
Improving the Solubility and Oral Bioavailability of a Novel Aromatic Aldehyde Antisickling Agent (PP10) for the Treatment of Sickle Cell Disease

 ,
,  ,
,  , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
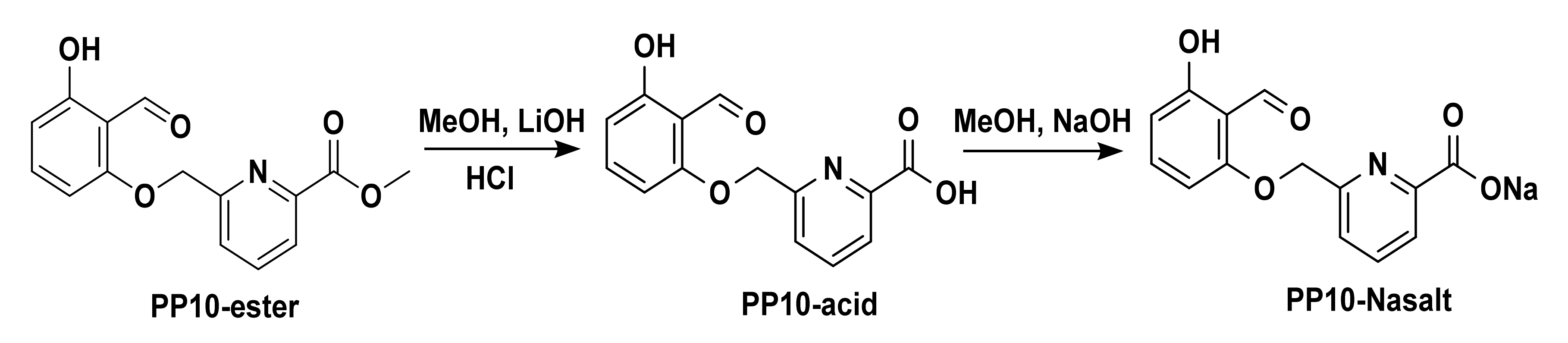
2.2. Synthesis of PP10, the Acid Form of PP10 (PP10-Acid), and Its Sodium Salt (PP10-NaSalt)
2.3. Stability of PP10 at Different pHs
2.4. In Vitro Antisickling, Hb Modification, and Hb Oxygen Equilibrium Studies of PP10 and Its Acid Analog with Sickle Whole Blood
2.5. Preparation of Polymeric-PP10 Binary Systems
2.6. Solubility Study of PP10 and the Polymeric-PP10 Binary Systems
2.7. Physicochemical Characterization of the Polymeric-PP10 Binary System
2.7.1. Differential Scanning Calorimetry (DSC)
2.7.2. Fourier Transform Infrared (FT-IR) Spectroscopy
