
A Quantitative Pharmacology Model of Exosome-Mediated Drug Efflux and Perturbation-Induced Synergy

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Effects of PTX, OME, and GW on Exosome Production/Excretion
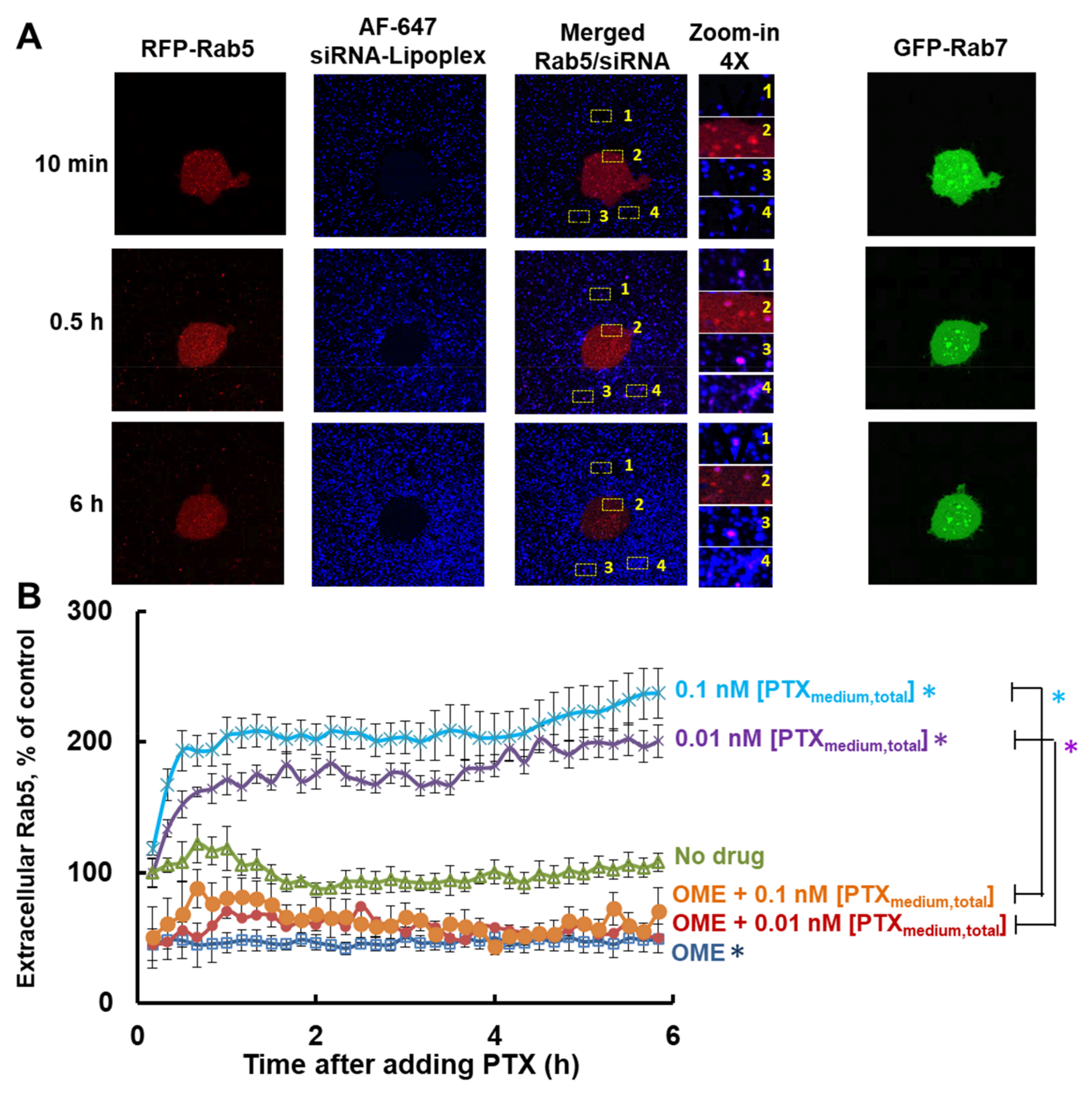
3.2. Effects of PTX and OME on Endocytic Transport
3.3. OME and GW Pretreatment Altered PTX Efflux
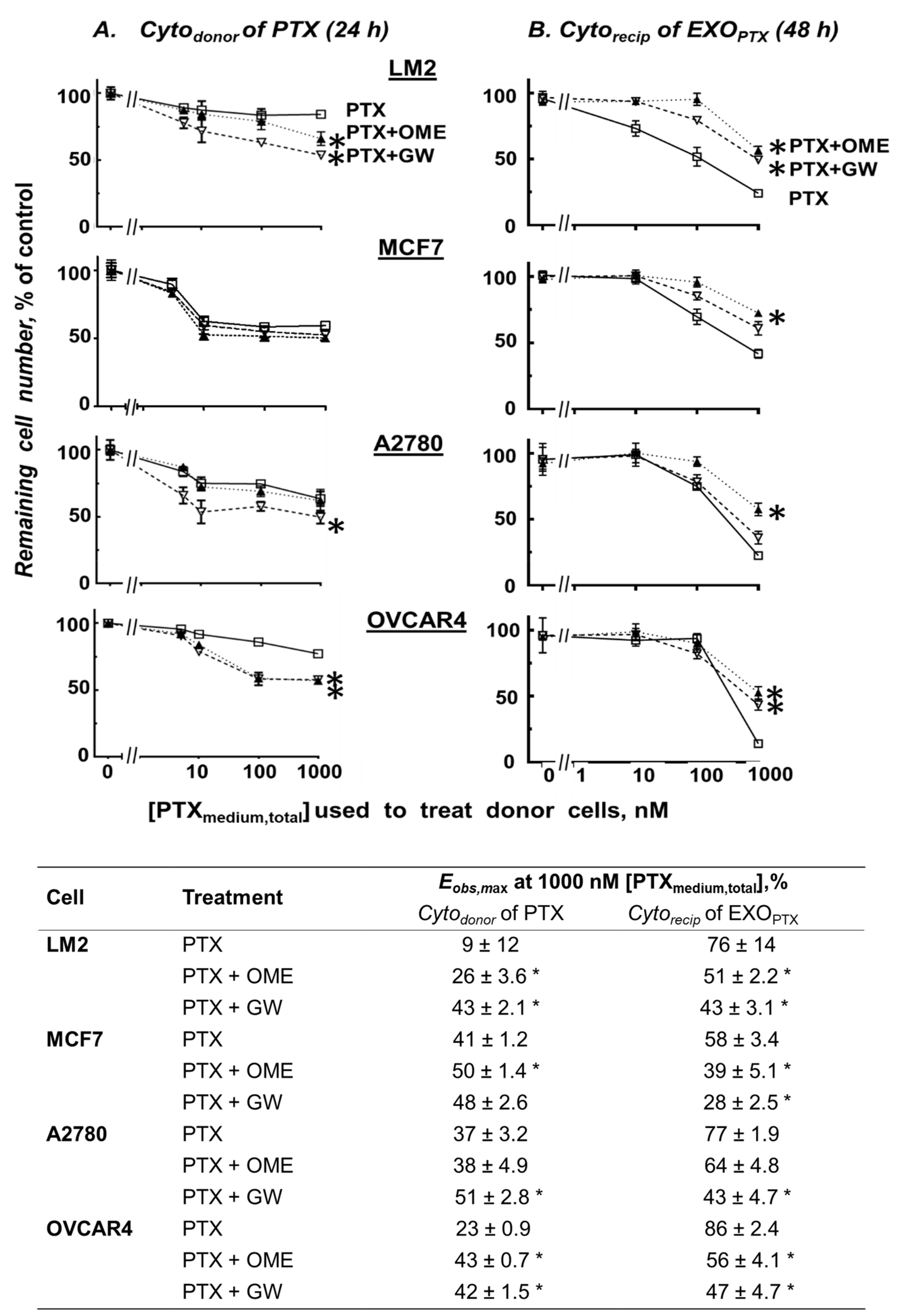
3.4. OME and GW Pretreatment Altered Cytodonor of PTX and Cytorecip of Exosomes Collected from Donor Cells Treated with a Drug (EXOdrug)
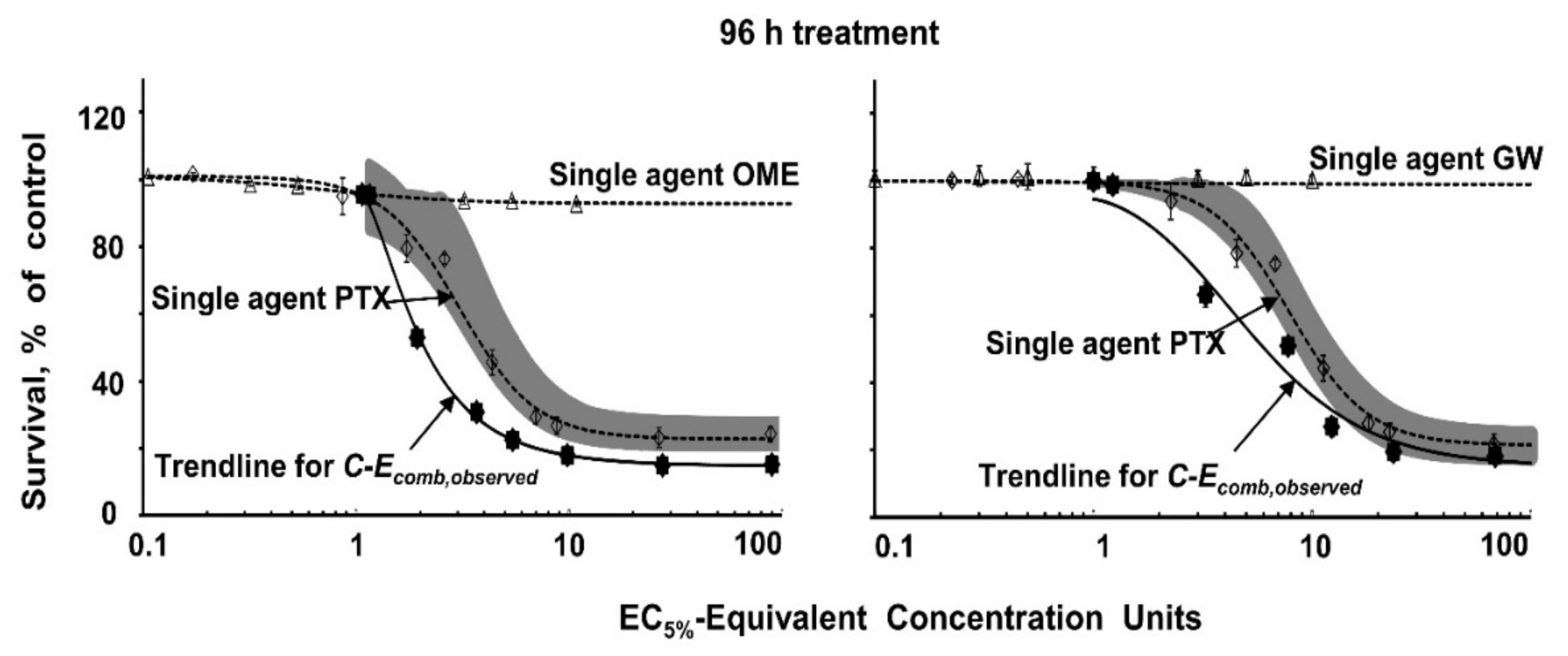
3.5. Analysis of Drug Interactivity
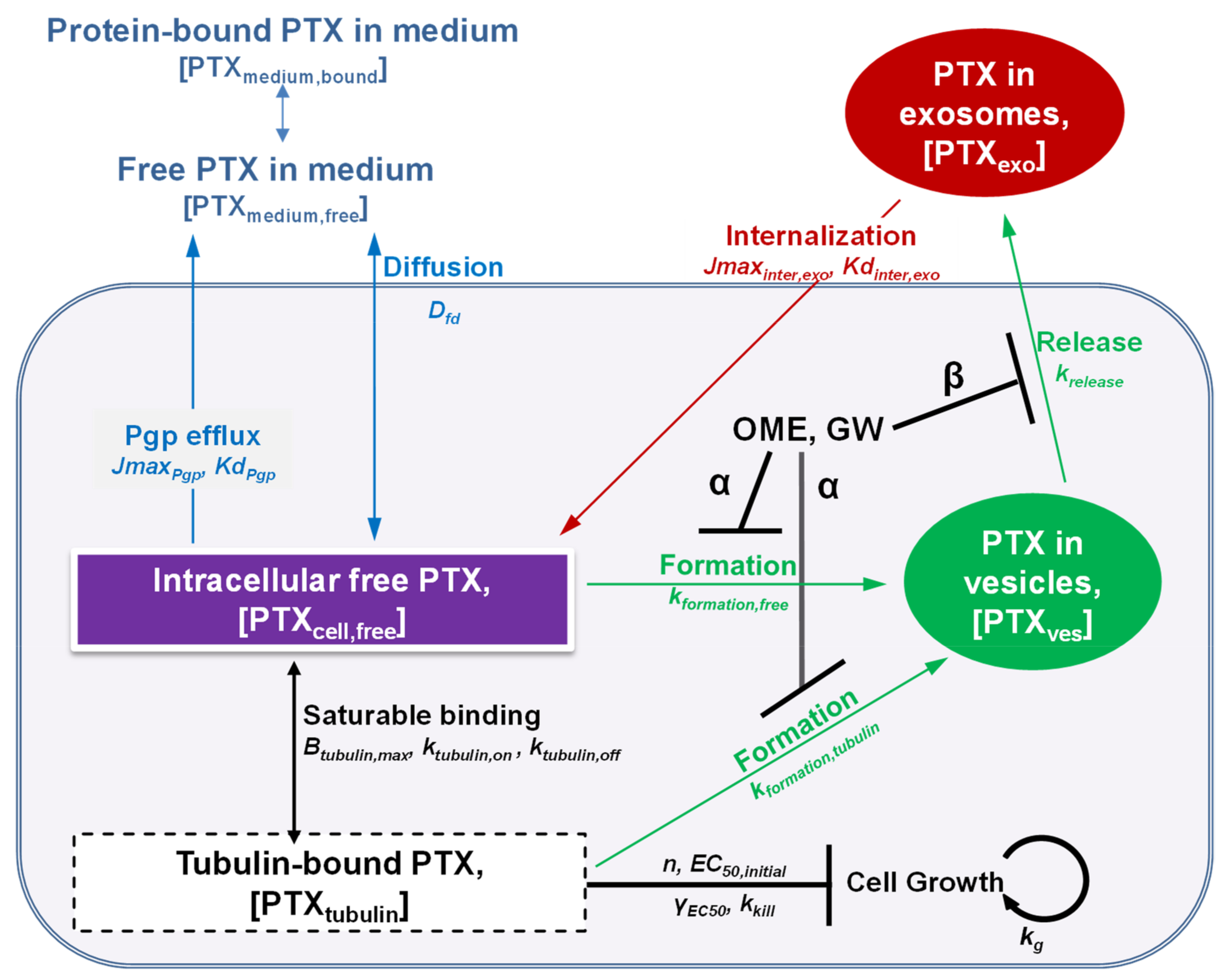
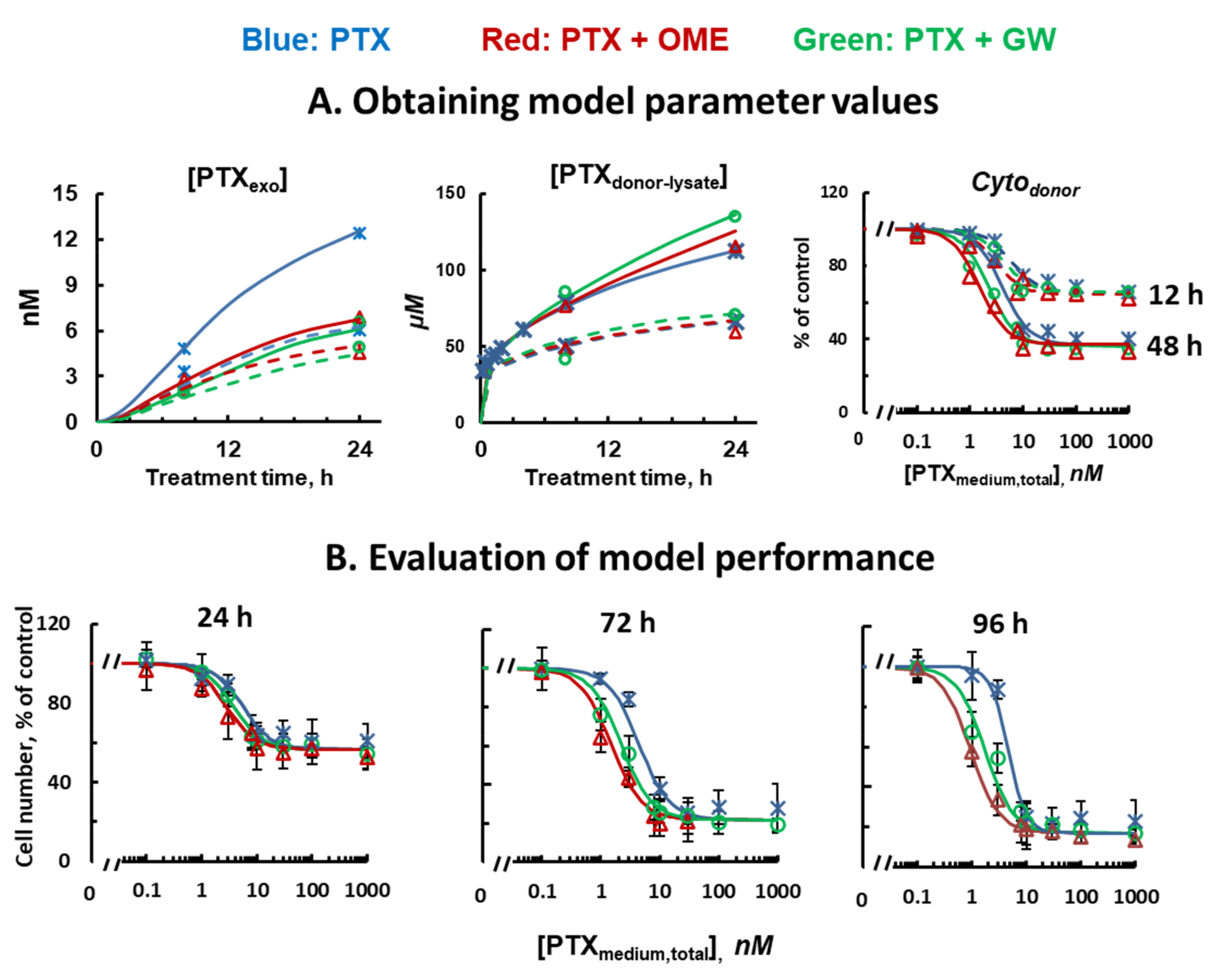
3.6. Quantitative Pharmacology Model and Evaluation of Model Performance
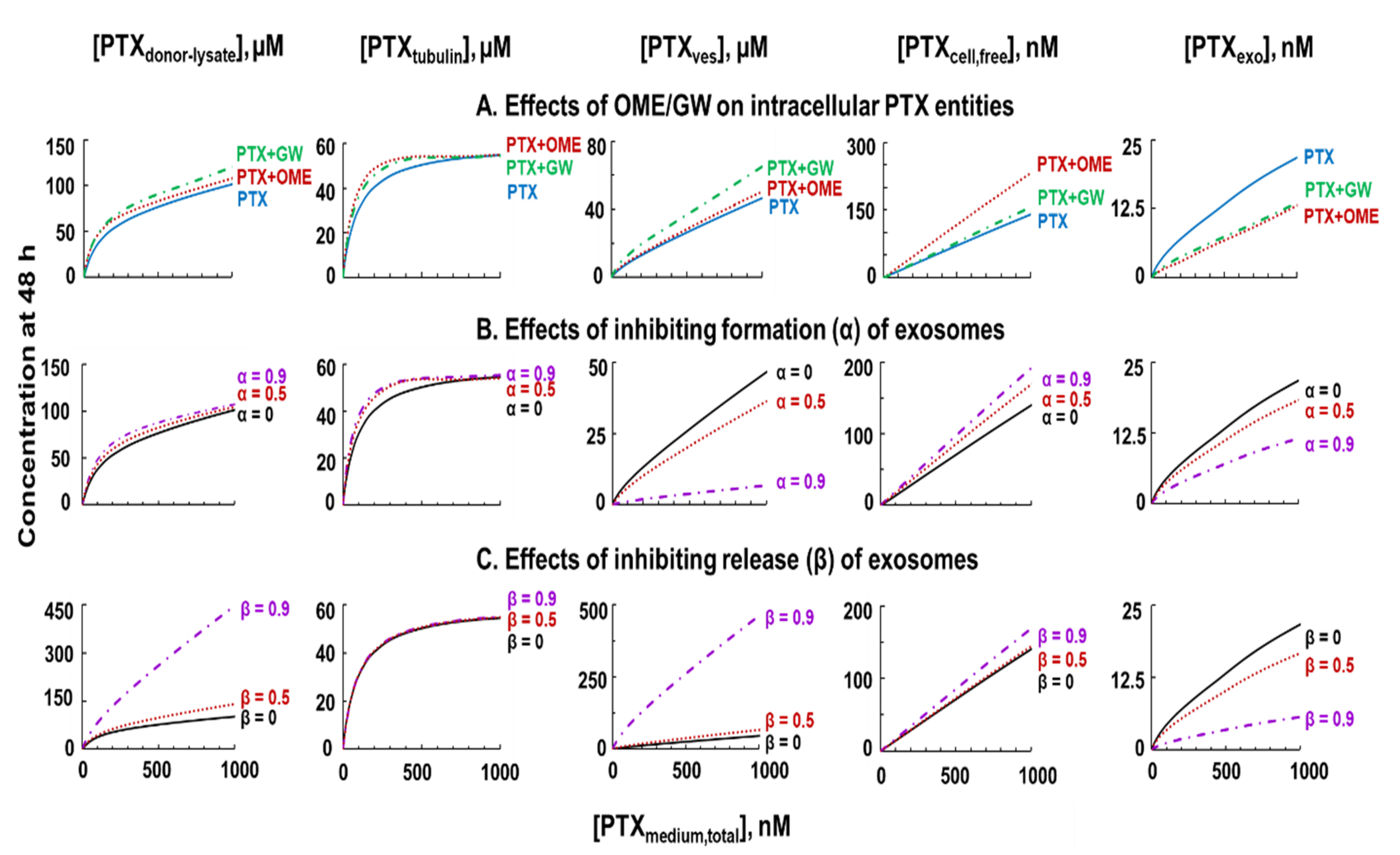
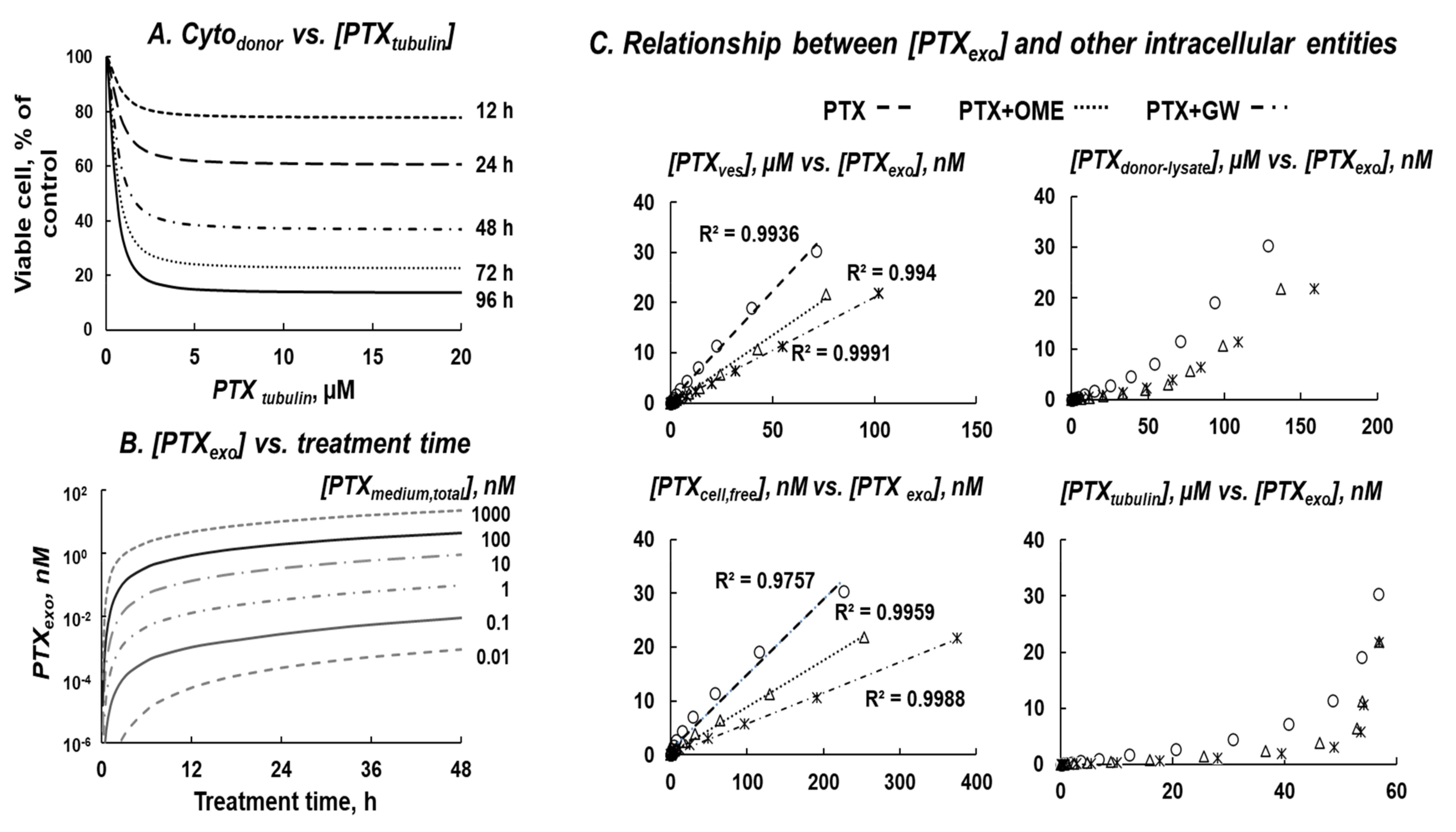
3.7. QP Model-Based Simulations to Quantify Non-Measurable PTX Entities and Intracellular Processes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Akuma, P.; Okagu, O.D.; Udenigwe, C.C. Naturally Occurring Exosome Vesicles as Potential Delivery Vehicle for Bioactive Compounds. Front. Sustain. Food Syst. 2019, 3. [Google Scholar] [CrossRef]
- Naslavsky, N.; Caplan, S. The enigmatic endosome—Sorting the ins and outs of endocytic trafficking. