
Development of Arteannuin B Sustained-Release Microspheres for Anti-Tumor Therapy by Integrated Experimental and Molecular Modeling Approaches

 , , ,
, , ,
Abstract
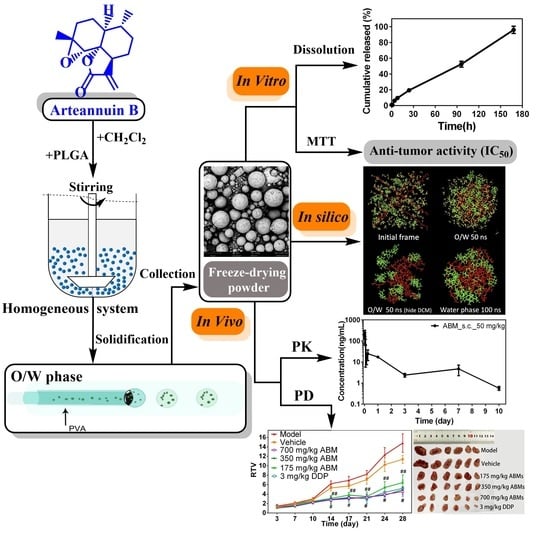
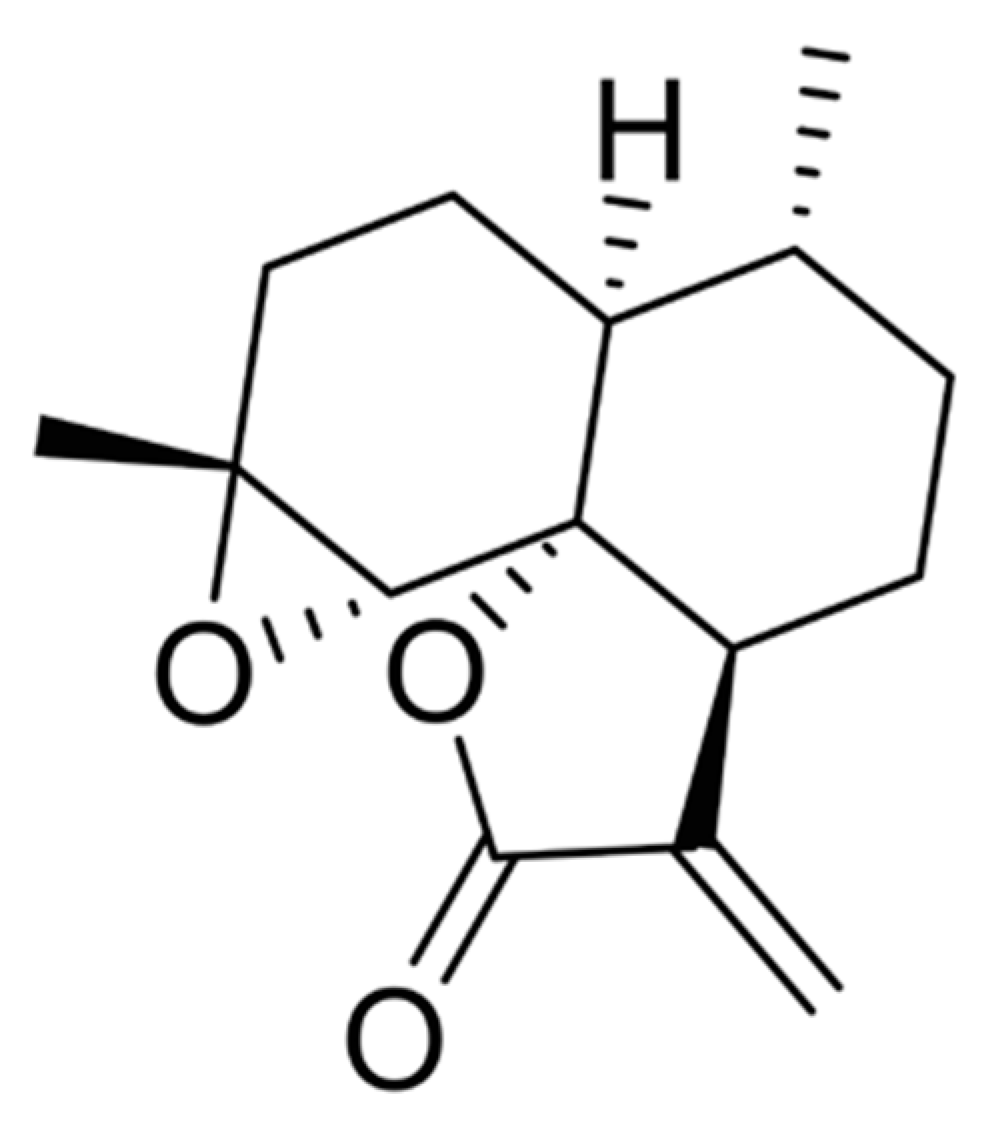
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Preparation of ABMs
2.3. Drug Loading and Encapsulation Efficiency of ABMs
2.4. Scanning Electron Microscope
2.5. Characterizations of Particle Size and Particle Size Distribution
2.6. Molecular Dynamics Simulations of ABMs Preparation Processes
2.7. Characterizations of In Vitro Release
2.8. Cell Culture
2.9. Anti-Tumor Cell Proliferation Activity
2.10. Animal Treatment
2.11. Administration and Blood Sampling in Rats
2.12. In Vivo Anti-Tumor Activity
2.13. Analysis of the Concentration of AB in Plasma
2.14. Statistical Analysis
3. Results and Discussion
3.1. Formulation Screening of ABMs
3.1.1. Screening of PLGA
3.1.2. The Stirring Rate of Emulsification
3.1.3. The Ratio between AB and PLGA
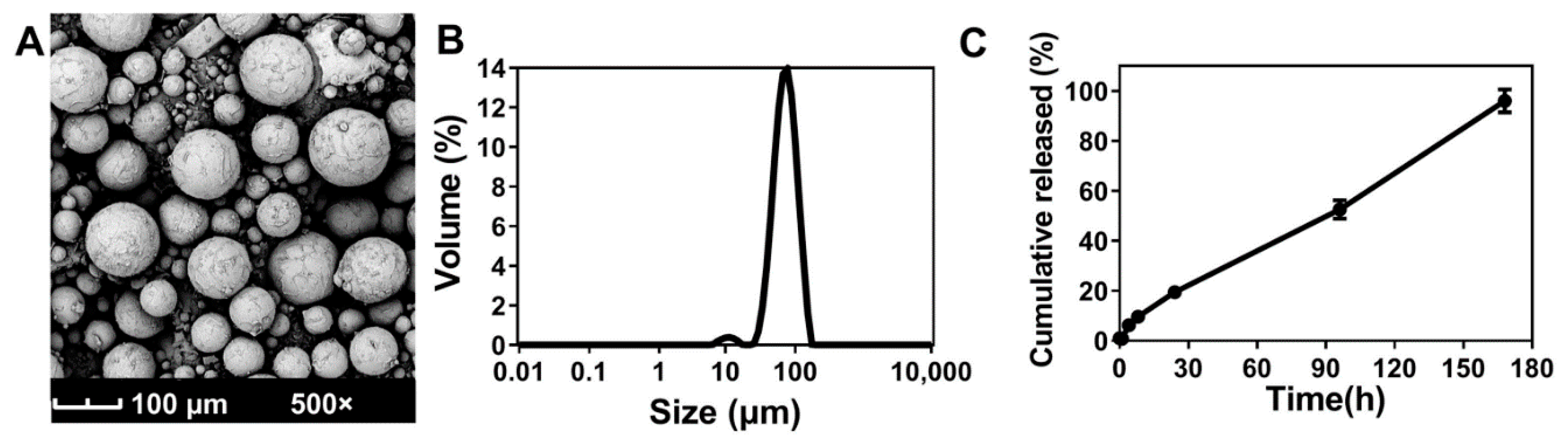
3.2. Characterization of ABMs
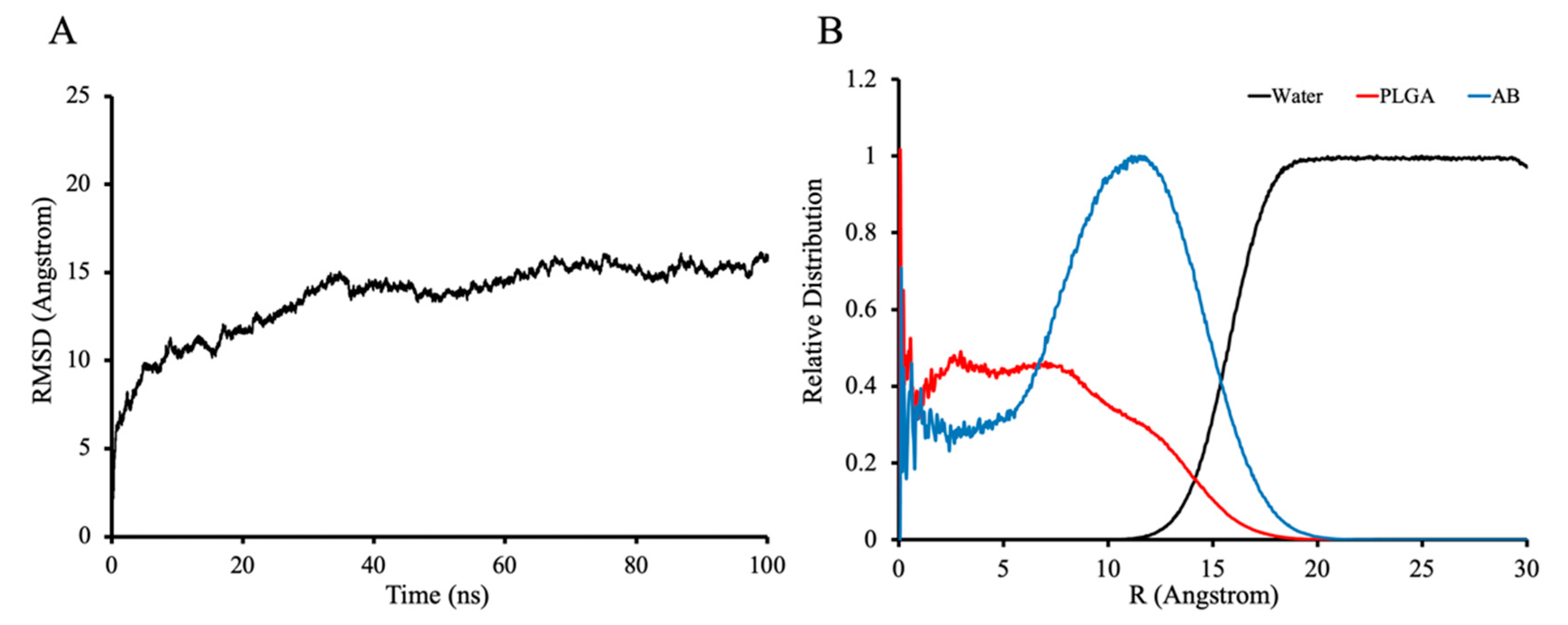
3.3. Molecular Dynamics Simulations of ABMs
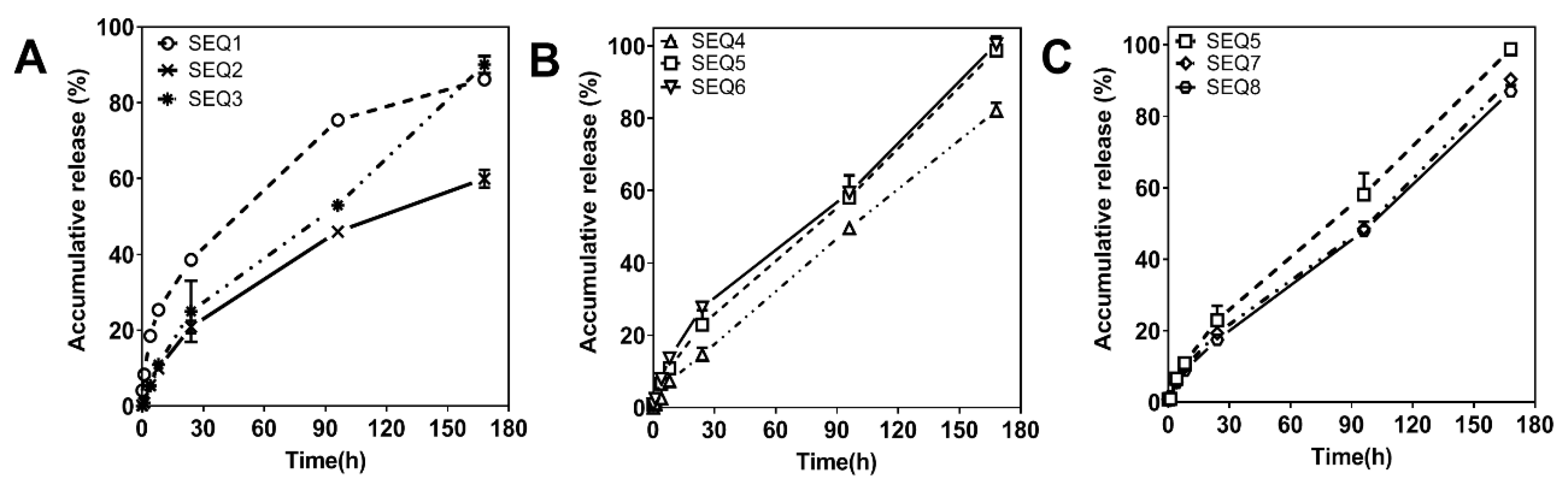
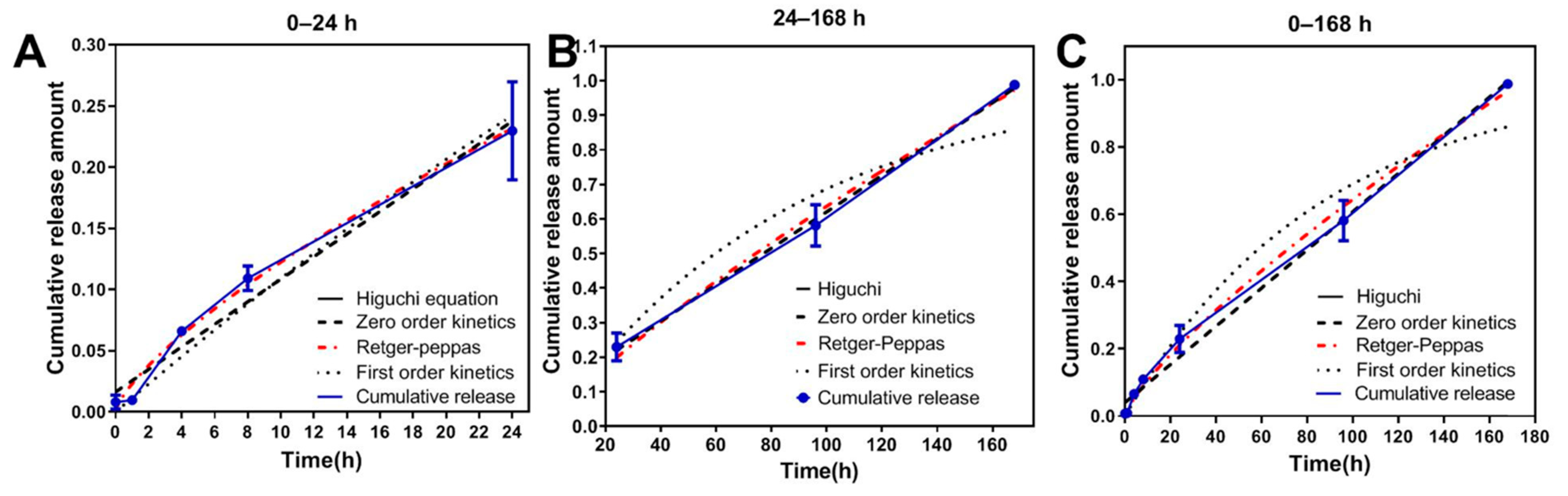
3.4. Mathematical Model Fitting of ABMs In Vitro Release Mechanism
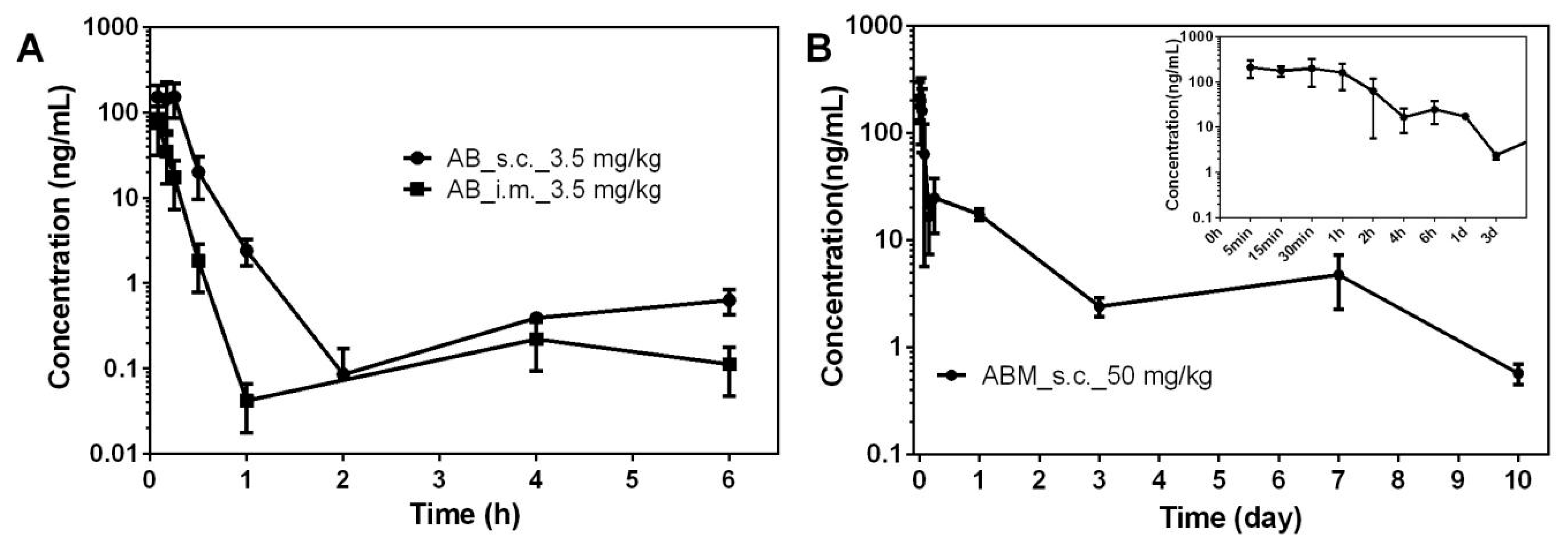
3.5. PK Study of ABMs
3.6. In Vitro Anti-Tumor Activity in Cell Experiments
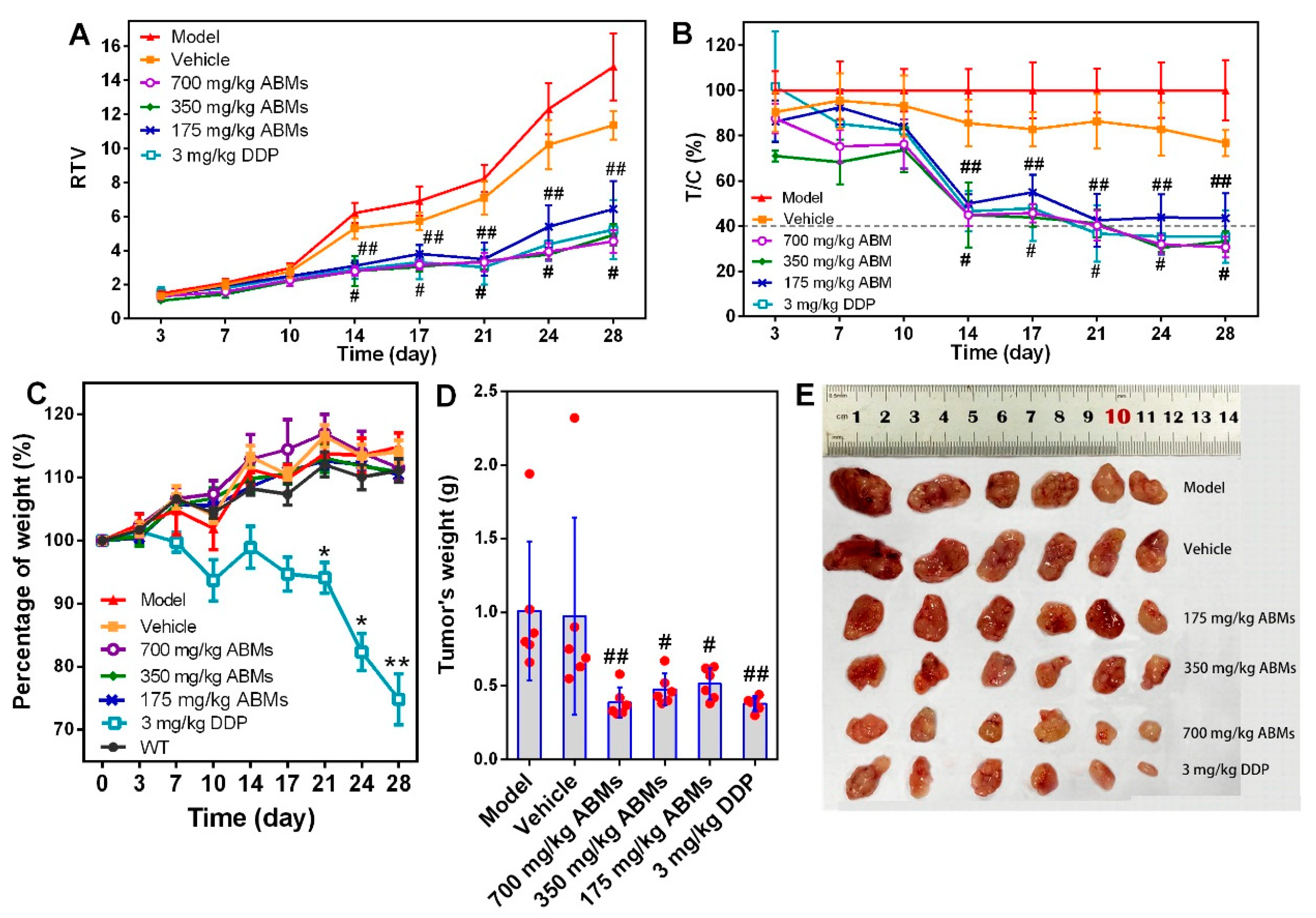
3.7. In Vivo Anti-Tumor Activity
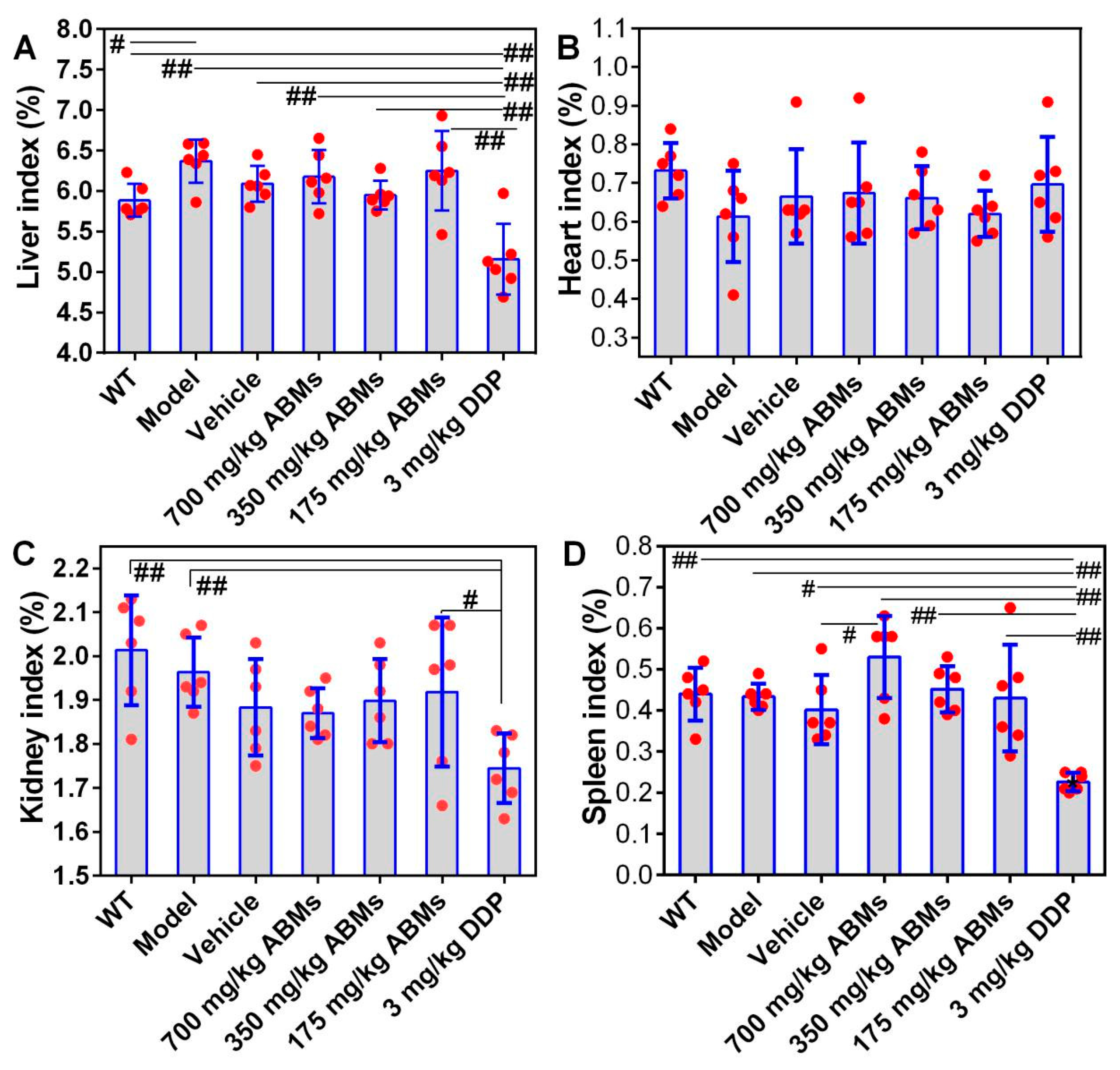
3.8. In Vivo Toxicity to Various Organs and Tumors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jeremić, D.; Jokić, A.; Behbud, A.; Stefanović, M. A new type of sesqiterpene lactone isolated from Artemisia annua L. arteannuin B. Tetrahedron Lett. 1973, 14, 3039–3042. [Google Scholar] [CrossRef]