2.7.3. X-ray Powder Diffraction (XRPD)
2.8. Formulation and Characterization of PP10 Oral Tablets
2.9. Preparation and Characterization of PP10 Intravenous Formulation
2.10. Pharmacokinetics Study
2.11. Statistical Analysis
3. Results and Discussion
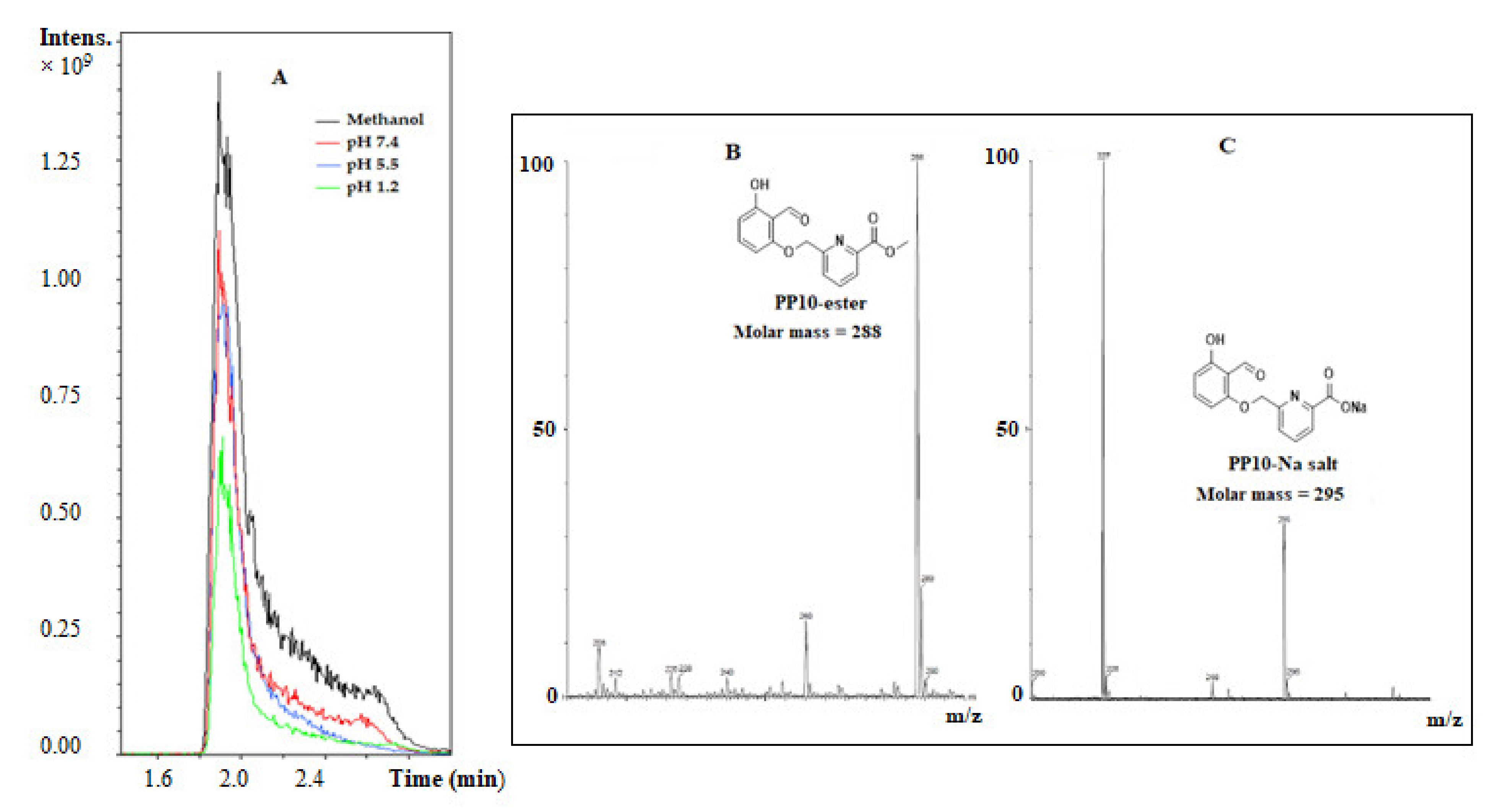
3.1. PP10 Hydrolyzed to the Acid Form (PP10-Acid) at Different Physiological pHs
3.2. PP10 Binary System with HP-β-CD Showed Improved Solubility
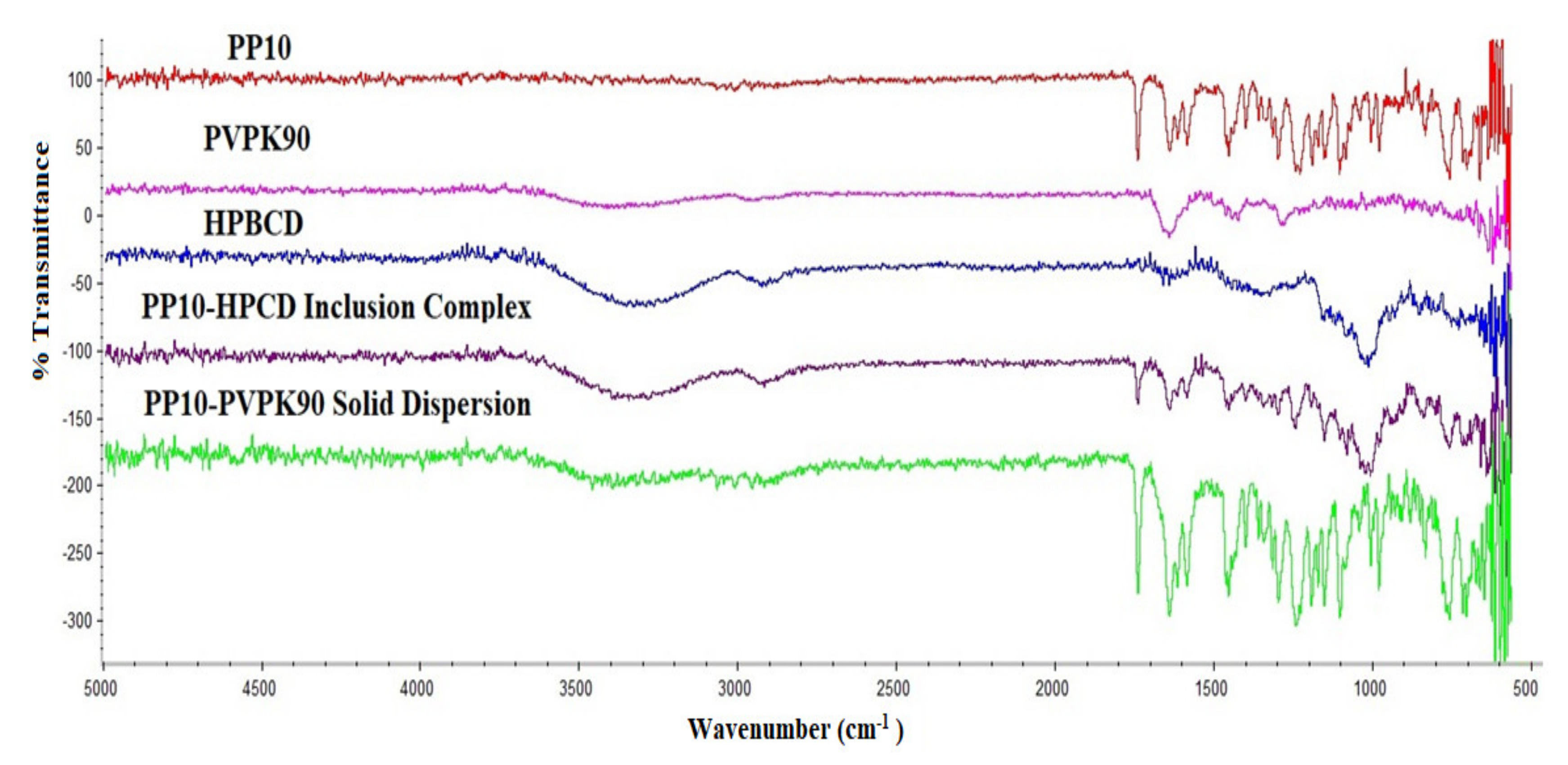
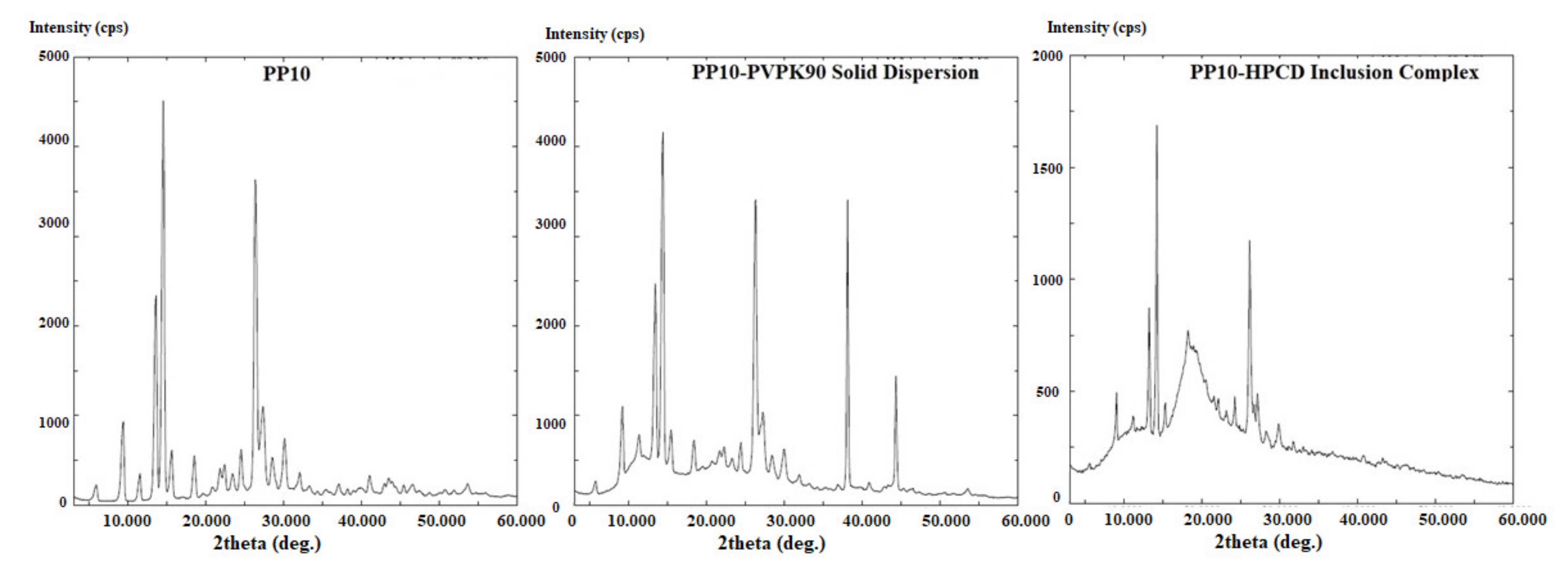
3.3. Physicochemical Characterization of the Binary Systems
3.4. Development of PP10 Oral Tablets
3.5. PP10 Showed Improved Intravenous Formulation with TPGS Micelle
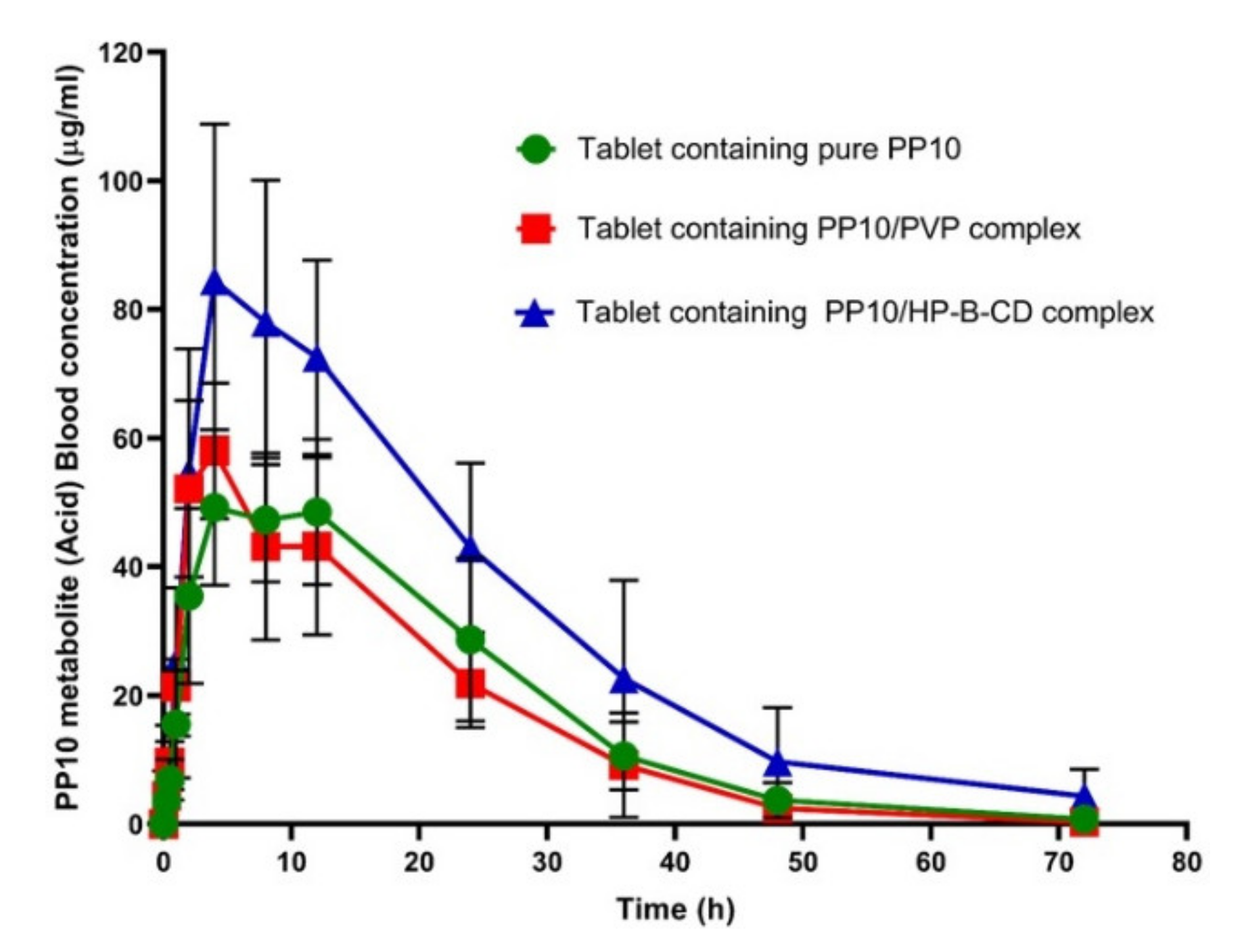
3.6. PP10 Formulation with HP-β-CD Binary System Showed Enhanced PK Properties and Bioavailability