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, L.; Ding, J.; Chen, C.; Wu, Z.J.; Liu, B.; Gao, Y.; Chen, W.; Liu, F.; Sun, W.; Li, X.F.; et al. Exosome-Transmitted lncARSR Promotes Sunitinib Resistance in Renal Cancer by Acting as a Competing Endogenous RNA. Cancer Cell 2016, 29, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Stone, L. Kidney cancer: Exosome transmission of sunitinib resistance. Nature reviews. Urology 2016, 13, 297. [Google Scholar] [CrossRef]
- Muller, L.; Mitsuhashi, M.; Simms, P.; Gooding, W.E.; Whiteside, T.L. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci. Rep. 2016, 6, 20254. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nature reviews. Immunology 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yeung, B.Z.; Cui, M.; Peer, C.J.; Lu, Z.; Figg, W.D.; Guillaume Wientjes, M.; Woo, S.; Au, J.L. Exosome is a mechanism of intercellular drug transfer: Application of quantitative pharmacology. J. Control Release 2017, 268, 147–158. [Google Scholar] [CrossRef]
- Avnet, S.; Lemma, S.; Cortini, M.; Pellegrini, P.; Perut, F.; Zini, N.; Kusuzaki, K.; Chano, T.; Grisendi, G.; Dominici, M.; et al. Altered pH gradient at the plasma membrane of osteosarcoma cells is a key mechanism of drug resistance. Oncotarget 2016, 7, 63408–63423. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Yu, S.; Zhou, L.; Shi, M.; Hu, Y.; Xu, X.; Shen, B.; Liu, S.; Yan, D.; Feng, J. Cisplatin-resistant lung cancer cell-derived exosomes increase cisplatin resistance of recipient cells in exosomal miR-100-5p-dependent manner. Int. J. Nanomed. 