- Dingan, D. Structural studies on artemisic acid. Mon. J. Sci. 1982, 12, 1359–1362. [Google Scholar]
- Yuan-Zhu, G.; Ya-Ping, Z.; Liang-Quan, L. Chemical transformations of arteannuin B. Acta Chim. Sin. Engl. Ed. 1983, 1, 28–36. [Google Scholar] [CrossRef]
- Qiang, W.; Cai, W.; Yang, Q.; Yang, L.; Dai, Y.; Zhao, Z.; Yin, J.; Li, Y.; Li, Q.; Wang, Y. Artemisinin B improves learning and memory impairment in AD dementia mice by suppressing neuroinflammation. Neuroscience 2018, 395, 1–12. [Google Scholar] [CrossRef]
- Navaratnam, V.; Mansor, S.M.; Sit, N.-W.; Grace, J.; Li, Q.; Olliaro, P. Pharmacokinetics of artemisinin-type compounds. Clin. Pharmacokinet. 2000, 39, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Balint, G.A. Artemisinin and its derivatives: An important new class of antimalarial agents. Pharmacol. Ther. 2001, 90, 261–265. [Google Scholar] [CrossRef]
- Paes, A.H.; Bakker, A.; Soe-Agnie, C.J. Impact of dosage frequency on patient compliance. Diabetes Care 1997, 20, 1512–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yujing, W. Research progress and prospect of microsphere product. Biol. Chem. Eng. 2017, 2, 80–83. [Google Scholar]
- Okada, H. One-and three-month release injectable microspheres of the LH-RH superagonist leuprorelin acetate. Adv. Drug Deliv. Rev. 1997, 28, 43–70. [Google Scholar] [CrossRef]
- Bohr, A.; Kristensen, J.; Stride, E.; Dyas, M.; Edirisinghe, M. Preparation of microspheres containing low solubility drug compound by electrohydrodynamic spraying. Int. J. Pharm. 2011, 412, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Wang, H.-T.; Liu, H.-Y.; Hong, H.; Lv, W.; Shang, Z.-H. Preparation of pingyangmycin PLGA microspheres and related in vitro/in vivo studies. Int. J. Pharm. 2010, 398, 130–136. [Google Scholar] [CrossRef]
