
Folic Acid-Targeted Paclitaxel-Polymer Conjugates Exert Selective Cytotoxicity and Modulate Invasiveness of Colon Cancer Cells

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
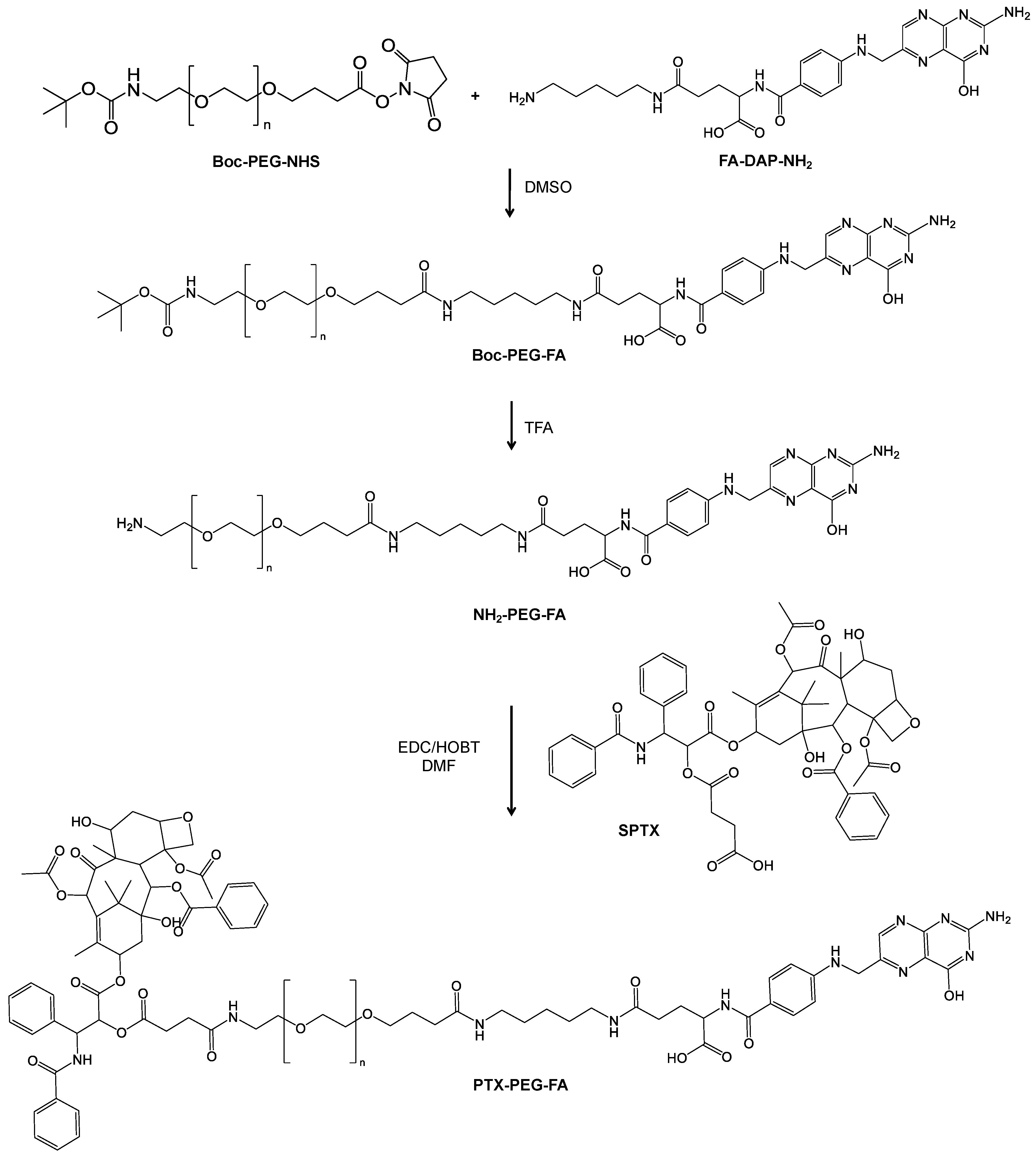
2.3. Synthesis of PTX-PEG, PTX-PEG-FA and PTX-PEG-(FA)3 Conjugates
2.4. Determination of Conjugated PTX
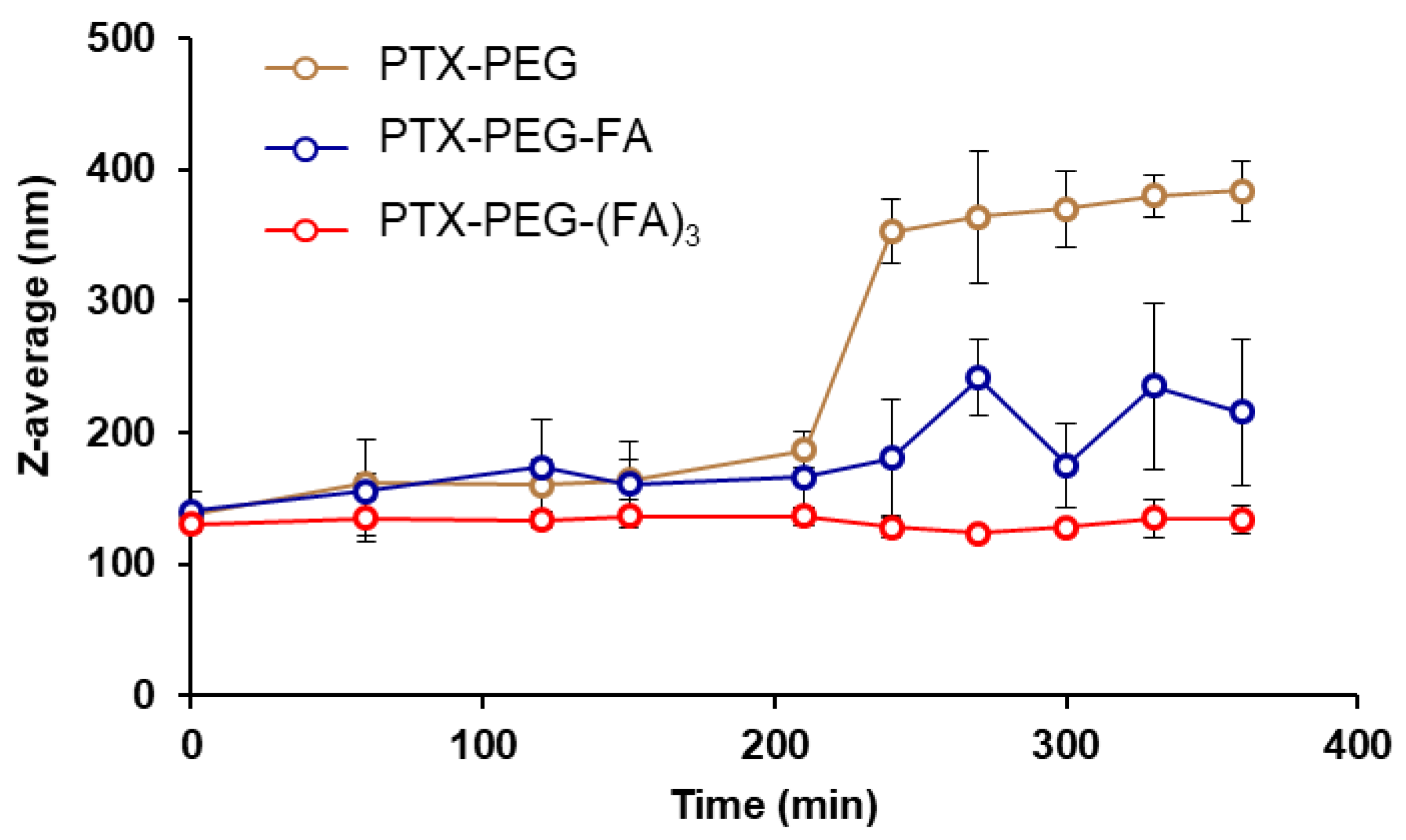
2.5. Dynamic Light Scattering (DLS) Analysis
2.6. Cell Cultures
2.7. Immunocytochemistry
2.8. Cell Viability Studies
2.9. Cell Cycle Analysis
2.10. Wound Healing Assay
2.11. mRNA Expression Analysis by qRT-PCR
2.12. ELISA Assay for VEGF Quantification
2.13. Pharmacokinetic Study in Mice
2.14. Statistical Analysis
3. Results
3.1. Synthesis and Characterizarion of PTX-PEG, PTX-PEG-FA and PTX-PEG-(FA)3 Conjugates
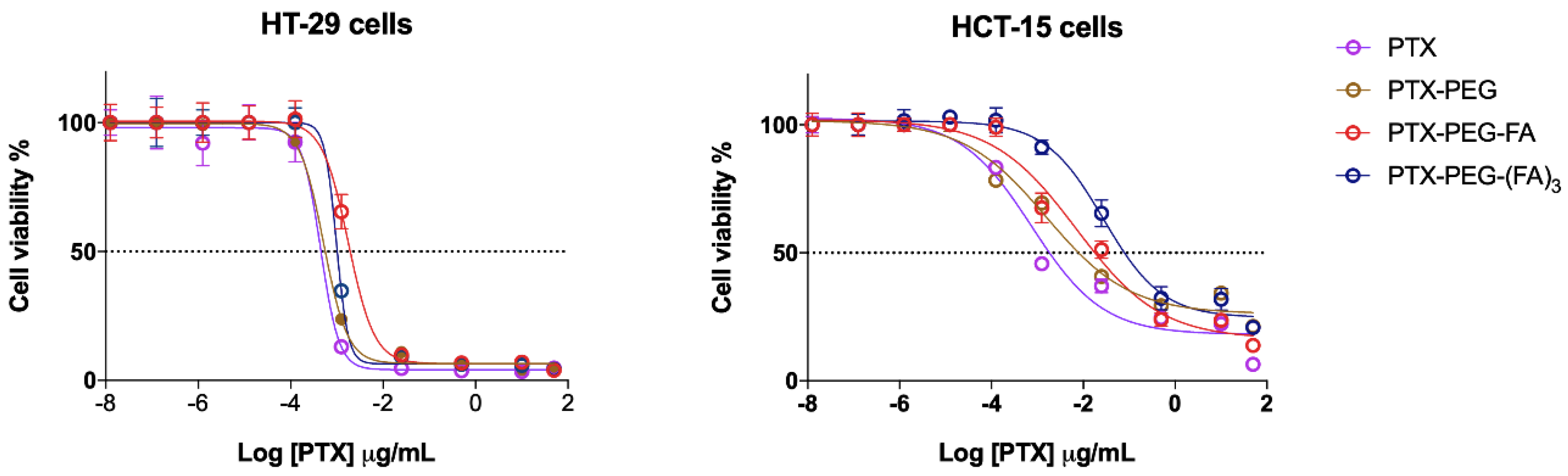
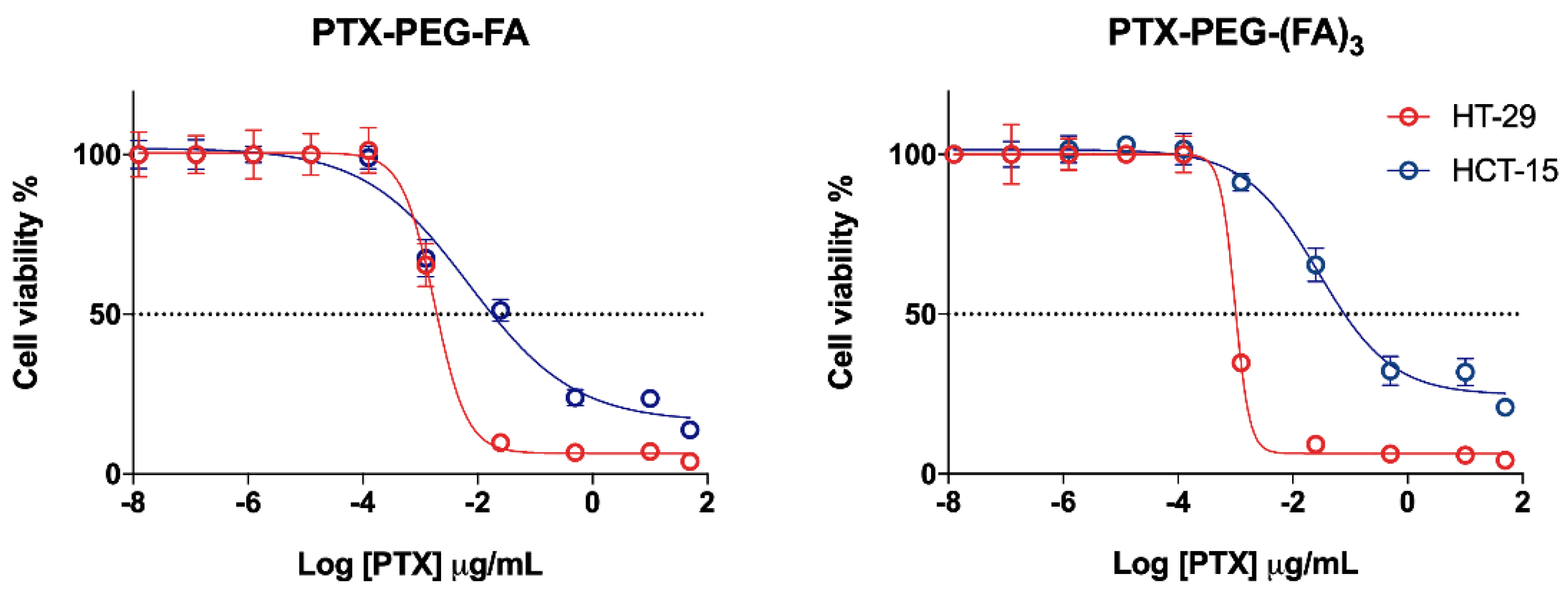
3.2. PTX-PEG-FA Conjugates Induced Cytotoxicity on FR-Postive HT-29 Cells
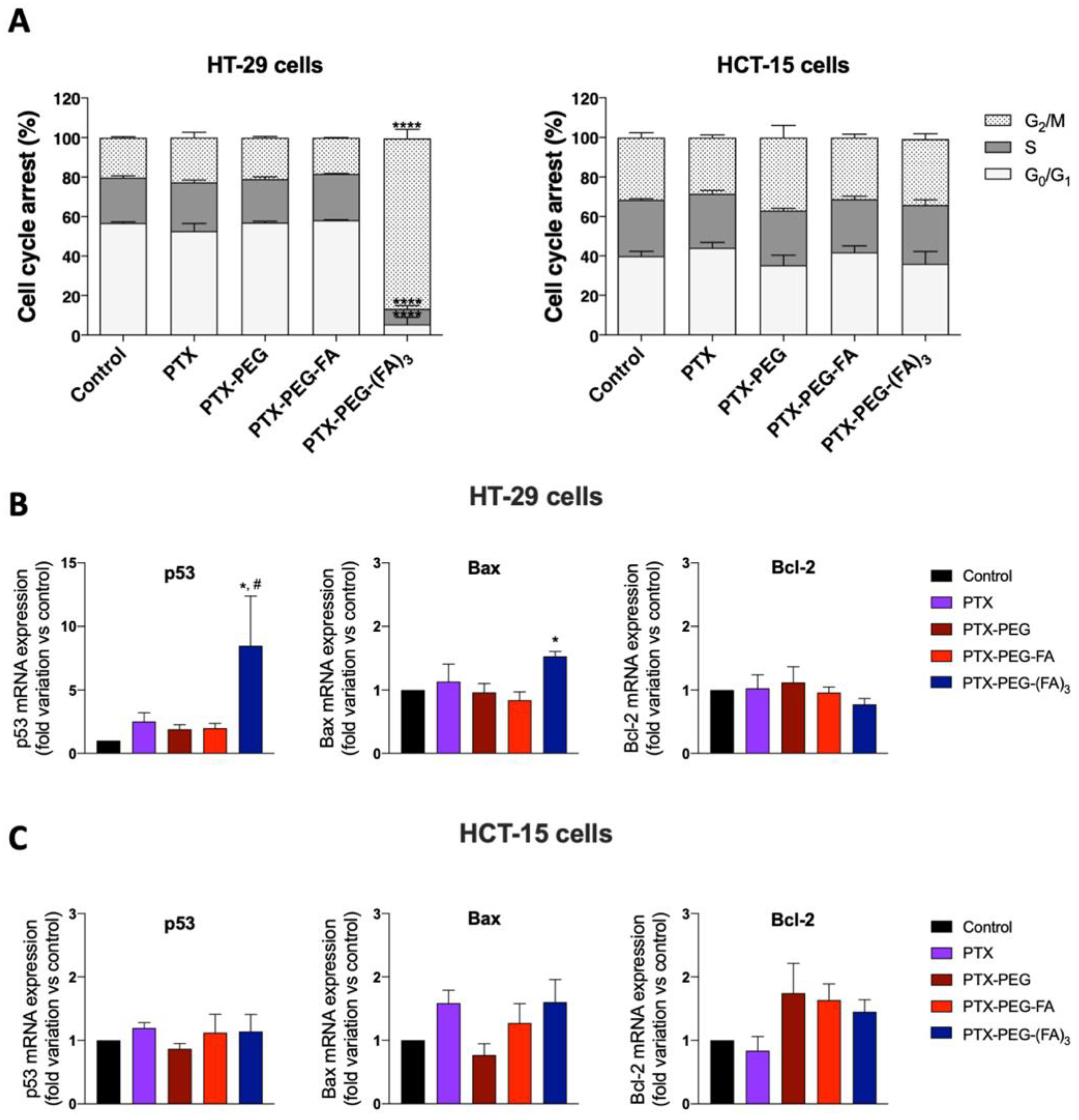
3.3. PTX-PEG-(FA)3 Induces G1/M Phase Cell Cycle Arrest in HT-29 Cells
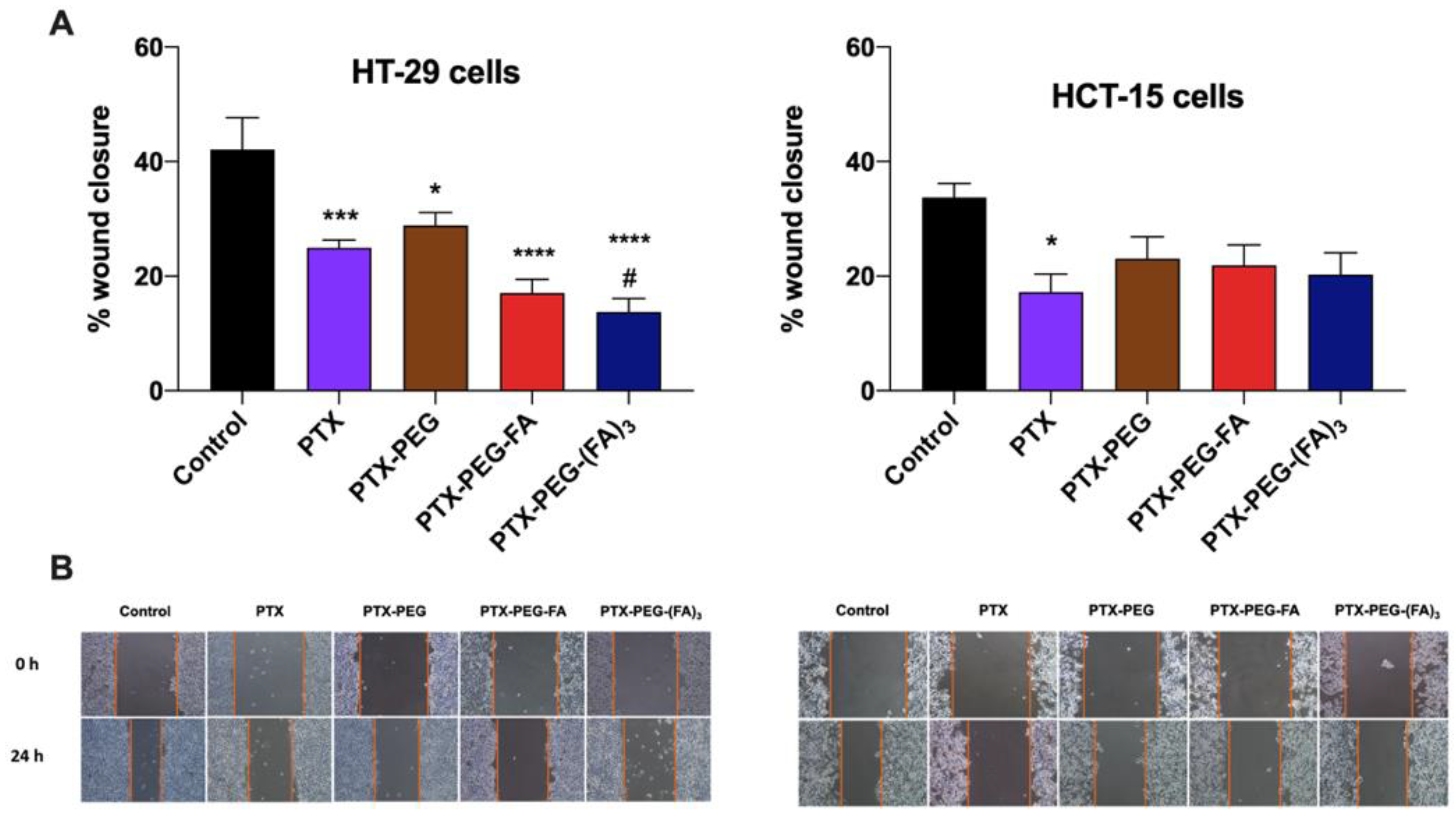
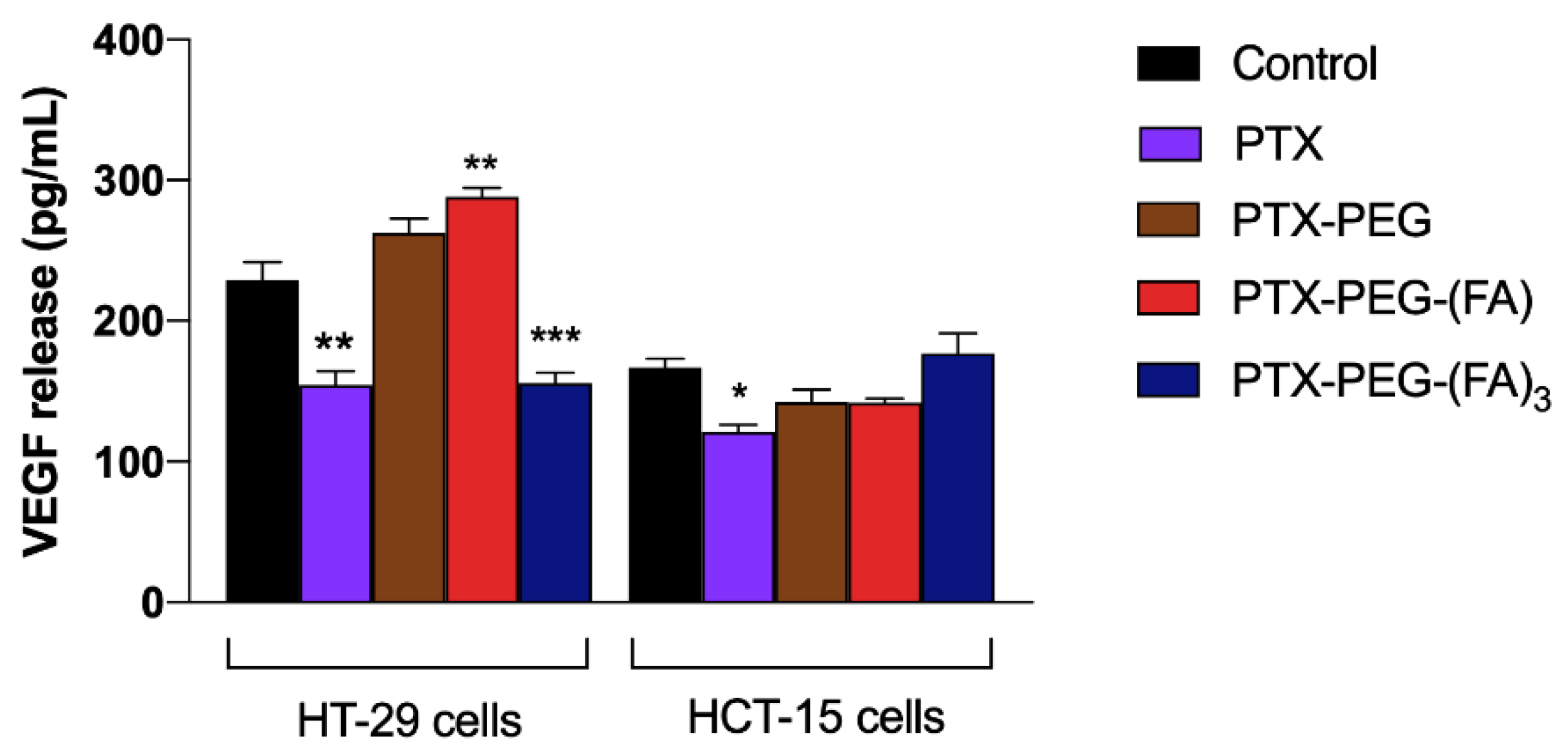
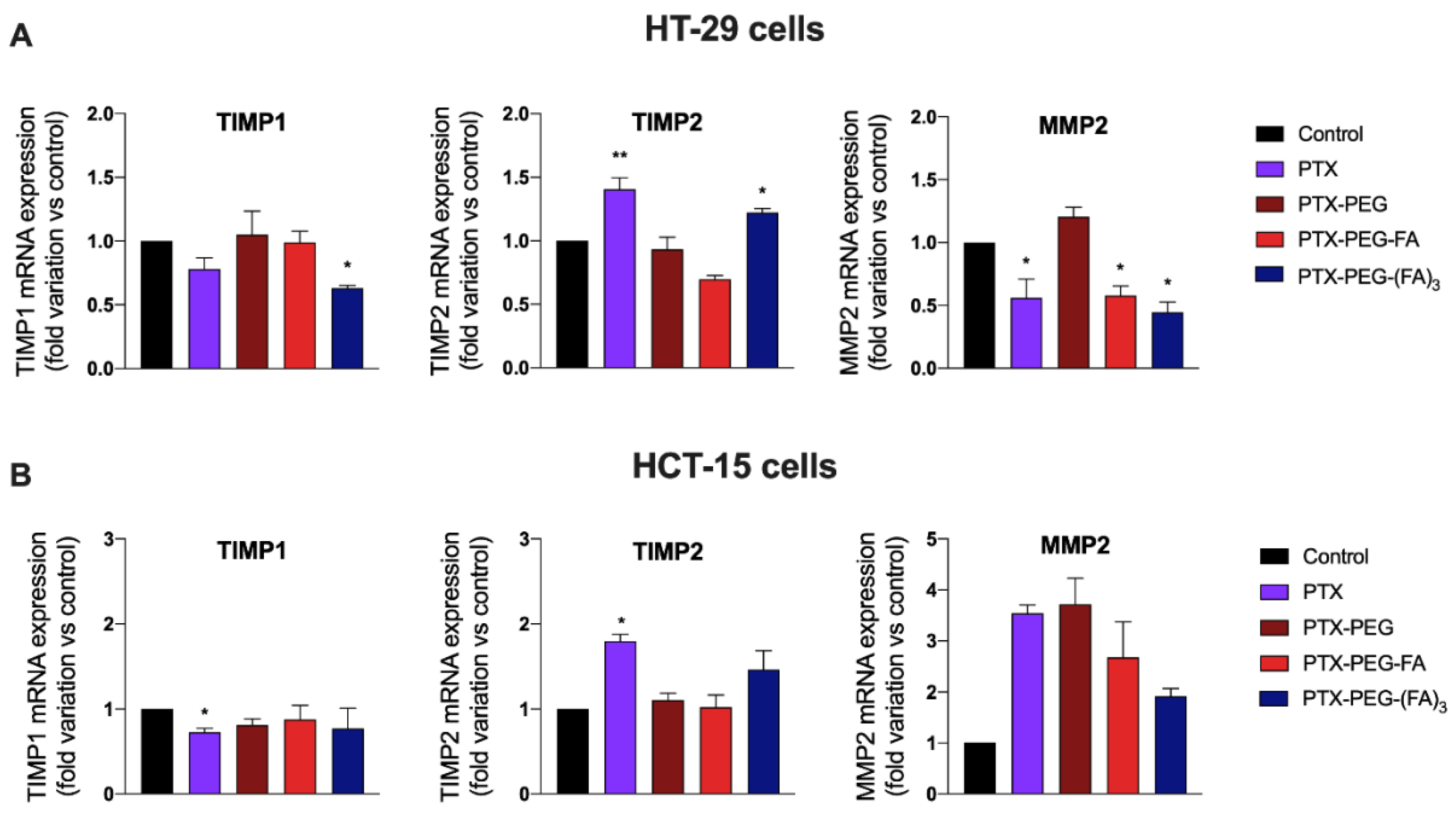
3.4. PTX-PEG-FA Conjugates Inhibit Migration of HT-29 Cells
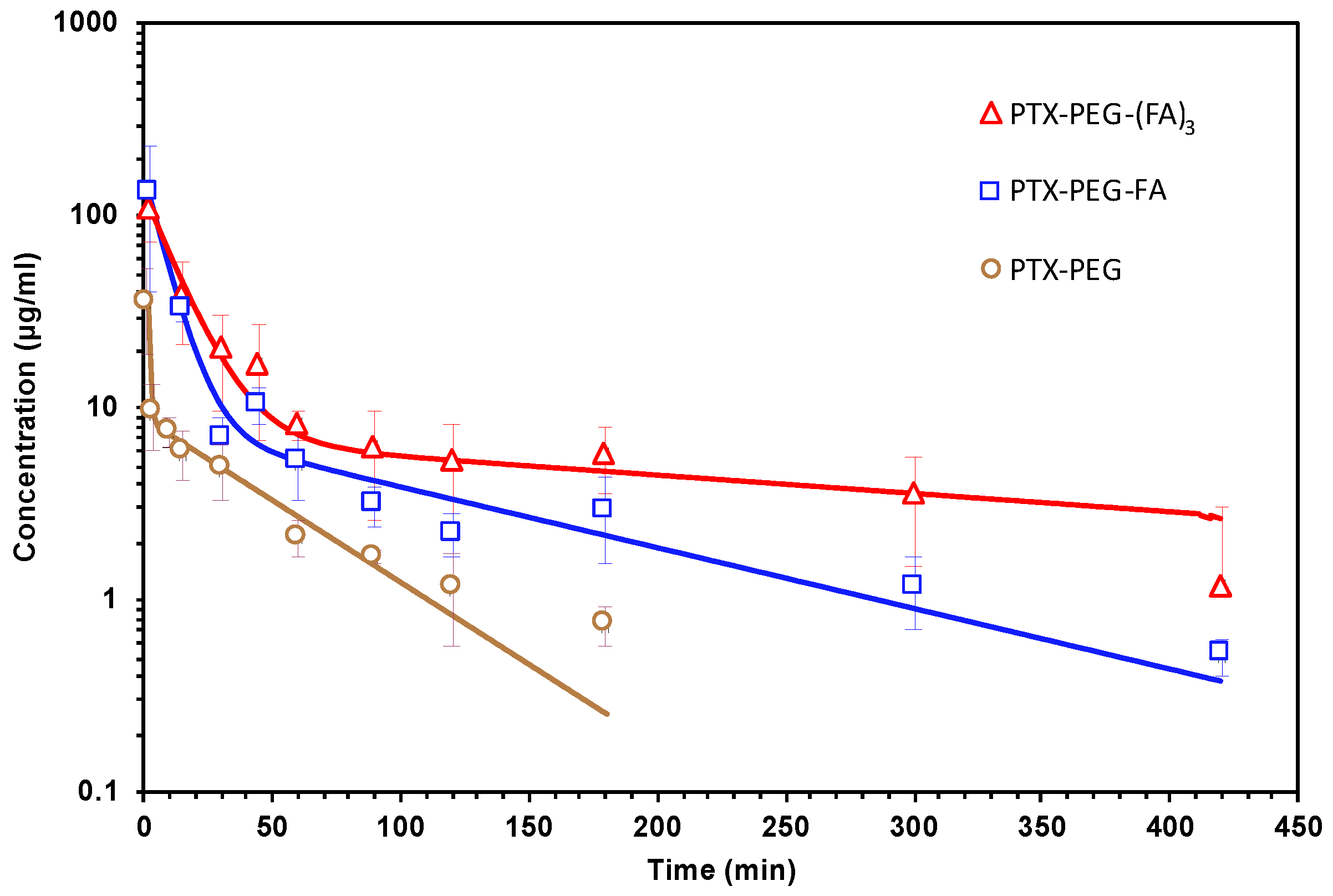
