
Poly(l-Lactic Acid)-co-poly(Butylene Adipate) New Block Copolymers for the Preparation of Drug-Loaded Long Acting Injectable Microparticles


 , ,
, ,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
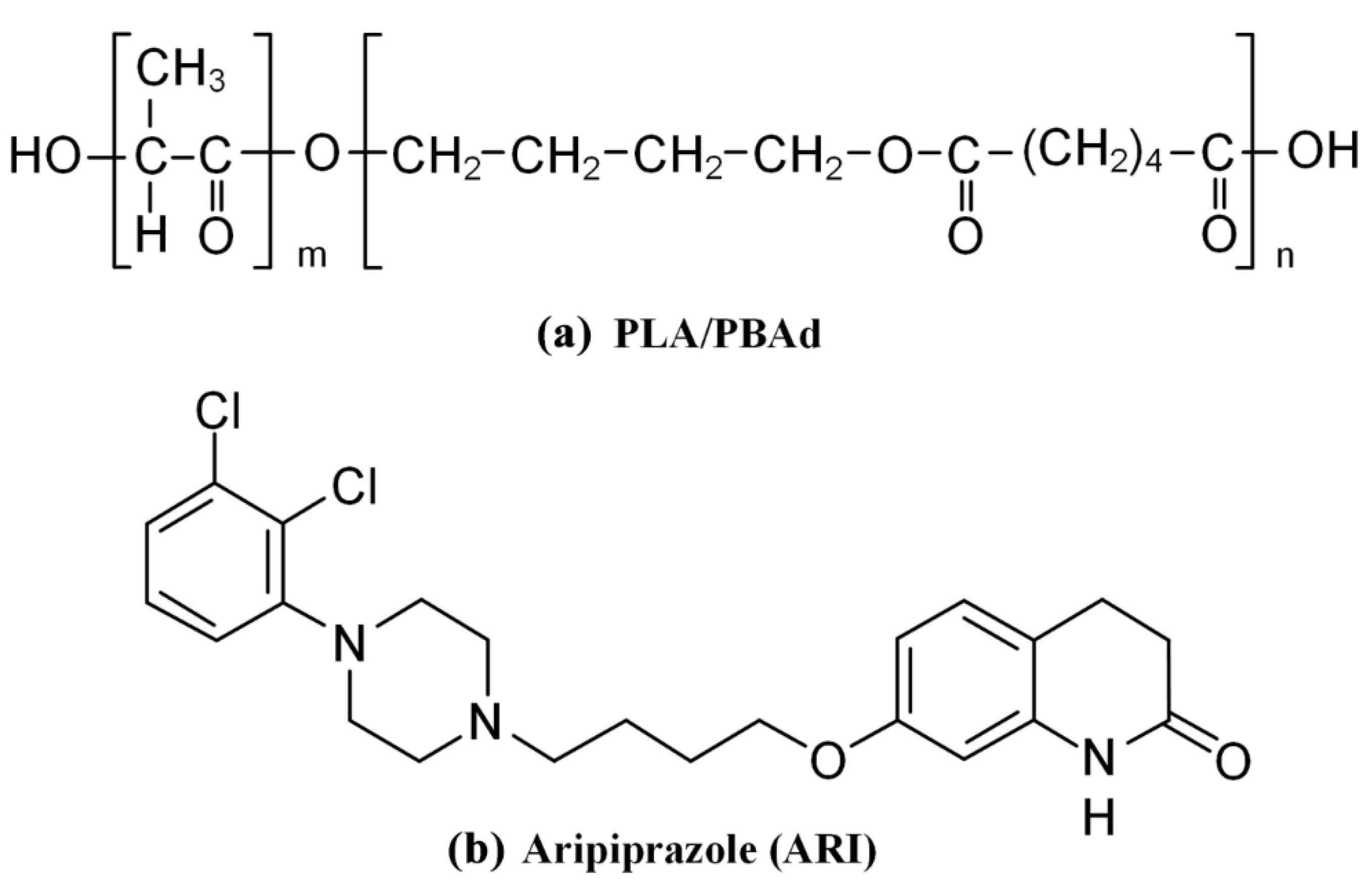
2.2. Synthesis of PBAd and PLA/PBAd Block Copolymers
2.3. Characterization of PLA/PBAd Block Copolymers
2.3.1. Enzymic Hydrolysis
2.3.2. Size-Exclusion Chromatography
2.3.3. Cytotoxicity Studies
2.4. Preparation of ARI MPs
2.5. Characterization of MPs
2.5.1. Differential Scanning Calorimetry (DSC)
2.5.2. Wide Angle Powder X-ray Diffractometry (pXRD)
2.5.3. Scanning Electron Microscopy (SEM)
2.5.4. Fourier-Transformed Infrared Spectroscopy (FTIR)
2.5.5. Yield, Encapsulation Efficiency and Drug Loading
2.5.6. In Vitro Dissolution Test
2.6. Statistical Analysis
3. Results and Discussion
3.1. Evaluation of neat PLA/PBAd Block Copolymers
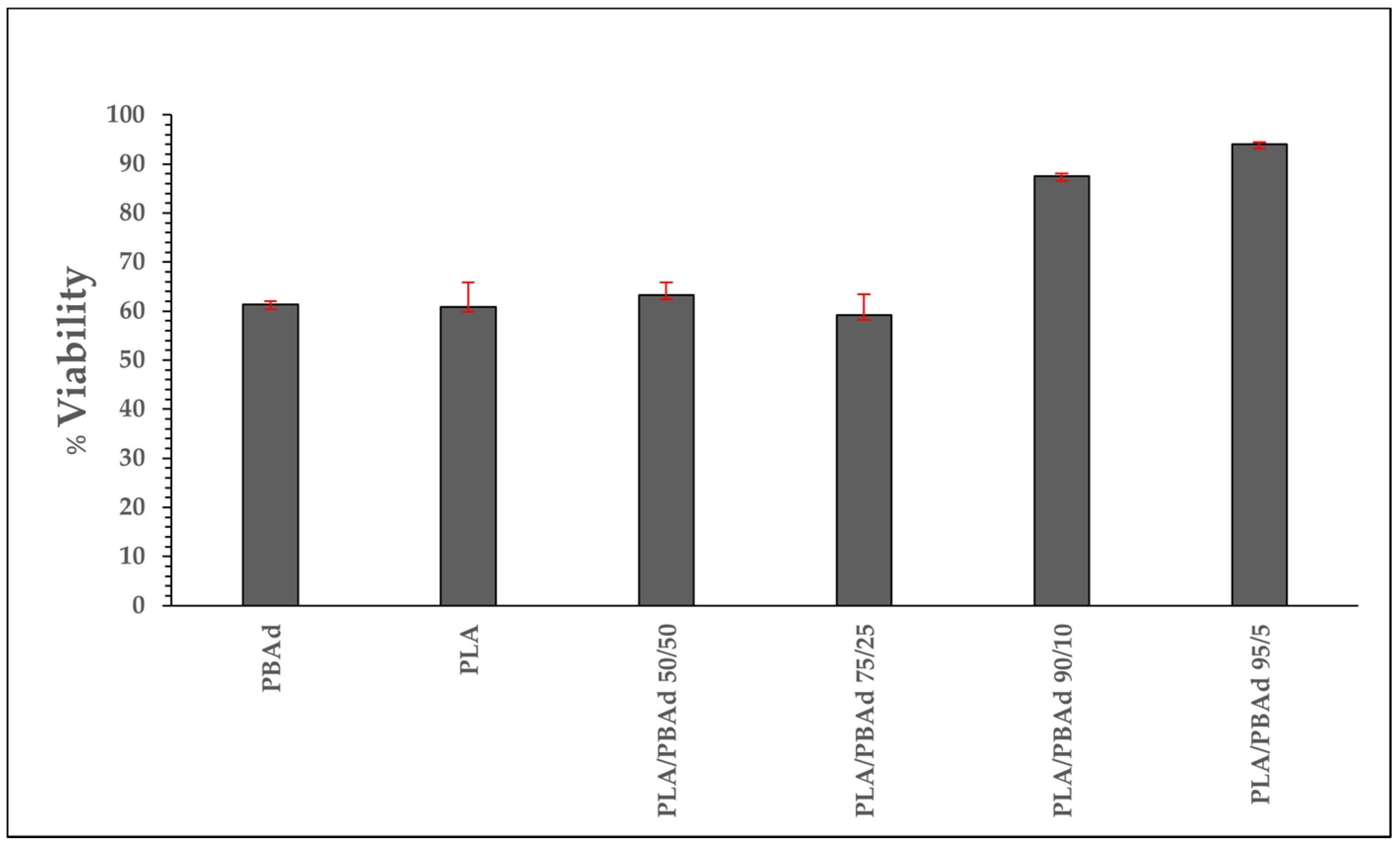
3.1.1. Cytotoxicity Results
3.1.2. Enzymatic Hydrolysis
3.2. Evaluation of ARI-loaded MPs
3.2.1. MPs Morphology Evaluation Via SEM
3.2.2. MPs Yield, Drug Loading and EE
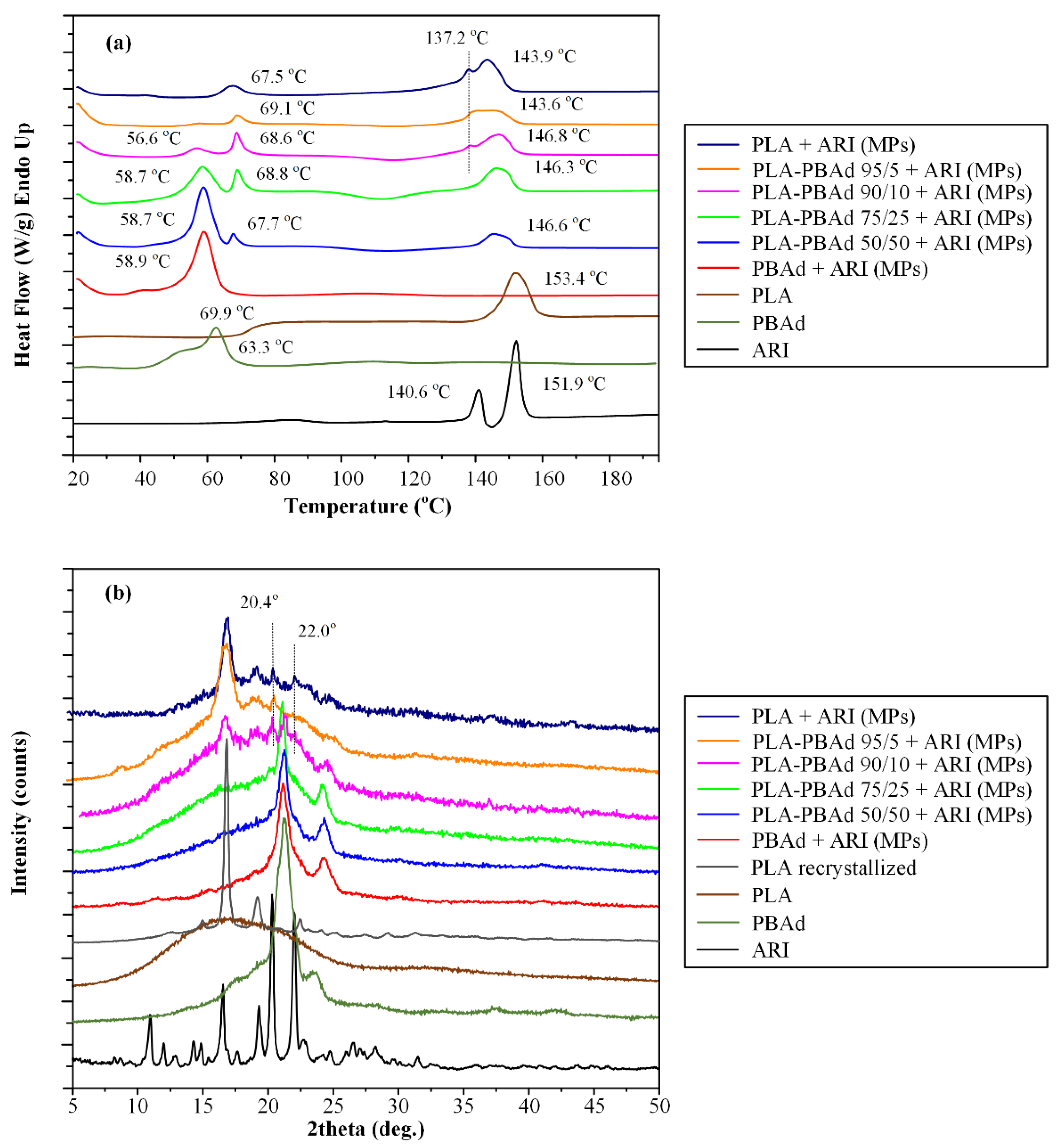
3.2.3. MPs’ Thermal Properties and Physical State Evaluation Via DSC
3.2.4. Physical State Verification Via pXRD
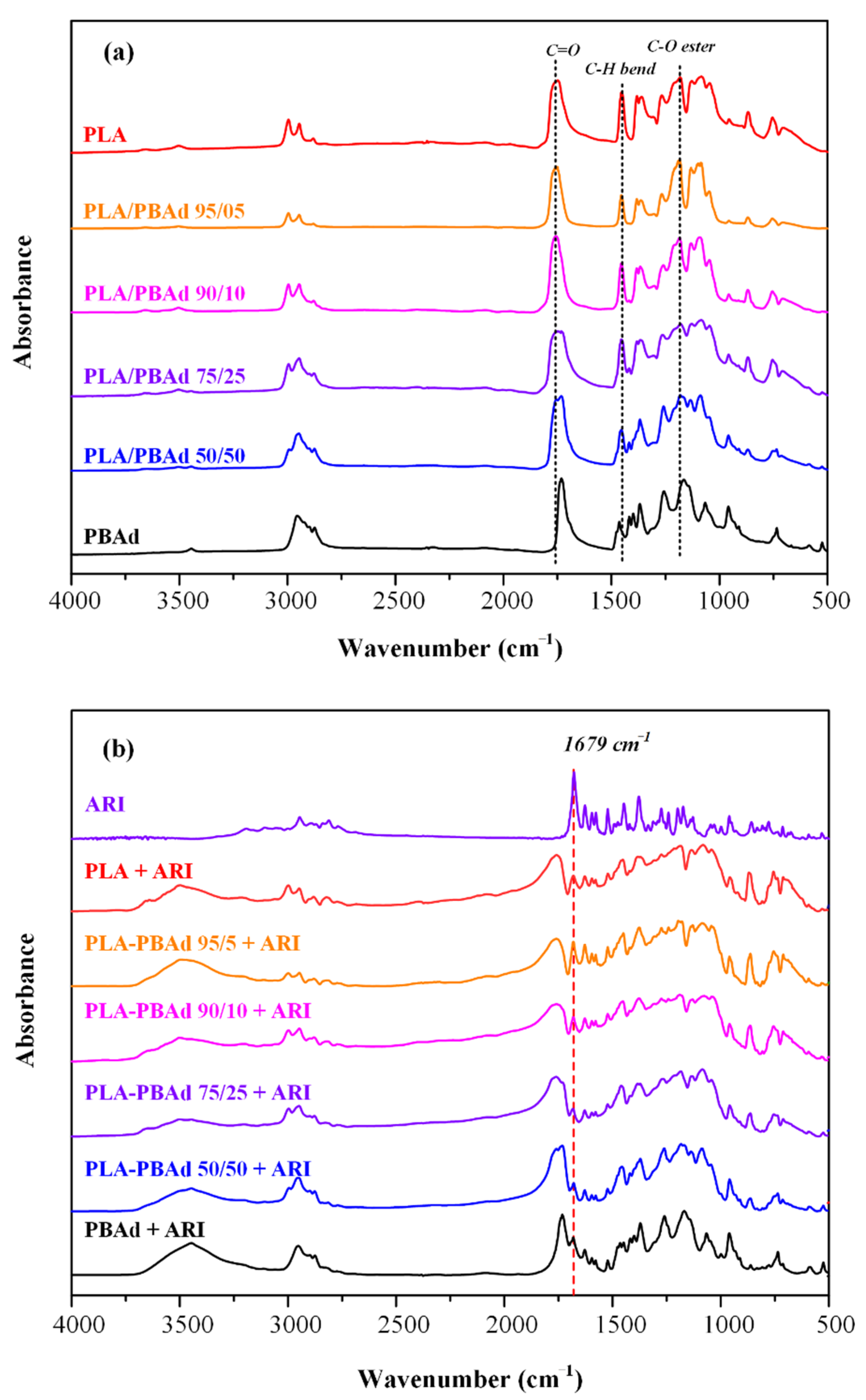
3.2.5. Evaluation of Molecular Interactions
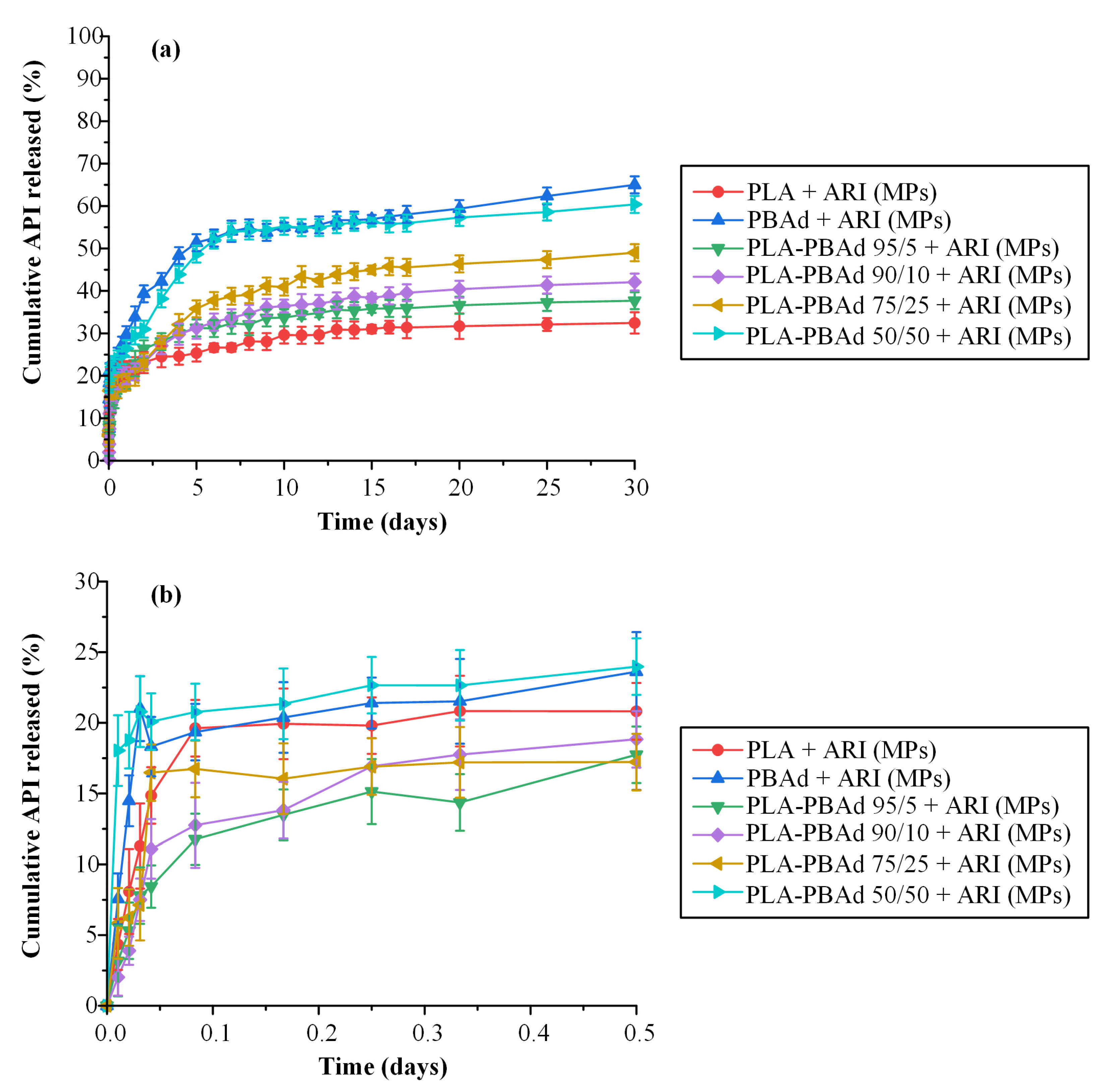
3.2.6. In Vitro Dissolution Profile
Morphology Evaluation after Dissolution Studies
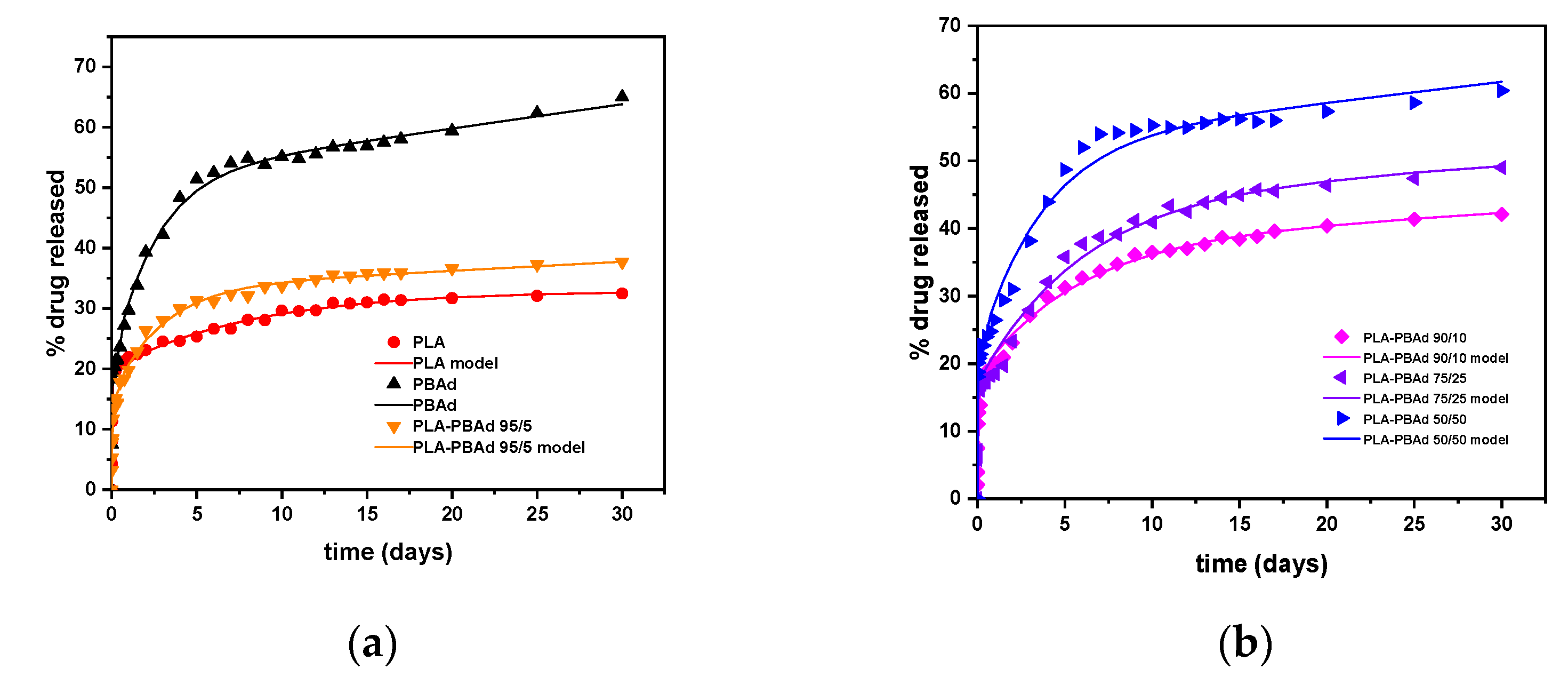
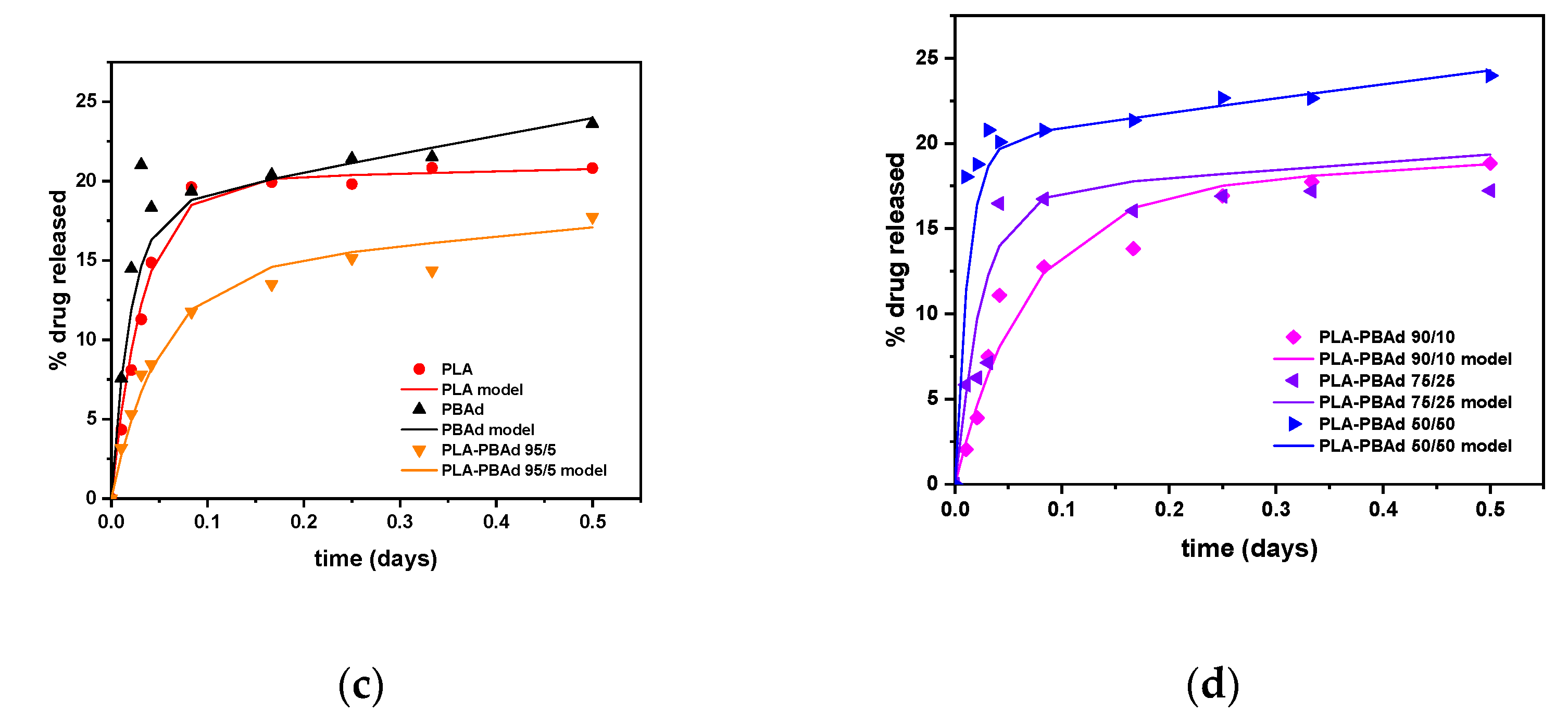
A Mechanistic Release Model
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Karp, J.M.; Langer, R. Development and therapeutic applications of advanced biomaterials. Curr. Opin. Biotechnol. 2007, 18, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Kenry; Liu, B. Recent advances in biodegradable conducting polymers and their biomedical applications. Biomacromolecules 2018, 19, 1783–1803. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Murphy, M.; Li, C.; Ting, K.; Soo, C.; Zheng, Z. Current development of biodegradable polymeric materials for biomedical applications. Drug Des. Dev. Ther. 2018, 12, 3117–3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molavi, F.; Barzegar-Jalali, M.; Hamishehkar, H. Polyester based polymeric nano and microparticles for pharmaceutical purposes: A review on formulation approaches. J. Control Release 2020, 320, 265–282. [Google Scholar] [CrossRef] [PubMed]