- Ng, S.-M.; Choi, J.-Y.; Han, H.-S.; Huh, J.-S.; Lim, J.O. Novel microencapsulation of potential drugs with low molecular weight and high hydrophilicity: Hydrogen peroxide as a candidate compound. Int. J. Pharm. 2010, 384, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.; Kohli, N.; Suri, C.R.; Sahoo, D.K. Molecular mechanism of improved structural integrity of protein in polymer based microsphere delivery system. Mol. Pharm. 2012, 9, 2403–2414. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Hiruta, Y.; Heinemann, D.; Heisterkamp, A.; Kanazawa, H.; Terakawa, M. Intracellular localization and delivery of plasmid DNA by biodegradable microsphere-mediated femtosecond laser optoporation. J. Biophotonics 2017, 10, 1723–1731. [Google Scholar] [CrossRef]
- Frede, A.; Neuhaus, B.; Knuschke, T.; Wadwa, M.; Kollenda, S.; Klopfleisch, R.; Hansen, W.; Buer, J.; Bruder, D.; Epple, M. Local delivery of siRNA-loaded calcium phosphate nanoparticles abates pulmonary inflammation. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 2395–2403. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.M.; McRae, G.I.; Vitale, K.M.; Kell, B.A. Controlled delivery of an LHRH analogue from biodegradable injectable microspheres. J. Control. Release 1985, 2, 187–195. [Google Scholar] [CrossRef]
- Alavi, M.; Webster, T.J. Recent progress and challenges for polymeric microsphere compared to nanosphere drug release systems: Is there a real difference? Bioorganic Med. Chem. 2021, 33, 116028. [Google Scholar] [CrossRef]