3.5. Pharmacokinetic Studies in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor Vascular Permeability and the EPR Effect in Macromolecular Therapeutics: A Review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Pasut, G. Grand Challenges in Nano-Based Drug Delivery. Front. Med. Technol. 2019, 1, 1. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.M.; Senter, P.D. Arming Antibodies: Prospects and Challenges for Immunoconjugates. Nat. Biotechnol. 2005, 23, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W. Antibody Drug Conjugates: Lessons from 20 Years of Clinical Experience. Ann. Oncol. 2016, 27, 2168–2172. [Google Scholar] [CrossRef]
- Grigoletto, A.; Maso, K.; Mero, A.; Rosato, A.; Schiavon, O.; Pasut, G. Drug and Protein Delivery by Polymer Conjugation. J. Drug. Deliv. Sci. Technol. 2016, 32, 132–141. [Google Scholar] [CrossRef]
- Canal, F.; Vicent, M.J.; Pasut, G.; Schiavon, O. Relevance of Folic Acid/Polymer Ratio in Targeted PEG-Epirubicin Conjugates. J. Control. Release 2010, 146, 388–399. [Google Scholar] [CrossRef]
- Pasut, G.; Canal, F.; Dalla Via, L.; Arpicco, S.; Veronese, F.M.; Schiavon, O. Antitumoral Activity of PEG-Gemcitabine Prodrugs Targeted by Folic Acid. J. Control. Release 2008, 127, 239–248. [Google Scholar] [CrossRef]
- Clementi, C.; Miller, K.; Mero, A.; Satchi-Fainaro, R.; Pasut, G. Dendritic Poly(Ethylene Glycol) Bearing Paclitaxel and Alendronate for Targeting Bone Neoplasms. Mol. Pharm. 