa, P.; Zhao, W.W.; Burr, A.M.; Hubbard, R.C.; Verburg, K.M.; Geis, G.S. Reduced incidence of gastroduodenal ulcers with celecoxib, a novel cyclooxygenase-2 inhibitor, compared to naproxen in patients with arthritis. Am. J. Gastroenterol. 2001, 96, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.S.; Weaver, A.L.; Graham, D.Y.; Kivitz, A.J.; Lipsky, P.E.; Hubbard, R.C.; Isakson, P.C.; Verburg, K.M.; Yu, S.S.; Zhao, W.W.; et al. Anti-inflammatory and upper gastrointestinal effects of celecoxib in rheumatoid arthritis: A randomized controlled trial. JAMA 1999, 282, 1921–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, P.; Zeidler, H.; Kvien, T.K.; Guslandi, M.; Naudin, R.; Stead, H.; Verburg, K.M.; Isakson, P.C.; Hubbard, R.C.; Geis, G.S. Celecoxib versus diclofenac in long-term management of rheumatoid arthritis: Randomized double-blind comparison. Lancet 1999, 354, 2106–2111. [Google Scholar] [CrossRef]
- NEDRUG. Celebrex® (Celecoxib) [Korean Prescribing Information]. Available online: https://nedrug.mfds.go.kr (accessed on 17 May 2021).
- Song, Y.W.; Choi, I.A.; Baek, H.-J.; Cho, C.S.; Lee, Y.A.; Chung, W.T.; Park, Y.E.; Lee, Y.J.; Park, Y.-B.; Lee, J.; et al. Comparison of the efficacy and safety profiles of a pelubiprofen versus celecoxib in patients with rheumatoid arthritis: A 6-week, multicenter, randomized, double-blind, phase III, non-inferiority clinical trial. BMC Musculoskelet. Disord. 2014, 15, 375. [Google Scholar] [CrossRef] [Green Version]
- Shin, B.J.; Kim, T.K.; Baik, J.S.; Shim, D.M. Comparison The Safety and The Efficacy between the Group of using Pelubiprofen Tab. and the Group of using Aceclofenac Tab. on Back Pain Patients-Multi Institution, Double Blind, Random Sample. J. Korean Soc. Spine Surg. 2012, 19, 38–46. [Google Scholar] [CrossRef]
- Kwak, S.S.; Lee, E.S.; Yoon, H.Y.; Kim, C.H.; Goo, Y.T.; Kang, M.J.; Lee, S.; Lee, B.S.; Jeon, H.R.; Oh, C.H.; et al. Immediate release tablet formulation of varenicline salicylate and comparative pharmacokinetic study in human volunteers. Drug Des. Devel. Ther. 2018, 12, 3377–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, B.D.; Conradi, R.A. Predictive relationships in the water solubility of salts of a nonsteroidal anti-inflammatory drug. J. Pharm. Sci. 1985, 74, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Wang, Y.W. Initial salt screening procedures for manufacturing ibuprofen. Drug Dev. Ind. Pharm. 2009, 35, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Mehlisch, D.R.; Ardia, A.; Pallotta, T. Analgesia with ibuprofen arginate versus conventional ibuprofen for patients with dysmenorrhea: A crossover trial. Curr. Ther. Res. Clin. Exp. 2003, 64, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.; Lee, S.J.; Ha, Y.-M.; Choi, Y.-S.; Kim, J.-W.; Park, S.-R.; Park, M.K. Pharmacokinetic and pharmacodynamic evaluation according to absorption differences in three formulations of ibuprofen. Drug Des. Devel. Ther. 2017, 11, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubatsch, I.; Ragnarsson, E.G.E.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef]
- Cho, S.; Lee, J.; Yoo, Y.; Cho, M.; Sohn, S.; Lee, B.-J. Improved Manufacturability and In Vivo Comparative Pharmacokinetics of Dapagliflozin Cocrystals in Beagle Dogs and Human Volunteers. Pharmaceutics 2021, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Marvanova, P.; Padrtova, T.; Pekarek, T.; Brus, J.; Czernek, J.; Mokry, P.; Humpa, O.; Oravec, M.; Jampilek, J. Synthesis and Characterization of New 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-Propoxybenzoates and Their Hydrochloride Salts. Molecules 2016, 21, 707. [Google Scholar] [CrossRef] [Green Version]
- Bookwala, M.; Thipsay, P.; Ross, S.; Zhang, F.; Bandari, S.; Repka, M.A. Preparation of a crystalline salt of indomethacin and tromethamine by hot melt extrusion technology. Eur. J. Pharm. Biopharm. 2018, 131, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Vadivelu, N.; Gowda, A.M.; Urman, R.D.; Jolly, S.; Kodumudi, V.; Maria, M.; Taylor, R.; Pergolizzi, J.V. Ketorolac tromethamine—Routes and clinical implications. Pain Pract. 2015, 15, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wu, G.; Wang, J.; Cui, Y.; Ma, J.; Zhu, Z.; Qiu, J.; Wu, J. Comparison of single-dose fosfomycin tromethamine and other antibiotics for lower uncomplicated urinary tract infection in women and asymptomatic bacteriuria in pregnant women: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2020, 56, 106018. [Google Scholar] [CrossRef] [PubMed]