- Xu, J.; Chen, Y.; Jiang, X.; Gui, Z.; Zhang, L. Development of hydrophilic drug encapsulation and controlled release using a modified nanoprecipitation method. Processes 2019, 7, 331. [Google Scholar] [CrossRef] [Green Version]
- Bazylińska, U.; Kulbacka, J.; Chodaczek, G. Nanoemulsion structural design in co-encapsulation of hybrid multifunctional agents: Influence of the smart PLGA polymers on the nanosystem-enhanced delivery and electro-photodynamic treatment. Pharmaceutics 2019, 11, 405. [Google Scholar] [CrossRef] [Green Version]
- Lei, H.; Gao, X.; Wu, W.D.; Wu, Z.; Chen, X.D. Aerosol-assisted fast formulating uniform pharmaceutical polymer microparticles with variable properties toward pH-sensitive controlled drug release. Polymers 2016, 8, 195. [Google Scholar] [CrossRef] [Green Version]
- Molavi, F.; Barzegar-Jalali, M.; Hamishehkar, H. Polyester based polymeric nano and microparticles for pharmaceutical purposes: A review on formulation approaches. J. Control. Release 2020, 320, 265–282. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhang, W.; Wang, R.; Wang, Y.; Ouyang, D. Research advances in molecular modeling in cyclodextrins. Curr. Pharm. Des. 2017, 23, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Su, Y.; Xiong, W.; Sun, X.; Ji, Y.; Yu, H.; Li, H.; Ouyang, D. Integrated computer-aided formulation design: A case study of andrographolide/cyclodextrin ternary formulation. Asian J. Pharm. Sci. 2021. [Google Scholar] [CrossRef]
- Zhao, Q.; Ye, Z.; Su, Y.; Ouyang, D. Predicting complexation performance between cyclodextrins and guest molecules by integrated machine learning and molecular modeling techniques. Acta Pharm. Sin. B 2019, 9, 1241–1252. [Google Scholar] [CrossRef]