2011, 8, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Clementi, C.; Polyak, D.; Eldar-Boock, A.; Benayoun, L.; Barshack, I.; Shaked, Y.; Pasut, G.; Satchi-Fainaro, R. Poly(Ethylene Glycol)-Paclitaxel-Alendronate Self-Assembled Micelles for the Targeted Treatment of Breast Cancer Bone Metastases. Biomaterials 2013, 34, 3795–3806. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Low, P.S. Folate-Mediated Targeting of Antineoplastic Drugs, Imaging Agents, and Nucleic Acids to Cancer Cells. J. Control. Release Off. J. Control. Release Soc. 1998, 53, 39–48. [Google Scholar] [CrossRef]
- Sudimack, J.; Lee, R.J. Targeted Drug Delivery via the Folate Receptor. Adv. Drug. Deliv. Rev. 2000, 41, 147–162. [Google Scholar] [CrossRef]
- Rijnboutt, S.; Jansen, G.; Posthuma, G.; Hynes, J.B.; Schornagel, J.H.; Strous, G.J. Endocytosis of GPI-Linked Membrane Folate Receptor-Alpha. J. Cell. Biol. 1996, 132, 35–47. [Google Scholar] [CrossRef]
- Liang, X.; Luo, M.; Wei, X.-W.; Ma, C.-C.; Yang, Y.-H.; Shao, B.; Liu, Y.-T.; Liu, T.; Ren, J.; Liu, L.; et al. A Folate Receptor-Targeted Lipoplex Delivering Interleukin-15 Gene for Colon Cancer Immunotherapy. Oncotarget 2016, 7, 52207–52217. [Google Scholar] [CrossRef] [Green Version]
- Tiernan, J.P.; Perry, S.L.; Verghese, E.T.; West, N.P.; Yeluri, S.; Jayne, D.G.; Hughes, T.A. Carcinoembryonic Antigen Is the Preferred Biomarker for in Vivo Colorectal Cancer Targeting. Br. J. Cancer 2013, 108, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-L.; Chang, M.-C.; Huang, C.-Y.; Chiang, Y.-C.; Lin, H.-W.; Chen, C.