- Korzhikov, V.; Averianov, I.; Litvinchuk, E.; Tennikova, T.B. Polyester-based microparticles of different hydrophobicity: The patterns of lipophilic drug entrapment and release. J. Microencapsul. 2016, 33, 199–208. [Google Scholar] [CrossRef]
- Sokolovskaya, E.; Rahmani, S.; Misra, A.C.; Bräse, S.; Lahann, J. Dual-stimuli-responsive microparticles. ACS Appl. Mater. Interfaces 2015, 7, 9744–9751. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, K.; Patel, M.M.; Mehta, P.J. Long-acting injectables: Current perspectives and future promise. Crit. Rev. Ther. Drug Carr. Syst. 2019, 36, 137–181. [Google Scholar] [CrossRef]
- Meyer, J.M. Converting oral to long-acting injectable antipsychotics: A guide for the perplexed. CNS Spectr. 2017, 22, 14–28. [Google Scholar] [CrossRef] [Green Version]
- Pacchiarotti, I.; Tiihonen, J.; Kotzalidis, G.D.; Verdolini, N.; Murru, A.; Goikolea, J.M.; Valentí, M.; Aedo, A.; Vieta, E. Long-acting injectable antipsychotics (LAIs) for maintenance treatment of bipolar and schizoaffective disorders: A systematic review. European Neuropsychopharmacol. 2019, 29, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Otte, A.; Soh, B.K.; Yoon, G.; Yu, D.; Yun, Y.; et al. Injectable, long-acting PLGA formulations: Analyzing PLGA and understanding microparticle formation. J. Control. Release 2019, 304, 125–134. [Google Scholar] [CrossRef]
- Wan, F.; Yang, M. Design of PLGA-based depot delivery systems for biopharmaceuticals prepared by spray drying. Int. J. Pharm. 2016, 498, 82–95. [Google Scholar] [CrossRef]
- Lee, B.K.; Yun, Y.; Park, K. PLA micro- and nano-particles. Adv. Drug Deliv. Rev. 2016, 107, 176–191. [Google Scholar] [CrossRef] [Green Version]
- Cun, D.; Foged, C.; Yang, M.; Frøkjær, S.; Nielsen, H.M. Preparation and characterization of poly(dl-lactide-co-glycolide) nanoparticles for siRNA delivery. Int. J. Pharm. 2010, 390, 70–75. [Google Scholar] [CrossRef]