2017, 12, 3721–3733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol. Cancer Ther. 2005, 4, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharm. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullan, J.E.; Confeld, M.I.; Osborn, J.K.; Kim, J.; Sarkar, K.; Mallik, S. Exosomes as Drug Carriers for Cancer Therapy. Mol. Pharm. 2019, 16, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wu, D.; Ma, X.; Wang, J.; Hou, W.; Zhang, W. Exosomes as drug carriers for cancer therapy and challenges regarding exosome uptake. Biomed. Pharm. 2020, 128, 110237. [Google Scholar] [CrossRef]
- Oliveira, C.; Calmeiro, J.; Carrascal, M.A.; Falcao, A.; Gomes, C.; Miguel Neves, B.; Teresa Cruz, M. Exosomes as new therapeutic vectors for pancreatic cancer treatment. Eur. J. Pharm. Biopharm. 2021, 161, 4–14. [Google Scholar] [CrossRef]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Kandimalla, R.; Wallen, M.; Tyagi, N.; Wilcher, S.; Yan, J.; Schultz, D.J.; Spencer, W.; et al. Exosome-mediated delivery of RNA and DNA for gene therapy. Cancer Lett. 2021, 505, 58–72. [Google Scholar] [CrossRef]
- Au, J.L.; Yeung, B.Z.; Wientjes, M.G.; Lu, Z.; Wientjes, M.G. Delivery of cancer therapeutics to extracellular and intracellular targets: Determinants, barriers, challenges and opportunities. Adv. Drug Deliv. Rev. 2016, 97, 280–301. [Google Scholar] [CrossRef] [Green Version]