-A.; Hsieh, C.-Y.; Cheng, W.-F. Serous Ovarian Carcinoma Patients with High Alpha-Folate Receptor Had Reducing Survival and Cytotoxic Chemo-Response. Mol. Oncol. 2012, 6, 360–369. [Google Scholar] [CrossRef] [Green Version]
- D’Angelica, M.; Ammori, J.; Gonen, M.; Klimstra, D.S.; Low, P.S.; Murphy, L.; Weiser, M.R.; Paty, P.B.; Fong, Y.; DeMatteo, R.P.; et al. Folate Receptor-α Expression in Resectable Hepatic Colorectal Cancer Metastases: Patterns and Significance. Mod. Pathol. 2011, 24, 1221–1228. [Google Scholar] [CrossRef] [Green Version]
- Pasut, G.; Greco, F.; Mero, A.; Mendichi, R.; Fante, C.; Green, R.J.; Veronese, F.M. Polymer-Drug Conjugates for Combination Anticancer Therapy: Investigating the Mechanism of Action. J. Med. Chem. 2009, 52, 6499–6502. [Google Scholar] [CrossRef]
- Santucci, L.; Mencarelli, A.; Renga, B.; Pasut, G.; Veronese, F.; Zacheo, A.; Germani, A.; Fiorucci, S. Nitric Oxide Modulates Proapoptotic and Antiapoptotic Properties of Chemotherapy Agents: The Case of NO-Pegylated Epirubicin. FASEB J. 2006, 20, 765–767. [Google Scholar] [CrossRef]
- Frión-Herrera, Y.; Gabbia, D.; Scaffidi, M.; Zagni, L.; Cuesta-Rubio, O.; De Martin, S.; Carrara, M. The Cuban Propolis Component Nemorosone Inhibits Proliferation and Metastatic Properties of Human Colorectal Cancer Cells. Int. J. Mol. Sci. 2020, 21, 1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frión-Herrera, Y.; Gabbia, D.; Cuesta-Rubio, O.; De Martin, S.; Carrara, M. Nemorosone Inhibits the Proliferation and Migration of Hepatocellular Carcinoma Cells. Life Sci. 2019, 235, 116817. [Google Scholar] [CrossRef] [PubMed]