- Crowe, A.; Wright, C. The impact of P-glycoprotein mediated efflux on absorption of 11 sedating and less-sedating antihistamines using Caco-2 monolayers. Xenobiotica 2012, 42, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Nahas, G.G.; Sutin, K.M.; Fermon, C.; Streat, S.; Wiklund, L.; Wahlander, S.; Yellin, P.; Brasch, H.; Kanchuger, M.; Capan, L.; et al. Guidelines for the treatment of acidaemia with THAM. Drugs 1998, 55, 191–224. [Google Scholar] [CrossRef] [PubMed]
- Mauleón, D.; Artigas, R.; García, M.L.; Carganico, G. Preclinical and clinical development of dexketoprofen. Drugs 1996, 52, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Song, W.H.; Yeom, D.W.; Lee, D.H.; Lee, K.M.; Yoo, H.J.; Chae, B.R.; Song, S.H.; Choi, Y.W. In situ intestinal permeability and in vivo oral bioavailability of celecoxib in supersaturating self-emulsifying drug delivery system. Arch. Pharm. Res. 2014, 37, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zou, D.; Zhang, Y.; Zhang, Q.; Feng, Y.; Guo, Y.; Liu, Y.; Zhang, X.; Cheng, G.; Wang, C.; et al. Pharmaceutical salts/cocrystals of enoxacin with dicarboxylic acids: Enhancing in vitro antibacterial activity of enoxacin by improving the solubility and permeability. Eur. J. Pharm. Biopharm. 2020, 154, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Luoma, J.T.; Hilleman, D. A Review of Currently Available Fenofibrate and Fenofibric Acid Formulations. Cardiol. Res. 2013, 4, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charalabidis, A.; Sfouni, M.; Bergström, C.; Macheras, P. The Biopharmaceutics Classification System (BCS) and the Biopharmaceutics Drug Disposition Classification System (BDDCS): Beyond guidelines. Int. J. Pharm. 2019, 566, 264–281. [Google Scholar] [CrossRef]
- Vogt, M.; Kunath, K.; Dressman, J.B. Dissolution enhancement of fenofibrate by micronization, cogrinding and spray-drying: Comparison with commercial preparations. Eur. J. Pharm. Biopharm. 2008, 68, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Bosselmann, S.; Williams, R.O. Has nanotechnology led to improved therapeutic outcomes? Drug Dev. Ind. Pharm. 2012, 38, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Trilipix® (fenofibric acid delayed release capsules). In Full Prescribing Information; Abbott Laboratories: North Chicago, IL, USA, 2011.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Assignments | PEL | PEL-T |
| a, a’, a” | 7.44–7.36 (m, 5H) | 7.46–7.38 (m, 5H) |
| b’, b | 3.78–3.72 (m, 1H) | 3.66–3.61 (m, 7H) |
| c | 2.85–2.82 (m, 2H) | 2.88–2.85 (m, 2H) |
| d | 2.50 (t, 2H) | 2.51 (t, 2H) |
| e | 1.94–1.89 (m, 2H) | 1.96–1.91 (m, 2H) |
| f | 1.78–1.73 (m, 2H) | 1.81–1.76 (m, 2H) |
| g | 1.47 (d, 3H) | 1.45 (d, 3H) |
| h | 4.94 (s, 1H) | |
| Dissolution Media | PEL (mg/mL) | PEL-T (mg/mL) |
|---|---|---|
| pH 1.2 | 0.03 ± 0.01 | 654.29 ± 4.53 |
| pH 4.0 | 0.07 ± 0.04 | 355.36 ± 5.65 |
| pH 6.8 | 4.05 ± 0.14 | 436.67 ± 14.15 |
| water | 0.13 ± 0.03 | 851.60 ± 18.36 |
| Direction | PEL | PEL-T | p-Value |
|---|---|---|---|
| A to B | 12.61 ± 1.44 | 15.35 ± 0.91 | 0.018 |
| B to A | 15.57 ± 4.60 | 11.65 ± 2.89 | 0.081 |
| Efflux ratio | 1.24 | 0.76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.Y.; Oh, D.H.; Park, S.-W.; Chae, B.R.; Kim, C.W.; Han, S.H.; Shin, H.J.; Yeom, S.B.; Lee, D.Y.; Park, M.K.; et al. Development of Pelubiprofen Tromethamine with Improved Gastrointestinal Safety and Absorption. Pharmaceutics 2021, 13, 745. https://doi.org/10.3390/pharmaceutics13050745
Park JY, Oh DH, Park S-W, Chae BR, Kim CW, Han SH, Shin HJ, Yeom SB, Lee DY, Park MK, et al. Development of Pelubiprofen Tromethamine with Improved Gastrointestinal Safety and Absorption. Pharmaceutics. 2021; 13(5):745. https://doi.org/10.3390/pharmaceutics13050745
Chicago/Turabian StylePark, Ji Yeon, Dong Ho Oh, Sang-Wook Park, Bo Ram Chae, Chul Woo Kim, Sang Heon Han, Hyeon Jong Shin, Soo Bin Yeom, Da Yeong Lee, Min Kyu Park, and et al. 2021. "Development of Pelubiprofen Tromethamine with Improved Gastrointestinal Safety and Absorption" Pharmaceutics 13, no. 5: 745. https://doi.org/10.3390/pharmaceutics13050745
APA StylePark, J. Y., Oh, D. H., Park, S. -W., Chae, B. R., Kim, C. W., Han, S. H., Shin, H. J., Yeom, S. B., Lee, D. Y., Park, M. K., Park, S. -E., Park, J. -B., & Lee, K. -T. (2021). Development of Pelubiprofen Tromethamine with Improved Gastrointestinal Safety and Absorption. Pharmaceutics, 13(5), 745. https://doi.org/10.3390/pharmaceutics13050745