- Au, J.L.; Abbiati, R.A.; Wientjes, M.G.; Lu, Z. Target Site Delivery and Residence of Nanomedicines: Application of Quantitative Systems Pharmacology. Pharm. Rev. 2019, 71, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Essandoh, K.; Yang, L.; Wang, X.; Huang, W.; Qin, D.; Hao, J.; Wang, Y.; Zingarelli, B.; Peng, T.; Fan, G.C. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim. Biophys. Acta 2015, 1852, 2362–2371. [Google Scholar] [CrossRef] [Green Version]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federici, C.; Petrucci, F.; Caimi, S.; Cesolini, A.; Logozzi, M.; Borghi, M.; D’Ilio, S.; Lugini, L.; Violante, N.; Azzarito, T.; et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS ONE 2014, 9, e88193. [Google Scholar] [CrossRef] [Green Version]
- Kuh, H.J.; Jang, S.H.; Wientjes, M.G.; Au, J.L. Computational model of intracellular pharmacokinetics of paclitaxel. The Journal of pharmacology and experimental therapeutics. J. Pharmacol. Exp. Ther. 2000, 293, 761–770. [Google Scholar] [PubMed]
- Au, J.L.; Li, D.; Gan, Y.; Gao, X.; Johnson, A.L.; Johnston, J.; Millenbaugh, N.J.; Jang, S.H.; Kuh, H.J.; Chen, C.T.; et al. Pharmacodynamics of immediate and delayed effects of paclitaxel: Role of slow apoptosis and intracellular drug retention. Cancer Res. 1998, 58, 2141–2148. [Google Scholar]
- Jang, S.H.; Wientjes, M.G.; Au, J.L. Kinetics of P-glycoprotein-mediated efflux of paclitaxel. J. Pharmacol. Exp. Ther. 2001, 298, 1236–1242. [Google Scholar] [PubMed]