- Gabbia, D.; Pozzo, L.; Zigiotto, G.; Roverso, M.; Sacchi, D.; Dalla Pozza, A.; Carrara, M.; Bogialli, S.; Floreani, A.; Guido, M.; et al. Dexamethasone Counteracts Hepatic Inflammation and Oxidative Stress in Cholestatic Rats via CAR Activation. PLoS ONE 2018, 13, e0204336. [Google Scholar] [CrossRef] [PubMed]
- Gabbia, D.; Roverso, M.; Guido, M.; Sacchi, D.; Scaffidi, M.; Carrara, M.; Orso, G.; Russo, F.P.; Floreani, A.; Bogialli, S.; et al. Western Diet-Induced Metabolic Alterations Affect Circulating Markers of Liver Function before the Development of Steatosis. Nutrients 2019, 11, 1602. [Google Scholar] [CrossRef] [Green Version]
- Orlando, R.; De Martin, S.; Andrighetto, L.; Floreani, M.; Palatini, P. Fluvoxamine Pharmacokinetics in Healthy Elderly Subjects and Elderly Patients with Chronic Heart Failure. Br. J. Clin. Pharmacol. 2010, 69, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.-E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int. J. Mol. Sci. 2020, 22, 130. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Fan, F.; Wang, R.; Ye, X.; Xia, L.; Boulbes, D.; Ellis, L.M. Intracrine VEGF Signalling Mediates Colorectal Cancer Cell Migration and Invasion. Br. J. Cancer 2017, 117, 848–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, A.; Raufman, J.