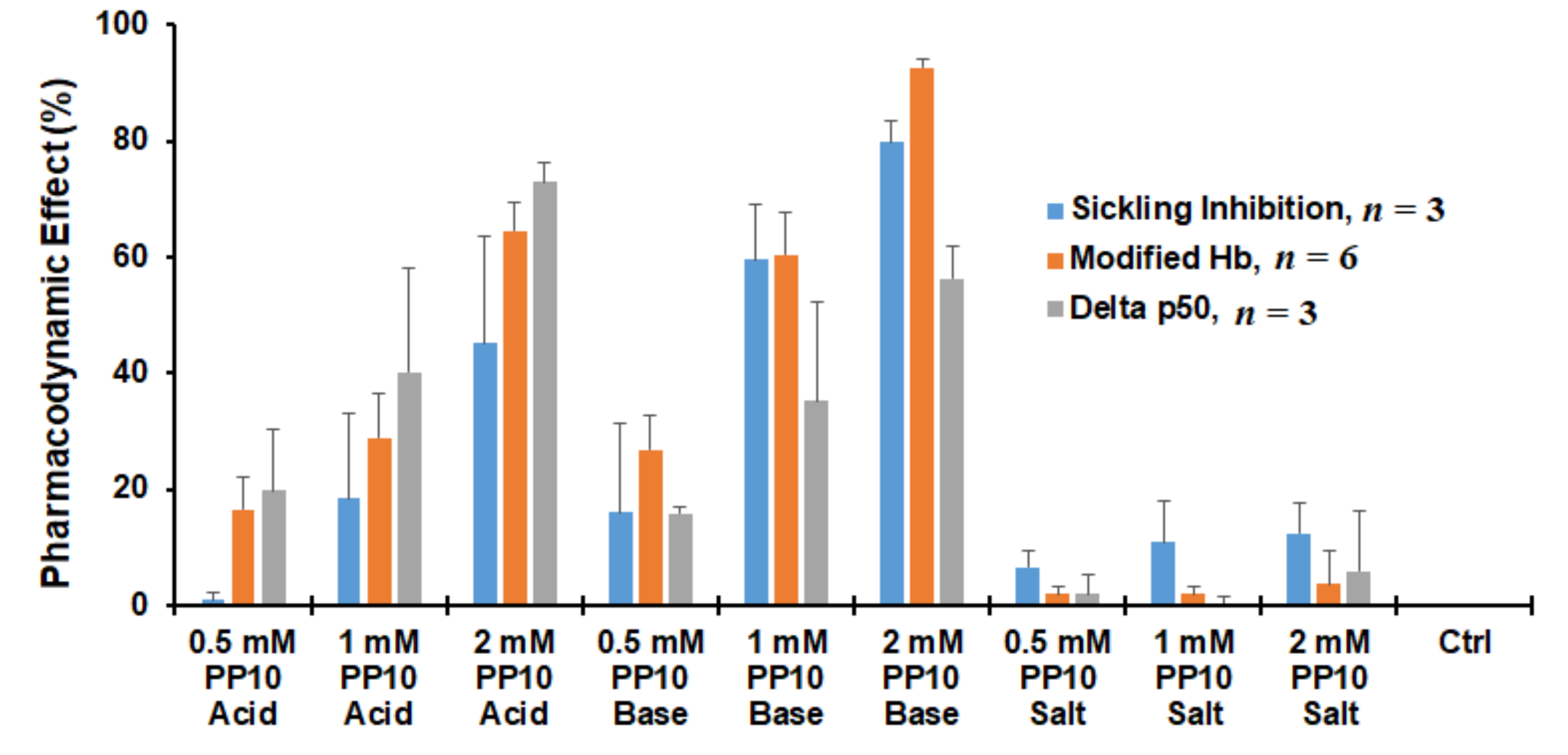
3.7. Both Parent PP10 and the Acid Form (PP10-Acid) Exhibit Biological Activity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aliyu, Z.Y.; Gordeuk, V.; Sachdev, V.; Babadoko, A.; Mamman, A.I.; Akpanpe, P.; Attah, E.; Suleiman, Y.; Aliyu, N.; Yusuf, J.; et al. Prevalence and risk factors for pulmonary artery systolic hypertension among sickle cell disease patients in Nigeria. Am. J. Hematol. 2008, 83, 485–490. Available online: https://pubmed.ncbi.nlm.nih.gov/18306362/ (accessed on 13 July 2021). [CrossRef] [PubMed] [Green Version]
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle Cell Disease. N. Engl. J. Med. 2017, 377, 302–305. Available online: https://pubmed.ncbi.nlm.nih.gov/28723338/ (accessed on 13 July 2021). [CrossRef] [PubMed] [Green Version]