- Desai, K.-G.H.; Schwendeman, S.P. Active self-healing encapsulation of vaccine antigens in PLGA microspheres. J. Control. Release 2013, 165, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Yuan, W.; Wu, F.; Chen, J.; He, M.; Jin, T. Formulating erythropoietin-loaded sustained-release PLGA microspheres without protein aggregation. J. Control. Release 2008, 130, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Jensen, D.M.K.; Cun, D.; Maltesen, M.J.; Frokjaer, S.; Nielsen, H.M.; Foged, C. Spray drying of siRNA-containing PLGA nanoparticles intended for inhalation. J. Control. Release 2010, 142, 138–145. [Google Scholar] [CrossRef]
- Park, W.; Na, K. Polyelectrolyte complex of chondroitin sulfate and peptide with lower pI value in poly(lactide-co-glycolide) microsphere for stability and controlled release. Colloids Surf. B Biointerfaces 2009, 72, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-m.; Yang, F.; Yang, Y.-q.; Song, F.-l.; Xu, A.-l. Recombinant interferon-alpha2b poly (lactic-co-glycolic acid) microspheres: Pharmacokinetics-pharmacodynamics study in rhesus monkeys following intramuscular administration. Acta Pharmacol. Sin. 2008, 29, 1370–1375. [Google Scholar] [CrossRef] [Green Version]
- Ungaro, F.; d’Emmanuele di Villa Bianca, R.; Giovino, C.; Miro, A.; Sorrentino, R.; Quaglia, F.; La Rotonda, M.I. Insulin-loaded PLGA/cyclodextrin large porous particles with improved aerosolization properties: In vivo deposition and hypoglycaemic activity after delivery to rat lungs. J. Control. Release 2009, 135, 25–34. [Google Scholar] [CrossRef]
- Kumar, R.; Palmieri, M.J. Points to consider when establishing drug product specifications for parenteral microspheres. AAPS J. 2010, 12, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Kim, S.; Park, K. Issues in long-term protein delivery using biodegradable microparticles. J. Control. Release 2010, 146, 241–260. [Google Scholar] [CrossRef]
- Nanaki, S.; Barmpalexis, P.; Papakonstantinou, Z.; Christodoulou, E.; Kostoglou, M.; Bikiaris, D.N. Preparation of new risperidone depot microspheres based on novel biocompatible poly (alkylene adipate) polyesters as long-acting injectable formulations. J. Pharm. Sci. 2018, 107, 2891–2901. [Google Scholar] [CrossRef] [PubMed]
- Siafaka, P.I.; Barmbalexis, P.; Bikiaris, D.N. Novel electrospun nanofibrous matrices prepared from poly (lactic acid)/poly (butylene adipate) blends for controlled release formulations of an anti-rheumatoid agent. Eur. J. Pharm. Sci. 2016, 88, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Atkins, T.W. Fabrication of microcapsules using poly (ethylene adipate) and a blend of poly (ethylene adipate) with poly(hydroxybutyrate-hydroxyvalerate): Incorporation and release of bovine serum albumin. Biomaterials 1997, 18, 173–180. [Google Scholar] [CrossRef]