- Schwarz, G. Estimating the Dimension of a Model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Hu, G.; Chong, R.A.; Yang, Q.; Wei, Y.; Blanco, M.A.; Li, F.; Reiss, M.; Au, J.L.; Haffty, B.G.; Kang, Y. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell 2009, 15, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Wientjes, M.G.; Au, J.L. Evaluation of combination chemotherapy: Integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin. Cancer Res. 2004, 10, 7994–8004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Au, J.L.; Wientjes, M.G. Comparison of methods for evaluating drug-drug interaction. Front. Biosci. 2010, 2, 241–249. [Google Scholar]
- Zhao, L.; Au, J.L.; Wientjes, M.G. Method to Assess Interactivity of Drugs with Nonparallel Concentration-Effect Relationships. Curr. Cancer Drug Targets 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharm. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Lu, Z.; Yeung, B.Z.; Wientjes, M.G.; Cole, D.J.; Au, J.L. Tumor priming enhances siRNA delivery and transfection in intraperitoneal tumors. J. Control Release 2014, 178, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Shukla, R.S.; Jain, A.; Zhao, Z.; Cheng, K. Intracellular trafficking and exocytosis of a multi-component siRNA nanocomplex. Nanomedicine 2016, 12, 1323–1334. [Google Scholar] [CrossRef] [Green Version]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular uptake of nanoparticles: Journey inside the cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Wientjes, M.G.; Au, J.L.