- Akinsheye, I.; Klings, E.S. Sickle Cell Anemia and Vascular Dysfunction: The Nitric Oxide Connection. J. Cell Physiol. 2010, 224, 620–625. Available online: https://pubmed.ncbi.nlm.nih.gov/20578237/ (accessed on 13 July 2021). [CrossRef]
- Belcher, J.D.; Bryant, C.J.; Nguyen, J.; Bowlin, P.R.; Kielbik, M.C.; Bischof, J.C.; Hebbel, R.P.; Vercellotti, G.M. Transgenic Sickle Mice Have Vascular Inflammation. Blood 2003, 101, 3953–3959. Available online: https://pubmed.ncbi.nlm.nih.gov/12543857/ (accessed on 13 July 2021). [CrossRef] [Green Version]
- De Franceschi, L. Pathophisiology of Sickle Cell Disease and New Drugs for the Treatment. Mediterr. J. Hematol. Infect. Dis. 2009, 1, e2009024. Available online: https://pubmed.ncbi.nlm.nih.gov/21415994/ (accessed on 13 July 2021). [CrossRef]
- Sinha, C.B.; Bakshi, N.; Ross, D.; Krishnamurti, L. From Trust to Skepticism: An In-Depth Analysis across Age Groups of Adults with Sickle Cell Disease on Their Perspectives Regarding Hydroxyurea. PLoS ONE 2018, 13, e0199375. Available online: https://pubmed.ncbi.nlm.nih.gov/29949647/ (accessed on 13 July 2021). [CrossRef]
- Mvalo, T.; Topazian, H.; Kamthunzi, P.; Chen, J.; Kambalame, I.; Mafunga, P.; Mumba, N.; Chiume-Chiphaliwali, M.; Paseli, K.; Key, N.; et al. Increasing Hydroxyurea Use in Children with Sickle Cell Disease at Kamuzu Central Hospital, Malawi. Blood Adv. 2018, 2 (Suppl. 1), 30–32. Available online: https://pubmed.ncbi.nlm.nih.gov/30504195/ (accessed on 13 July 2021). [CrossRef] [PubMed]
- Kaufman, M.B. Pharmaceutical Approval Update. Pharm. Ther. 2017, 42, 620. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3776487/ (accessed on 13 July 2021).