- Brunner, C.T.; Baran, E.T.; Pinho, E.D.; Reis, R.L.; Neves, N.M. Performance of biodegradable microcapsules of poly (butylene succinate), poly (butylene succinate-co-adipate) and poly (butylene terephthalate-co-adipate) as drug encapsulation systems. Colloids Surf. B Biointerfaces 2011, 84, 498–507. [Google Scholar] [CrossRef] [Green Version]
- Zorba, T.; Chrissafis, K.; Paraskevopoulos, K.M.; Bikiaris, D.N. Synthesis, characterization and thermal degradation mechanism of three poly (alkylene adipate)s: Comparative study. Polym. Degrad. Stab. 2007, 92, 222–230. [Google Scholar] [CrossRef]
- Atkins, T.W. Biodegradation of poly (ethylene adipate) microcapsules in physiological media. Biomaterials 1998, 19, 61–67. [Google Scholar] [CrossRef]
- Karava, V.; Siamidi, A.; Vlachou, M.; Christodoulou, E.; Zamboulis, A.; Bikiaris, D.N.; Kyritsis, A.; Klonos, P.A. Block copolymers based on poly (butylene adipate) and poly (l-lactic acid) for biomedical applications: Synthesis, structure and thermodynamical studies. Soft Matter. 2021, 17, 2439–2453. [Google Scholar] [CrossRef]
- Burris, K.D.; Molski, T.F.; Xu, C.; Ryan, E.; Tottori, K.; Kikuchi, T.; Yocca, F.D.; Molinoff, P.B. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J. Pharmacol. Exp. Ther. 2002, 302, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.M.; Sanchez, R.; Perry, P.P.; Jin, N.; Johnson, B.R.; Forbes, R.A.; McQuade, R.D.; Carson, W.H.; Fleischhacker, W.W. Aripiprazole intramuscular depot as maintenance treatment in patients with schizophrenia: A 52-week, multicenter, randomized, double-blind, placebo-controlled study. J. Clin. Psychiatry 2012, 73, 617–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motiwala, F.B.; Siscoe, K.S.; El-Mallakh, R.S. Review of depot aripiprazole for schizophrenia. Patient Prefer. Adherence 2013, 7, 1181–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanaki, S.; Barmpalexis, P.; Iatrou, A.; Christodoulou, E.; Kostoglou, M.; Bikiaris, D.N. Risperidone controlled release microspheres based on poly (lactic acid)-poly (propylene adipate) novel polymer blends appropriate for long acting injectable formulations. Pharmaceutics 2018, 10, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Jeong, H.; Rho, J.; Shin, J.