- Gao, H.; Wang, W.; Dong, J.; Ye, Z.; Ouyang, D. An integrated computational methodology with data-driven machine learning, molecular modeling and PBPK modeling to accelerate solid dispersion formulation design. Eur. J. Pharm. Biopharm. 2021, 158, 336–346. [Google Scholar] [CrossRef]
- Gao, H.; Ye, Z.; Dong, J.; Gao, H.; Yu, H.; Li, H.; Ouyang, D. Predicting drug/phospholipid complexation by the lightGBM method. Chem. Phys. Lett. 2020, 747, 137354. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Burgess, D.J. Evaluation of in vivo–in vitro release of dexamethasone from PLGA microspheres. J. Control. Release 2008, 127, 137–145. [Google Scholar] [CrossRef]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, K.; Zhang, J.; Jiang, H.; Liu, J.; Hao, Y.; Yasuda, H.; Ichimaru, A.; Yamamoto, K. In vitro and in vivo studies of cyclosporin A-loaded microspheres based on copolymers of lactide and ɛ-caprolactone: Comparison with conventional PLGA microspheres. Int. J. Pharm. 2005, 295, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Na, X.; Zhou, W.; Li, T.; Hong, D.; Li, J.; Ma, G. Preparation of double-emulsion-templated microspheres with controllable porous structures by premix membrane emulsification. Particuology 2019, 44, 22–27. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, L.; Tao, A.; Bera, H.; Tang, X.; Cun, D.; Yang, M. Formulation and in vitro characterization of long-acting PLGA injectable microspheres encapsulating a peptide analog of LHRH. J. Mater. Sci. Technol. 2021, 63, 133–144. [Google Scholar] [CrossRef]