- Sadaf, A.; Quinn, C.T. L-glutamine for sickle cell disease: Knight or pawn? Exp. Biol. Med. 2020, 245, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 429–439. Available online: https://pubmed.ncbi.nlm.nih.gov/27959701/ (accessed on 13 July 2021). [CrossRef] [PubMed]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. Available online: https://pubmed.ncbi.nlm.nih.gov/31199090/ (accessed on 13 July 2021). [CrossRef]
- Oder, E.; Safo, M.K.; Abdulmalik, O.; Kato, G.J. New Developments in Anti-Sickling Agents: Can Drugs Directly Prevent the Polymerization of Sickle Haemoglobin In Vivo? Br. J. Haematol. 2016, 175, 24–30. Available online: https://pubmed.ncbi.nlm.nih.gov/27605087/ (accessed on 13 July 2021). [CrossRef] [PubMed] [Green Version]
- Safo, M.K.; Ahmed, M.H.; Ghatge, M.S.; Boyiri, T. Hemoglobin-Ligand Binding: Understanding Hb Function and Allostery on Atomic Level. Biochim. Biophys. Acta 2011, 1814, 797–809. Available online: https://pubmed.ncbi.nlm.nih.gov/21396487/ (accessed on 13 July 2021). [CrossRef]
- Safo, M.K.; Kato, G.J. Therapeutic Strategies to alter the Oxygen Affinity of Sickle Hemoglobin. Hematol. Oncol. Clin. N. Am. 2014, 28, 217–231. Available online: https://pubmed.ncbi.nlm.nih.gov/24589263/ (accessed on 13 July 2021). [CrossRef] [PubMed] [Green Version]
- Aggarwal, A.; Jain, S. Physicochemical Characterization and Dissolution Study of Solid Dispersions of Ketoconazole with Nicotinamide. Chem. Pharm. Bull. 2011, 59, 629–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, T.A. Formulation and Clinical Investigation of Optimized Vinpocetine Lyoplant-Tabs: New Strategy in Development of Buccal Solid Dosage Form. Drug Des. Devel. Ther. 2019, 13, 205–220. [Google Scholar] [CrossRef] [Green Version]
- Tran, P.; Pyo, Y.C.; Kim, D.H.; Lee, S.E.; Kim, J.K.; Park, J.S. Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics 2019, 11, 132. Available online: https://pubmed.ncbi.nlm.nih.gov/30893899/ (accessed on 30 September 2020). [CrossRef] [Green Version]
- Pagare, P.P.; Ghatge, M.S.; Chen, Q.; Musayev, F.N.; Venitz, J.; Abdulmalik, O.; Zhang, Y.; Safo, M.K. Exploration of Structure-Activity Relationship of Aromatic Aldehydes Bearing Pyridinylmethoxy-Methyl Esters as Novel Antisickling Agents. J. Med. Chem. 2020, 63, 14724–14739. Available online: https://pubmed.ncbi.nlm.nih.gov/33205981/ (accessed on 13 July 2021). [CrossRef]
- Dufu, K.; Yalcin, O.; Ao-Ieong, E.S.; Hutchaleelala, A.; Xu, Q.; Li, Z.; Vlahakis, N.; Oksenberg, D.; Lehrer-Graiwer, J.; Cabrales, P. GBT1118, A Potent Allosteric Modifier of Hemoglobin O 2 Affinity, increases Tolerance to Severe Hypoxia in Mice. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H381–H391. Available online: https://pubmed.ncbi.nlm.nih.gov/28526710/ (accessed on 13 July 2021). [CrossRef] [Green Version]
- Metcalf, B.; Chuang, C.; Dufu, K.; Patel, M.P.; Silva-Garcia, A.; Johnson, C.; Lu, Q.; Partridge, J.R.; Patskovska, L.; Patskovsky, Y.; et al. Discovery of GBT440, An Orally Bioavailable R-State Stabilizer of Sickle Cell Hemoglobin. ACS Med. Chem. Lett. 2017, 8, 321–326. Available online: https://pubmed.ncbi.nlm.nih.gov/28337324/ (accessed on 13 July 2021). [CrossRef]
- Oksenberg, D.; Dufu, K.; Patel, M.P.; Chuang, C.; Li, Z.; Xu, Q.; Silva-Garcia, A.; Zhou, C.; Hutchaleelaha, A.; Patskovska, L.; et al. GBT440 Increases Haemoglobin Oxygen Affinity, Reduces Sickling and Prolongs RBC Half-Life in a Murine Model of Sickle Cell Disease. Br. J. Haematol. 2016, 175, 141–153. Available online: https://pubmed.ncbi.nlm.nih.gov/27378309/ (accessed on 13 July 2021). [CrossRef]