-Y.; Lee, D.Y.; Hwang, T.; Kim, K.J. Mechanical properties and cytotoxicity of PLA/PCL films. Biomed. Eng. Lett. 2018, 8, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Balla, E.; Daniilidis, V.; Karlioti, G.; Kalamas, T.; Stefanidou, M.; Bikiaris, N.D.; Vlachopoulos, A.; Koumentakou, I.; Bikiaris, D.N. Poly (lactic Acid): A Versatile Biobased Polymer for the Future with Multifunctional Properties—from Monomer Synthesis, Polymerization Techniques and Molecular Weight Increase to PLA Applications. Polymers 2021, 13, 1822. [Google Scholar] [CrossRef]
- Cordewener, F.W.; van Geffen, M.F.; Joziasse, C.A.P.; Schmitz, J.P.; Bos, R.R.M.; Rozema, F.R.; Pennings, A.J. Cytotoxicity of poly(96l/4d-lactide): The influence of degradation and sterilization. Biomaterials 2000, 21, 2433–2442. [Google Scholar] [CrossRef]
- Nanaki, S.G.; Pantopoulos, K.; Bikiaris, D.N. Synthesis of biocompatible poly(ɛ-caprolactone)- block-poly (propylene adipate) copolymers appropriate for drug nanoencapsulation in the form of core-shell nanoparticles. Int. J. Nanomed. 2011, 6, 2981–2995. [Google Scholar] [CrossRef] [Green Version]
- Karavelidis, V.; Giliopoulos, D.; Karavas, E.; Bikiaris, D. Nanoencapsulation of a water soluble drug in biocompatible polyesters. Effect of polyesters melting point and glass transition temperature on drug release behavior. Eur. J. Pharm. Sci. 2010, 41, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Díaz, E.; Puerto, I.; Sandonis, I.; Ribeiro, S.; Lanceros-Mendez, S. Hydrolytic degradation and cytotoxicity of poly (lactic-co-glycolic acid)/multiwalled carbon nanotubes for bone regeneration. J. Appl. Polym. Sci. 2020, 137, 48439. [Google Scholar] [CrossRef]
- Elmowafy, E.M.; Tiboni, M.; Soliman, M.E. Biocompatibility, biodegradation and biomedical applications of poly (lactic acid)/poly (lactic-co-glycolic acid) micro and nanoparticles. J. Pharm. Investig. 2019, 49, 347–380. [Google Scholar] [CrossRef]
- Abou-Zeid, D.-M.; Müller, R.-J.; Deckwer, W.-D. Biodegradation of Aliphatic Homopolyesters and Aliphatic−Aromatic Copolyesters by Anaerobic Microorganisms. Biomacromolecules 2004, 5, 1687–1697. [Google Scholar] [CrossRef]