- Malik, S.A.; Ng, W.H.; Bowen, J.; Tang, J.; Gomez, A.; Kenyon, A.J.; Day, R.M. Electrospray synthesis and properties of hierarchically structured PLGA TIPS microspheres for use as controlled release technologies. J. Colloid Interface Sci. 2016, 467, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, S.; Wang, Y.; Wang, B.; Deng, J.; Zhu, L.; Cao, Y. Formulation of porous poly (lactic-co-glycolic acid) microparticles by electrospray deposition method for controlled drug release. Mater. Sci. Eng. C 2014, 39, 113–119. [Google Scholar] [CrossRef]
- Amoyav, B.; Benny, O. Microfluidic based fabrication and characterization of highly porous polymeric microspheres. Polymers 2019, 11, 419. [Google Scholar] [CrossRef] [Green Version]
- Tamani, F.; Bassand, C.; Hamoudi, M.; Danede, F.; Willart, J.; Siepmann, F.; Siepmann, J. Mechanistic explanation of the (up to) 3 release phases of PLGA microparticles: Diprophylline dispersions. Int. J. Pharm. 2019, 572, 118819. [Google Scholar] [CrossRef]
- Allison, S.D. Analysis of initial burst in PLGA microparticles. Expert Opin. Drug Deliv. 2008, 5, 615–628. [Google Scholar] [CrossRef]
- Laracuente, M.-L.; Marina, H.Y.; McHugh, K.J. Zero-order drug delivery: State of the art and future prospects. J. Control. Release 2020. [Google Scholar] [CrossRef]
- Peppas, N. Analysis of Fickian and non-Fickian drug release from polymers. Pharm. Acta Helv. 1985, 60, 110–111. [Google Scholar]
- Yan, H.; Ding, D.; Zhang, Z.; Sun, E.; Song, J.; Jia, X. Study on sustained release preparations of Epimedium component. Zhongguo Zhongyao Za Zhi China J. Chin. Mater. Med. 2015, 40, 1484–1488. [Google Scholar]
- Zhao, Y.-N.; Xu, X.; Wen, N.; Song, R.; Meng, Q.; Guan, Y.; Cheng, S.; Cao, D.; Dong, Y.; Qie, J. A drug carrier for sustained zero-order release of peptide therapeutics. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Weidner, J. Drug delivery. Drug Discov. Today 2001, 6, 216–217. [Google Scholar] [CrossRef]