-P.; Xie, G. The Role of Matrix Metalloproteinases in Colorectal Cancer. Cancers 2014, 6, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Das Gupta, T.K.; Beattie, C.W. P28-Mediated Activation of P53 in G2 –M Phase of the Cell Cycle Enhances the Efficacy of DNA Damaging and Antimitotic Chemotherapy. Cancer Res. 2016, 76, 2354–2365. [Google Scholar] [CrossRef] [Green Version]
- Demidenko, Z.N.; Kalurupalle, S.; Hanko, C.; Lim, C.; Broude, E.; Blagosklonny, M.V. Mechanism of G1-like Arrest by Low Concentrations of Paclitaxel: Next Cell Cycle P53-Dependent Arrest with Sub G1 DNA Content Mediated by Prolonged Mitosis. Oncogene 2008, 27, 4402–4410. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Xu, S.; Zhang, H.; Wang, Y.; Xiao, C.; Jiang, T.; Wu, L.; Zhang, T.; Sun, X.; Zhong, L.; et al. TIMP1 Is a Prognostic Marker for the Progression and Metastasis of Colon Cancer through FAK-PI3K/AKT and MAPK Pathway. J. Exp. Clin. Cancer Res. 2016, 35, 148. [Google Scholar] [CrossRef] [Green Version]
- Kryczka, J.; Stasiak, M.; Dziki, L.; Mik, M.; Dziki, A.; Cierniewski, C.S. Matrix Metalloproteinase-2 Cleavage of the Β1 Integrin Ectodomain Facilitates Colon Cancer Cell Motility. J. Biol. Chem. 2012, 287, 36556–36566. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Liu, Y.; Feng, H.; Bian, X.; Zhao, W.; Yang, Z.; Gu, B.; Li, Z.; Liu, Y. CD133 Affects the Invasive Ability of HCT116 Cells by Regulating TIMP-2. Am. J. Pathol. 2013, 182, 565–576. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequence | Reverse Sequence | RefSeq | Product Length (Bp) |