- Abdulmalik, O.; Pagare, P.P.; Huang, B.; Xu, G.G.; Ghatge, M.S.; Xu, X.; Chen, Q.; Anabaraonye, N.; Musayev, F.N.; Omar, A.M.; et al. VZHE-039, A Novel Antisickling Agent that Prevents Erythrocyte Sickling under Both Hypoxic and Anoxic Conditions. Sci. Rep. 2020, 10, 1–14. Available online: https://pubmed.ncbi.nlm.nih.gov/33219275/ (accessed on 13 July 2021). [CrossRef]
- Burchall, G.; Maxwell, E. Haemoglobin Stanleyville II Modifies Sickle Disease Phenotype. Pathology 2010, 42, 310–312. Available online: https://pubmed.ncbi.nlm.nih.gov/20350235/ (accessed on 13 July 2021). [CrossRef]
- Rhoda, M.-D.; Martin, J.; Blouquit, Y.; Garel, M.-C.; Edelstein, S.J.; Rosa, J. Sickle Cell Hemoglobin Fiber Formation Strongly Inhibited by the Stanleyville II Mutation (alpha 78 Asn leads to Lys). Biochem. Biophys. Res. Commun. 1983, 111, 8–13. Available online: https://pubmed.ncbi.nlm.nih.gov/6681956/ (accessed on 13 July 2021). [CrossRef]
- Abdulmalik, O.; Safo, M.K.; Chen, Q.; Yang, J.; Brugnara, C.; Ohene-Frempong, K.; Abraham, D.J.; Asakura, T. 5-hydroxymethyl-2-furfural modifies intracellular sickle haemoglobin and inhibits sickling of red blood cells. Br. J. Haematol. 2005, 128, 552–561. Available online: https://pubmed.ncbi.nlm.nih.gov/15686467/ (accessed on 13 July 2021). [CrossRef] [PubMed]
- Safo, M.K.; Abdulmalik, O.; Danso-Danquah, R.; Burnett, J.C.; Nokuri, S.; Joshi, G.S.; Musayev, F.N.; Asakura, T.; Abraham, D.J. Structural Basis for the Potent Antisickling Effect of a Novel Class of Five-Membered Heterocyclic Aldehydic Compounds. J. Med. Chem. 2004, 47, 4665–4676. Available online: https://pubmed.ncbi.nlm.nih.gov/15341482/ (accessed on 13 July 2021). [CrossRef] [PubMed]
- US Pharmacopoeial Convention Inc. The United States Pharmacopeia TNF. USP 28/NF 23; US Pharmacopoeial Convention Inc.: Rockville, MD, USA, 2005. [Google Scholar]
- Ahmed, T.A.; Bawazir, A.O.; Alharbi, W.S.; Safo, M.K. Enhancement of Simvastatin ex vivo Permeation from Mucoadhesive Buccal Films Loaded with Dual Drug Release Carriers. Int. J. Nanomed. 2020, 15, 4001–4020. [Google Scholar] [CrossRef]
- Ahmed, T.A.; Suhail, M.A.A.; Hosny, K.M.; Abd-Allah, F.I. Clinical Pharmacokinetic Study for the Effect of Glimepiride Matrix Tablets Developed by Quality by Design Concept. Drug Dev. Ind. Pharm. 2018, 44, 66–81. [Google Scholar] [CrossRef]
- Shinde, V.R.; Shelake, M.R.; Shetty, S.S.; Chavan-Patil, A.B.; Pore, Y.V.; Late, S.G. Enhanced solubility and dissolution rate of lamotrigine by inclusion complexation and solid dispersion technique. J. Pharm. Pharmacol. 2008, 60, 1121–1129. [Google Scholar] [CrossRef]
- Brun, H.; Paul, M.; Razzouq, N.; Binhas, M.; Gibaud, S.; Astier, A. Cyclodextrin Inclusion Complexes of the Central Analgesic Drug Nefopam. Drug Dev. Ind. Pharm. 2006, 32, 1123–1134. [Google Scholar] [CrossRef]
- Ceschel, G.C.; Mora, P.C.; Borgia, S.L.; Maffei, P.; Ronchi, C. Skin permeation study of dehydroepiandrosterone (DHEA) compared with its α-cyclodextrin complex form. J. Pharm. Sci. 2002, 91, 2399–2407. [Google Scholar] [CrossRef]
- Badr-Eldin, S.M.; Ahmed, T.A.; Ismail, H.R. Aripiprazole-cyclodextrin binary systems for dissolution enhancement: Effect of preparation technique, cyclodextrin type and molar ratio. Iran. J. Basic Med. Sci. 2013, 16, 1223–1231. [Google Scholar]
- Ding, J.; Li, J.; Mao, S. Development and Evaluation of Vinpocetine Inclusion Complex for Brain Targeting. Asian J. Pharm. Sci. 2015, 10, 114–120. Available online: https://core.ac.uk/download/pdf/82054298.pdf (accessed on 22 August 2017). [CrossRef] [Green Version]
- Nicolescu, C.; Aramă, C.; Nedelcu, A.; Monciu, C.-M. Phase Solubility Studies of the Inclusion Complexes of Repaglinide with β-cyclodextrin and β-cyclodextrin Derivatives. Farmacia 2010, 5858, 620–628. Available online: http://www.revistafarmacia.ro/20105/issue52010art09-620-628.pdf (accessed on 27 July 2017).