-S. Delivery of nanomedicines to extracellular and intracellular compartments of a solid tumor. Adv. Drug Deliv. Rev. 2012, 64, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.Y.; Jeon, H.K.; Hong, J.E.; Cho, Y.J.; Ryu, J.Y.; Choi, J.J.; Lee, S.H.; Yoon, G.; Kim, W.Y.; Do, I.G.; et al. Proton pump inhibitors enhance the effects of cytotoxic agents in chemoresistant epithelial ovarian carcinoma. Oncotarget 2015, 6, 35040–35050. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.Y.; Zhang, J.; Wang, J.L.; Sun, S.; Wang, Z.H.; Wang, L.P.; Zhang, Q.L.; Lv, F.F.; Cao, E.Y.; Shao, Z.M.; et al. Intermittent high dose proton pump inhibitor enhances the antitumor effects of chemotherapy in metastatic breast cancer. J. Exp. Clin. Cancer Res. 2015, 34, 85. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.W.; Zhao, F.; Wang, J.Y.; Wang, H.Y.; Ge, S.H.; Wang, X.; Zhang, L.; Liu, R.; Ba, Y.; Li, H.L.; et al. Tumor microenvironment interruption: A novel anti-cancer mechanism of Proton-pump inhibitor in gastric cancer by suppressing the release of microRNA-carrying exosomes. Am. J. Cancer Res. 2017, 7, 1913–1925. [Google Scholar] [PubMed]
- Azzarito, T.; Venturi, G.; Cesolini, A.; Fais, S. Lansoprazole induces sensitivity to suboptimal doses of paclitaxel in human melanoma. Cancer Lett. 2015, 356, 697–703. [Google Scholar] [CrossRef]