|---|---|---|---|---|
| p53 | GAGACCTGTGGGAAGCG | CGGGGACAGCATCAAAT | NM_001126118.2 | 123 |
| Bax | CACTGAAGCGACTGATGTCCC | CCGCCACAAAGATGGTCAC | NM_001291428.2 | 91 |
| Bcl2 | TGTGTGTGGAGAGCGTCAA | CAGCCCAGACTCACATCACCA | NM_000657.3 | 148 |
| TIMP-1 | GCTGTGAGGAATGCACAGTGTTT | GGACTGGAAGCCCTTTTCAGA | NM_003254.3 | 116 |
| TIMP-2 | TCTGTGACTTCATCGTGCCC | ATGTAGCACGGGATCATGGG | NM_003255.5 | 121 |
| MMP-2 | CATCCAGACTTCCTCAGGCGG | GGTCCTGGCAATCCCTTTGTATG | NM_004530.6 | 75 |
| GAPDH | ACATCAAGAAGGTGGTGAAGCA | GTCAAAGGTGGAGGAGTGGTT | NM_002046.7 | 119 |
| Conjugate | % of Free PTX | FA Loading (mol%) | PTX Loading (w/w%) | Hydrodynamic Diameter (nm) |
|---|---|---|---|---|
| PTX-PEG | 1.33 | - | 9.11 ± 0.1 | 137.4 ± 1.8 |
| PTX-PEG-FA | 1.47 | 1 | 10.27 ± 1.02 | 140.4 ± 0.18 |
| PTX-PEG-(FA)3 | 0.79 | 3 | 8.53 ± 0.2 | 130.1 ± 2.28 |
| Cell Line | PTX | PTX-PEG | PTX-PEG-FA | PTX-PEG-(FA)3 |
|---|---|---|---|---|
| HT-29 | 0.444 ± 0.123 | 0.518 ± 0.241 | 1.766 ± 0.791 # | 0.979 ± 0.151 |
| HCT-15 | 0.750 ± 0.265 | 1.580 ± 0.454 * | 7.153 ± 1.423 **,# | 27.69 ± 4.12 ***,####,§§§ |
| Sample | t½α (min) | t½β (min) | AUC 0-inf (µg min/mL) | CL (mL/min) | Vd (mL) |
|---|---|---|---|---|---|
| PTX-PEG | 0.46 | 35.2 | 1566.3 | 0.15 | 6.6 |