- Zuidema, J.; Kadir, F.; Titulaer, H.; Oussoren, C. Release and absorption rates of intramuscularly and subcutaneously injected pharmaceuticals (II). Int. J. Pharm. 1994, 105, 189–207. [Google Scholar] [CrossRef]
- McLennan, D.N.; Porter, C.J.; Charman, S.A. Subcutaneous drug delivery and the role of the lymphatics. Drug Discov. Today Technol. 2005, 2, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, X.; Li, B.; Hou, Y.; Cai, Z.; Yang, J.; Li, Y. Paclitaxel-loaded PLGA microspheres with a novel morphology to facilitate drug delivery and antitumor efficiency. RSC Adv. 2018, 8, 3274–3285. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Formulas | 0–24 h | 24–168 h | 0–168 h | |||
|---|---|---|---|---|---|---|---|
| Fitting Equation Results | R2 | Fitting Equation Results | R2 | Fitting Equation Results | R2 | ||
| Zero-order kinetic | Q = Kt + b | Q = 0.009220 × t + 0.01629 | 0.9726 | Q = 0.005265 × t + 0.09424 | 0.9983 | Q = 0.005676 × t + 0.04047 | 0.9929 |
| First-order kinetic | ln(1−Q) = −Kt | ln(1−Q) = −0.01156 × t | 0.9672 | ln(1−Q) = −0.01161 × t | 0.9122 | ln(1−Q) = −0.01172 × t | 0.9679 |
| Higuchi | Q = Kt1/2 | Q = 0.04259 × t1/2 | 0.9375 | Q = 0.06814 × t1/2 | 0.8985 | Q = 0.06670 × t1/2 | 0.9461 |
| Hixcon-Crowell | (100−Q)1/3 = −Kt | (100−Q)1/3 = 0.1958 × t | −116,8000 | (100−Q)1/3 = 0.02885 × t | −56,346 | (100−Q)1/3 = 0.02885 × t | −68,825 |
| Retger-Peppas | Q = Ktn | Q = 0.02285 × t0.7288 | 0.9915 | Q = 0.01490 × t0.8154 | 0.9917 | Q = 0.01763 × t0.7815 | 0.9959 |
| Sample | Pathway | Dose | t1/2 | Cmax | AUClast |
|---|---|---|---|---|---|
| (mg/kg) | (day) | (ng/mL) | (day·ng/mL) | ||
| ABMs | SC | 50 | 2.820 ± 0.350 | 262.000 ± 146.860 | 74.774 ± 5.441 |