- Li, M.; Nguyen, L.; Subramaniyan, B.; Bio, M.; Peer, C.J.; Kendrick, J.; Figg, W.D.; Woo, S.; You, Y. PBPK modeling-based optimization of site-specific chemo-photodynamic therapy with far-red light-activatable paclitaxel prodrug. J. Control Release 2019, 308, 86–97. [Google Scholar] [CrossRef]
- Li, M.; Thapa, P.; Rajaputra, P.; Bio, M.; Peer, C.J.; Figg, W.D.; You, Y.; Woo, S. Quantitative modeling of the dynamics and intracellular trafficking of far-red light-activatable prodrugs: Implications in stimuli-responsive drug delivery system. J. Pharmacokinet. Pharmacodyn. 2017, 44, 521–536. [Google Scholar] [CrossRef]
- Wozniak, K.M.; Vornov, J.J.; Wu, Y.; Nomoto, K.; Littlefield, B.A.; DesJardins, C.; Yu, Y.; Lai, G.; Reyderman, L.; Wong, N.; et al. Sustained Accumulation of Microtubule-Binding Chemotherapy Drugs in the Peripheral Nervous System: Correlations with Time Course and Neurotoxic Severity. Cancer Res. 2016, 76, 3332–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineda, J.J.; Miller, M.A.; Song, Y.; Kuhn, H.; Mikula, H.; Tallapragada, N.; Weissleder, R.; Mitchison, T.J. Site occupancy calibration of taxane pharmacology in live cells and tissues. Proc. Natl. Acad. Sci. USA 2018, 115, E11406–E11414. [Google Scholar] [CrossRef] [Green Version]
- NIH White Paper by the QSP Workshop Group. Available online: https://www.nigms.nih.gov/training/documents/systemspharmawpsorger2011.pdf (accessed on 12 June 2021).