| PTX-PEG-FA | 5.62 | 95.4 | 2438.5 | 0.12 | 8.2 |
| PTX-PEG-(FA)3 | 8.94 | 313.8 | 4740.78 | 0.08 | 7.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigoletto, A.; Martinez, G.; Gabbia, D.; Tedeschini, T.; Scaffidi, M.; Martin, S.D.; Pasut, G. Folic Acid-Targeted Paclitaxel-Polymer Conjugates Exert Selective Cytotoxicity and Modulate Invasiveness of Colon Cancer Cells. Pharmaceutics 2021, 13, 929. https://doi.org/10.3390/pharmaceutics13070929
Grigoletto A, Martinez G, Gabbia D, Tedeschini T, Scaffidi M, Martin SD, Pasut G. Folic Acid-Targeted Paclitaxel-Polymer Conjugates Exert Selective Cytotoxicity and Modulate Invasiveness of Colon Cancer Cells. Pharmaceutics. 2021; 13(7):929. https://doi.org/10.3390/pharmaceutics13070929
Chicago/Turabian StyleGrigoletto, Antonella, Gabriele Martinez, Daniela Gabbia, Tommaso Tedeschini, Michela Scaffidi, Sara De Martin, and Gianfranco Pasut. 2021. "Folic Acid-Targeted Paclitaxel-Polymer Conjugates Exert Selective Cytotoxicity and Modulate Invasiveness of Colon Cancer Cells" Pharmaceutics 13, no. 7: 929. https://doi.org/10.3390/pharmaceutics13070929
APA StyleGrigoletto, A., Martinez, G., Gabbia, D., Tedeschini, T., Scaffidi, M., Martin, S. D., & Pasut, G. (2021). Folic Acid-Targeted Paclitaxel-Polymer Conjugates Exert Selective Cytotoxicity and Modulate Invasiveness of Colon Cancer Cells. Pharmaceutics, 13(7), 929. https://doi.org/10.3390/pharmaceutics13070929