| AB | SC | 3.5 | 0.048 ± 0.003 | 235.667 ± 100.431 | 2.538 ± 0.842 |
| IM | 3.5 | 0.096 ± 0.052 | 74.500 ± 34.155 | 0.618 ± 0.131 |
| Time (h) | 24 | 48 | 72 | 96 |
|---|---|---|---|---|
| AB | 211.37 ± 63.17 | 53.10 ± 22.52 | 27.52 ± 6.71 | 16.17 ± 4.78 |
| ABMs | 290.59 ± 40.06 | 38.40 ± 9.58 | 11.13 ± 10.00 | 3.94 ± 1.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Huang, W.; Wang, N.; Ouyang, D.; Xiao, L.; Zhang, S.; Ou, X.; He, T.; Yu, R.; Song, L. Development of Arteannuin B Sustained-Release Microspheres for Anti-Tumor Therapy by Integrated Experimental and Molecular Modeling Approaches. Pharmaceutics 2021, 13, 1236. https://doi.org/10.3390/pharmaceutics13081236
Wang Y, Huang W, Wang N, Ouyang D, Xiao L, Zhang S, Ou X, He T, Yu R, Song L. Development of Arteannuin B Sustained-Release Microspheres for Anti-Tumor Therapy by Integrated Experimental and Molecular Modeling Approaches. Pharmaceutics. 2021; 13(8):1236. https://doi.org/10.3390/pharmaceutics13081236
Chicago/Turabian StyleWang, Yanqing, Weijuan Huang, Nannan Wang, Defang Ouyang, Lifeng Xiao, Sirui Zhang, Xiaozheng Ou, Tingsha He, Rongmin Yu, and Liyan Song. 2021. "Development of Arteannuin B Sustained-Release Microspheres for Anti-Tumor Therapy by Integrated Experimental and Molecular Modeling Approaches" Pharmaceutics 13, no. 8: 1236. https://doi.org/10.3390/pharmaceutics13081236
APA StyleWang, Y., Huang, W., Wang, N., Ouyang, D., Xiao, L., Zhang, S., Ou, X., He, T., Yu, R., & Song, L. (2021). Development of Arteannuin B Sustained-Release Microspheres for Anti-Tumor Therapy by Integrated Experimental and Molecular Modeling Approaches. Pharmaceutics, 13(8), 1236. https://doi.org/10.3390/pharmaceutics13081236