- Bikiaris, D.N.; Nianias, N.P.; Karagiannidou, E.G.; Docoslis, A. Effect of different nanoparticles on the properties and enzymatic hydrolysis mechanism of aliphatic polyesters. Polym. Degrad. Stab. 2012, 97, 2077–2089. [Google Scholar] [CrossRef]
- Marten, E.; Müller, R.-J.; Deckwer, W.-D. Studies on the enzymatic hydrolysis of polyesters I. Low molecular mass model esters and aliphatic polyesters. Polym. Degrad. Stab. 2003, 80, 485–501. [Google Scholar] [CrossRef]
- Qiu, Z.; Ikehara, T.; Nishi, T. Poly(hydroxybutyrate)/poly (butylene succinate) blends: Miscibility and nonisothermal crystallization. Polymer 2003, 44, 2503–2508. [Google Scholar] [CrossRef]
- Tsuji, H.; Tezuka, Y. Alkaline and enzymatic degradation of L-lactide copolymers, 1. Amorphous-made films of L-lactide copolymers with D-lactide, glycolide, and epsilon-caprolactone. Macromol. Biosci. 2005, 5, 135–148. [Google Scholar] [CrossRef]
- Jiménez, A.; Peltzer, M.; Ruseckaite, R. Poly (Lactic Acid) Science and Technology: Processing, Properties, Additives and Applications; Royal Society of Chemistry: London, UK, 2014. [Google Scholar]
- Łaszcz, M.; Witkowska, A. Studies of phase transitions in the aripiprazole solid dosage form. J. Pharm. Biomed. Anal. 2016, 117, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Brittain, H.G. Chapter 1—aripiprazole: Polymorphs and solvatomorphs. In Profiles of Drug Substances, Excipients and Related Methodology; Brittain, H.G., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 37, pp. 1–29. [Google Scholar]
- Crank, J.; Crank, E.P.J. The Mathematics of Diffusion; Clarendon Press: Oxford, UK, 1979. [Google Scholar]
- Tien, C. Adsorption Calculations and Modeling; Butterworth-Heinemann: Oxford, UK, 1994. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | SEC | DSC | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PBAd | PLA | ||||||||
| Sample | Mw (g/mol) | Mn (g/mol) | PDI | Tc (°C) | Tg (°C) | Tm (°C) | Tc (°C) | Tg (°C) | Tm (°C) |
| PLA | 130k | 73k | 1.79 | - | - | - | - | 55 | 149/155 |
| PLA/PBAd 95/05 | 98k | 61k | 1.60 | 21/32 | - | 53 | - | 53 | 148/155 |
| PLA/PBAd 90/10 | 97k | 59k | 1.65 | 15/32 | - | 52 | - | 54 | 148/155 |
| PLA/PBAd 75/25 | 95k | 57k | 1.66 | 2/31 | –64 | 52/55 | - | 54 | 147/154 |
| PLA/PBAd 50/50 | 98k | 57k | 1.73 | 28 | –60 | 52/55 | - | 54 | 148/154 |