- Jun, S.W.; Kim, M.-S.; Kim, J.-S.; Park, H.J.; Lee, S.; Woo, J.-S.; Hwang, S.-J. Preparation and characterization of simvastatin/hydroxypropyl-b-cyclodextrin inclusion complex using supercritical antisolvent (SAS) process. Eur. J. Pharm. Biopharm. 2007, 66, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Al-Gethmy, H.A.; Fahmy, U.A.; Alhakamy, N.A.; Ahmed, O.A.A.; El-Say, K.M. Optimization of the factors affecting the absorption of vardenafil from oral disintegrating tablets: A clinical pharmacokinetic investigation. Pharmaceutics 2019, 11, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Porter, W.; Merdan, T.; Li, L.C. Recent advances in intravenous delivery of poorly water-soluble compounds. Expert Opin. Drug Deliv. 2009, 6, 1261–1282. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quality Control Test | Tablet Containing Pure Drug | Tablet Containing PVPK90 | Tablet Containing HP-β-CD |
|---|---|---|---|
| Drug content (%) | 104.49 ± 3.71 | 102.34 ± 6.71 | 99.48 ± 6.94 |
| Tablet weight (mg) | 138 ± 0.01 | 137 ± 0.02 | 281 ± 0.02 |
| Tablet thickness (mm) | 1.72 ± 0.03 | 1.74 ± 0.02 | 3.32 ± 0.06 |
| Tablet hardness (N) | 49.67 ± 5.7 | 84.67 ± 7.8 | 204.33 ± 8.08 |
| Tablet friability (%) | 0.512 | 0.564 | 0.570 |
| Tablet disintegration (min) | 5.19 ± 0.13 | 5.84 ± 0.22 | 8.16 ± 0.08 |
| Parameter | Unit | Tablet Containing Pure PP10 | Tablet Containing PP10/PVP K-90 Complex | Tablet Containing PP10/HP-B-CD Complex |
|---|---|---|---|---|
| K | 1/h | 0.084 ± 0.028 | 0.098 ± 0.020 | 0.076 ± 0.056 |
| t½ | h | 10.122 ± 3.725 | 7.382 ± 1.661 | 12.316 ± 3.743 |
| Tmax | h | 8.667 ± 2.763 | 3.333 ± 1.033 | 6.667 ± 2.066 |
| Cmax | μg/ml | 53.711 ± 9.510 | 58.488 ± 10.056 | 93.319 ± 22.585 |
| AUC 0–t | μg/mL·h | 1329.989 ± 267.646 | 1204.287 ± 313.814 | 2266.320 ± 714.164 |
| AUC 0–inf | μg/mL·h | 1367.258 ± 242.548 | 1214.263 ± 312.306 | 2370.477 ± 810.456 |
| AUMC 0–inf | μg/mL·h2 | 26,141.480 ± 8606.141 | 19,384.575 ± 7600.453 | 54,948.412 ± 32,380.255 |
| MRT 0–inf | h | 19.054 ± 5.135 | 15.627 ± 2.959 | 21.396 ± 6.815 |
| Vd | (mg)/(μg/mL) | 1.113 ± 0.825 | 0.915 ± 0.268 | 0.731 ± 0.236 |
| Cl | (mg)/(μg/mL)/h | 0.075 ± 0.013 | 0.087 ± 0.021 | 0.048 ± 0.020 |
| Relative bioavailability | % | --- | 88.810 | 173.375 |
| Absolute bioavailability | % | 63.28 | 57.34 | 106.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, T.A.; El-Say, K.M.; Abd-Allah, F.I.; Omar, A.M.; El-Araby, M.E.; Muhammad, Y.A.; Pagare, P.P.; Zhang, Y.; Mohmmad, K.A.; Abdulmalik, O.; et al. Improving the Solubility and Oral Bioavailability of a Novel Aromatic Aldehyde Antisickling Agent (PP10) for the Treatment of Sickle Cell Disease. Pharmaceutics 2021, 13, 1148. https://doi.org/10.3390/pharmaceutics13081148
Ahmed TA, El-Say KM, Abd-Allah FI, Omar AM, El-Araby ME, Muhammad YA, Pagare PP, Zhang Y, Mohmmad KA, Abdulmalik O, et al. Improving the Solubility and Oral Bioavailability of a Novel Aromatic Aldehyde Antisickling Agent (PP10) for the Treatment of Sickle Cell Disease. Pharmaceutics. 2021; 13(8):1148. https://doi.org/10.3390/pharmaceutics13081148
Chicago/Turabian StyleAhmed, Tarek A., Khalid M. El-Say, Fathy I. Abd-Allah, Abdelsattar M. Omar, Moustafa E. El-Araby, Yosra A. Muhammad, Piyusha P. Pagare, Yan Zhang, Khadijah A. Mohmmad, Osheiza Abdulmalik, and et al. 2021. "Improving the Solubility and Oral Bioavailability of a Novel Aromatic Aldehyde Antisickling Agent (PP10) for the Treatment of Sickle Cell Disease" Pharmaceutics 13, no. 8: 1148. https://doi.org/10.3390/pharmaceutics13081148
APA StyleAhmed, T. A., El-Say, K. M., Abd-Allah, F. I., Omar, A. M., El-Araby, M. E., Muhammad, Y. A., Pagare, P. P., Zhang, Y., Mohmmad, K. A., Abdulmalik, O., & Safo, M. K. (2021). Improving the Solubility and Oral Bioavailability of a Novel Aromatic Aldehyde Antisickling Agent (PP10) for the Treatment of Sickle Cell Disease. Pharmaceutics, 13(8), 1148. https://doi.org/10.3390/pharmaceutics13081148