- Wang, Y.; Zhu, H.; Madabushi, R.; Liu, Q.; Huang, S.M.; Zineh, I. Model-Informed Drug Development: Current US Regulatory Practice and Future Considerations. Clin. Pharmacol. Ther. 2019, 105, 899–911. [Google Scholar] [CrossRef]
- Fang, L.; Kim, M.J.; Li, Z.; Wang, Y.; DiLiberti, C.E.; Au, J.; Hooker, A.; Ducharme, M.P.; Lionberger, R.; Zhao, L. Model-Informed Drug Development and Review for Generic Products: Summary of FDA Public Workshop. Clin. Pharm. Ther. 2018, 104, 27–30. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell | Exosome Recovered in Conditioned Medium | |||||
|---|---|---|---|---|---|---|
| No Drug, Number/106 Cells | % Change from Control | |||||
| +PTX | +OME | +GW | +OME + PTX | +GW + PTX | ||
| LM2 | 362 ± 29 | 69 ± 6.4 * | −26 ± 3.9 * | −26 ± 4.0 * | −11 ± 2.0 ** | −30 ± 7.7 * |
| MCF7 | 266 ± 17 | 43 ± 6.3 * | −43 ± 6.0 * | −21 ± 2.4 * | −12 ± 3.2 ** | −26 ± 7.4 * |
| A2780 | 227 ±39 | 55 ± 3.1 * | −1.9 ± 3.7 | −9.1 ± 2.7 | −6.9 ± 4.1 | −12 ± 4.5 |
| OVCAR4 | 265 ± 23 | 50 ± 2.9 * | −4.8 ± 2.5 | −9.6 ± 1.1 | −0.9 ± 4.2 | −2.5 ± 2.1 |
| pmol/106 Donor Cells | % Change Compared to Single Agent PTX | ||||||
|---|---|---|---|---|---|---|---|
| OME + PTX | GW + PTX | ||||||
| [PTXexo] | [PTXdonor-lysate] | [PTXexo] | [PTXdonor-lysate] | [PTXexo] | [PTXdonor-lysate] | ||
| Cell | A. Different cells treated with 1000 nM PTX for 24 h | ||||||
| LM2 | 6 ± 1 | 32 ± 1 | −25 ± 17 | +34 ± 8.1 | −79 ± 13 * | +66 ± 20 * | |
| MCF7 | 12 ± 1 | 76 ± 7 | −42 ± 9 * | +15 ± 8 | −49 ± 5 * | +23 ± 8 | |
| A2780 | 5 ± 1 | 52 ± 6 | −22 ± 2 | +131± 9 * | −34 ± 8 * | +105 ± 7 * | |
| OVCAR4 | 7 ± 2 | 72 ± 6 | −45 ± 5 * | +26 ± 10 | −74 ± 7 * | +97 ± 3 * | |
| [PTXmedium,total] | B. MCF7 cells treated with 300 or 1000 nM PTX for 8 or 24 h | ||||||
| 300 nM | 8 h | 21 ± 9 | 34 ± 13 | −36 ± 18 * | −4 ± 11 | −44 ± 14 * | +19 ± 9 |
| 24 h | 11 ± 6 | 45 ± 2 | −25 ± 13 | +11 ± 7 | −19 ± 6 | +7 ± 12 | |
| 1000 nM | 8 h | 19 ± 8 | 53 ± 9 | −41 ± 17 * | +3 ± 5 | −54 ± 19 * | +8 ± 3 |
| 24 h | 12 ± 1 | 76 ± 7 | −42 ± 9 * | +15 ± 8 | −49 ± 5 * | +23 ± 8 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Yeung, B.Z.; Wientjes, M.G.; Cui, M.; Peer, C.J.; Lu, Z.; Figg, W.D.; Woo, S.; Au, J.L.-S. A Quantitative Pharmacology Model of Exosome-Mediated Drug Efflux and Perturbation-Induced Synergy. Pharmaceutics 2021, 13, 997. https://doi.org/10.3390/pharmaceutics13070997
Wang J, Yeung BZ, Wientjes MG, Cui M, Peer CJ, Lu Z, Figg WD, Woo S, Au JL-S. A Quantitative Pharmacology Model of Exosome-Mediated Drug Efflux and Perturbation-Induced Synergy. Pharmaceutics. 2021; 13(7):997. https://doi.org/10.3390/pharmaceutics13070997
Chicago/Turabian StyleWang, Jin, Bertrand Z. Yeung, M. Guillaume Wientjes, Minjian Cui, Cody J. Peer, Ze Lu, William D. Figg, Sukyung Woo, and Jessie L.-S. Au. 2021. "A Quantitative Pharmacology Model of Exosome-Mediated Drug Efflux and Perturbation-Induced Synergy" Pharmaceutics 13, no. 7: 997. https://doi.org/10.3390/pharmaceutics13070997
APA StyleWang, J., Yeung, B. Z., Wientjes, M. G., Cui, M., Peer, C. J., Lu, Z., Figg, W. D., Woo, S., & Au, J. L. -S. (2021). A Quantitative Pharmacology Model of Exosome-Mediated Drug Efflux and Perturbation-Induced Synergy. Pharmaceutics, 13(7), 997. https://doi.org/10.3390/pharmaceutics13070997