| PBAd | 90k | 49k | 1.85 | 29 | –62 | 55 | - | - | - |
| Sample | Mw (g/mol) | Mn (g/mol) | % Weight Loss |
|---|---|---|---|
| PLA | 129.7k | 72.8k | 1.267 |
| PLA/PBAd 95/05 | 97.8k | 60.8k | 2.109 |
| PLA/PBAd 90/10 | 97.1k | 58.6k | 2.837 |
| PLA/PBAd 75/25 | 93.9k | 56.9k | 7.230 |
| PLA/PBAd 50/50 | 97.9k | 57.1k | 9.178 |
| PBAd | 88.9k | 47.9k | 10.912 |
| Sample | Average Particle Size (d50) (μm) | Yield (%) | Drug Loading (%) | EE (%) |
|---|---|---|---|---|
| PLA | 56.3 ± 15 | 77.73 ± 2.84 | 16.35 ± 1.75 | 42.73 ± 2.08 |
| PLA/PBAd 95/5 | 58.2 ± 15 | 89.32 ± 2.03 | 14.56 ± 1.86 | 44.84 ± 2.87 |
| PLA/PBAd 90/10 | 43.3 ± 10 | 92.51 ± 2.48 | 13.19 ± 2.83 | 38.17 ± 3.54 |
| PLA/PBAd 75/25 | 30.2 ± 10 | 98.60 ± 1.38 | 12.48 ± 2.16 | 39.78 ± 2.86 |
| PLA/PBAd 50/50 | 18.8 ± 5 | 97.40 ± 1.24 | 11.30 ± 3.14 | 32.67 ± 3.07 |
| PBAd | 21.3 ± 5 | 60.35 ± 3.68 | 17.48 ± 2.47 | 36.52 ± 4.23 |
| Sample | Remained ARI (%) |
|---|---|
| PLA | 66.28 ± 0.23 |
| PLA/PBAd 95/5 | 60.08 ± 2.12 |
| PLA/PBAd 90/10 | 58.42 ± 1.54 |
| PLA/PBAd 75/25 | 50.63 ± 2.03 |
| PLA/PBAd 50/50 | 38.94 ± 1.78 |
| PBAd | 34.78 ± 2.27 |
| Release Fitting Model | PLA | PBAd | PLA/PBAd | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 95/5 | 90/10 | 75/25 | 5050 | |||||||||
| R2 | k-Constant | R2 | k-Constant | R2 | k-Constant | R2 | k-Constant | R2 | k-Constant | R2 | k-Constant | |
| Fast-release phase | ||||||||||||
| Zero order | 0.60 | 2.77 d−1 | 0.77 | 8.03 d−1 | 0.68 | 4.61 d−1 | 0.67 | 4.32 d−1 | 0.82 | 5.54 d−1 | 0.80 | 7.43 d−1 |
| First order | <0.01 | 0.09 d−1 | 0.58 | 0.20 d−1 | 0.16 | 0.11 d−1 | 0.12 | 0.10 d−1 | 0.54 | 0.11 d−1 | 0.57 | 0.17d−1 |
| Higuchi | 0.31 | 14.84 d−1 | 0.93 | 25.45 d−1/2 | 0.83 | 16.45 d−1/2 | 0.81 | 16.03 d−1/2 | 0.94 | 16.67 d−1/2 | 0.93 | 22.95 d−1/2 |
| Hixson–Crowell | <0.01 | 0.05 d−1 | 0.69 | 0.06 d−1 | 0.08 | 0.03 d−1 | 0.04 | 0.03 d−1 | 0.48 | 0.03 d−1 | 0.49 | 0.05 d−1 |
| Korsmeyer–Peppas | 0.99 | 21.90 d−n | 0.99 | 29.93 d−n | 0.99 | 20.66 d−n | 0.98 | 20.35 d−n | 0.98 | 19.06 d−n | 0.98 | 26.70 d−n |
| Slow-release phase | ||||||||||||
| Zero order | 0.77 | 4.20 d−1 | 0.98 | 3.91 d−1 | 0.84 | 3.98 d−1 | 0.89 | 4.02 d−1 | 0.87 | 4.08 d−1 | 0.93 | 3.42 d−1 |
| First order | 0.69 | 0.07 d−1 | 0.71 | 0.03 d−1 | 0.75 | 0.06 d−1 | 0.77 | 0.06 d−1 | 0.74 | 0.06 d−1 | 0.80 | 0.04 d−1 |
| Higuchi | 0.60 | 16.64 d−1/2 | 0.53 | 10.58 d−1/2 | 0.66 | 16.08 d−1/2 | 0.66 | 15.21 d−1/2 | 0.62 | 14.60 d−1/2 | 0.66 | 12.79 d−1/2 |
| Hixson–Crowell | 0.74 | 0.02 d−1 | 0.76 | 0.01 d−1 | 0.81 | 0.02 d−1 | 0.83 | 0.02 d−1 | 0.79 | 0.01 d−1 | 0.85 | 0.01 d−1 |
| Korsmeyer–Peppas | 0.76 | 5.21 d−n | 0.98 | 0.61 d−n | 0.83 | 4.99 d−n | 0.88 | 3.89 d−n | 0.85 | 3.31 d−n | 0.93 | 2.62 d−n |
| Material | φ1 | φ2 | k1 (d−1) | k2 (d−1) | k3 (d−1) | D × 1017 (m2/s) |
|---|---|---|---|---|---|---|
| PLA | 20 | 13 | 30 | 0.12 | 0 | 7.4 |
| PLA-PBAd 95/5 | 14.5 | 18.25 | 20 | 0.36 | 0.15 | 23.5 |
| PLA-PBAd 90/10 | 17.5 | 20 | 15 | 0.18 | 0.15 | 6.5 |
| PLA-PBAd 75/25 | 16.25 | 28 | 40 | 0.17 | 0.15 | 3 |
| PLA-PBAd 50/50 | 20.5 | 31.7 | 80 | 0.28 | 0.3 | 1.9 |
| PBAd | 19 | 32 | 50 | 0.4 | 0.4 | 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karava, V.; Siamidi, A.; Vlachou, M.; Christodoulou, E.; Bikiaris, N.D.; Zamboulis, A.; Kostoglou, M.; Gounari, E.; Barmpalexis, P. Poly(l-Lactic Acid)-co-poly(Butylene Adipate) New Block Copolymers for the Preparation of Drug-Loaded Long Acting Injectable Microparticles. Pharmaceutics 2021, 13, 930. https://doi.org/10.3390/pharmaceutics13070930
Karava V, Siamidi A, Vlachou M, Christodoulou E, Bikiaris ND, Zamboulis A, Kostoglou M, Gounari E, Barmpalexis P. Poly(l-Lactic Acid)-co-poly(Butylene Adipate) New Block Copolymers for the Preparation of Drug-Loaded Long Acting Injectable Microparticles. Pharmaceutics. 2021; 13(7):930. https://doi.org/10.3390/pharmaceutics13070930
Chicago/Turabian StyleKarava, Vasiliki, Aggeliki Siamidi, Marilena Vlachou, Evi Christodoulou, Nikolaos D. Bikiaris, Alexandra Zamboulis, Margaritis Kostoglou, Eleni Gounari, and Panagiotis Barmpalexis. 2021. "Poly(l-Lactic Acid)-co-poly(Butylene Adipate) New Block Copolymers for the Preparation of Drug-Loaded Long Acting Injectable Microparticles" Pharmaceutics 13, no. 7: 930. https://doi.org/10.3390/pharmaceutics13070930
APA StyleKarava, V., Siamidi, A., Vlachou, M., Christodoulou, E., Bikiaris, N. D., Zamboulis, A., Kostoglou, M., Gounari, E., & Barmpalexis, P. (2021). Poly(l-Lactic Acid)-co-poly(Butylene Adipate) New Block Copolymers for the Preparation of Drug-Loaded Long Acting Injectable Microparticles. Pharmaceutics, 13(7), 930. https://doi.org/10.3390/pharmaceutics13070930