
Model-Informed Repurposing of Medicines for SARS-CoV-2: Extrapolation of Antiviral Activity and Dose Rationale for Paediatric Patients


Abstract
:1. Introduction
2. Materials and Methods
2.1. General Modelling and Simulation Methodology
2.2. Pre-Clinical Data, Modelling, and Extrapolation Assumptions
2.3. Pharmacokinetic Modelling and Paediatric Extrapolation
2.4. Prediction of Drug Exposure at the Target Organ
2.5. Clinical Trial Simulations (CTS)
2.6. Go/No-Go Criteria
2.7. Safety Thresholds
3. Results
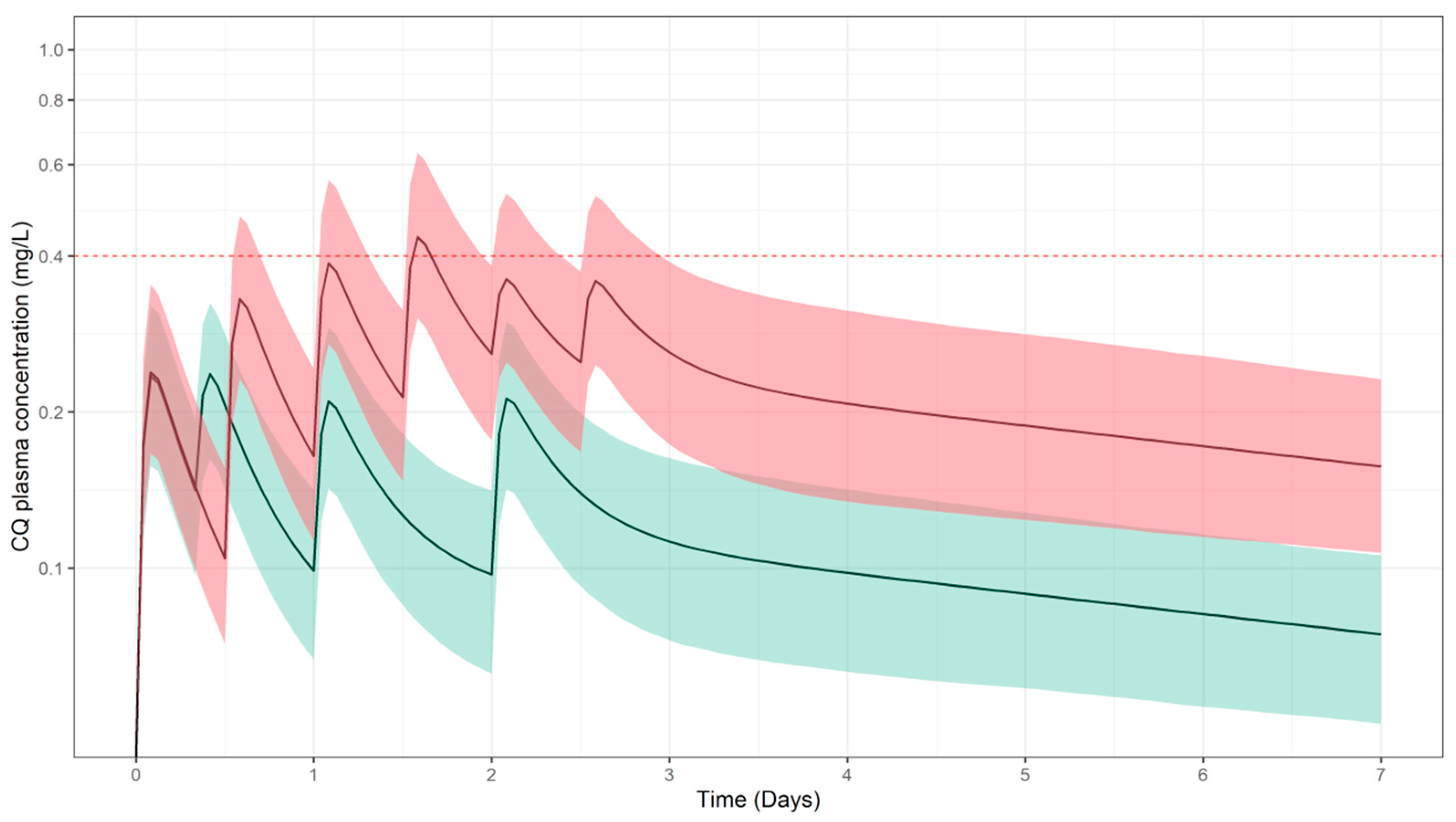
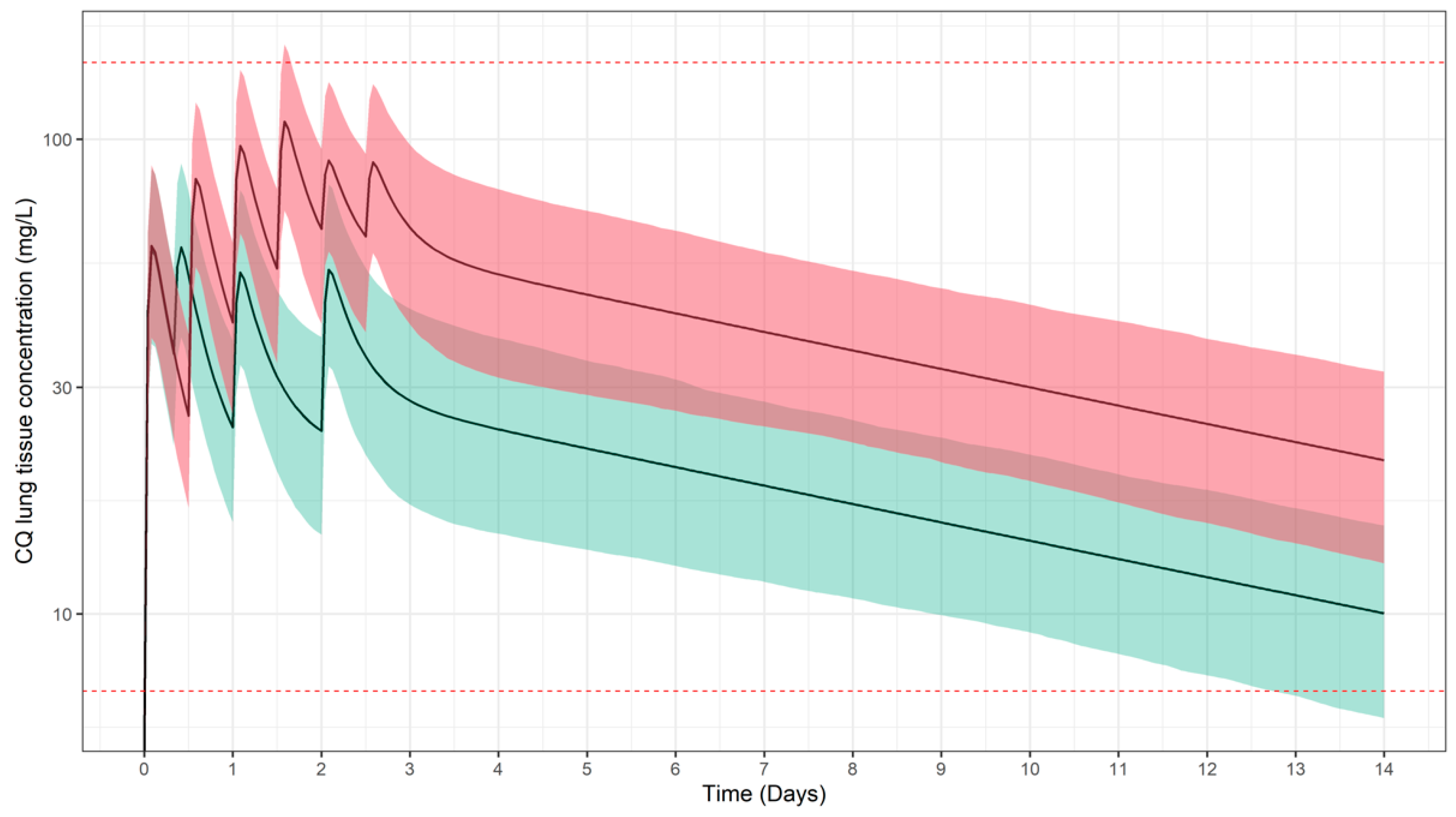
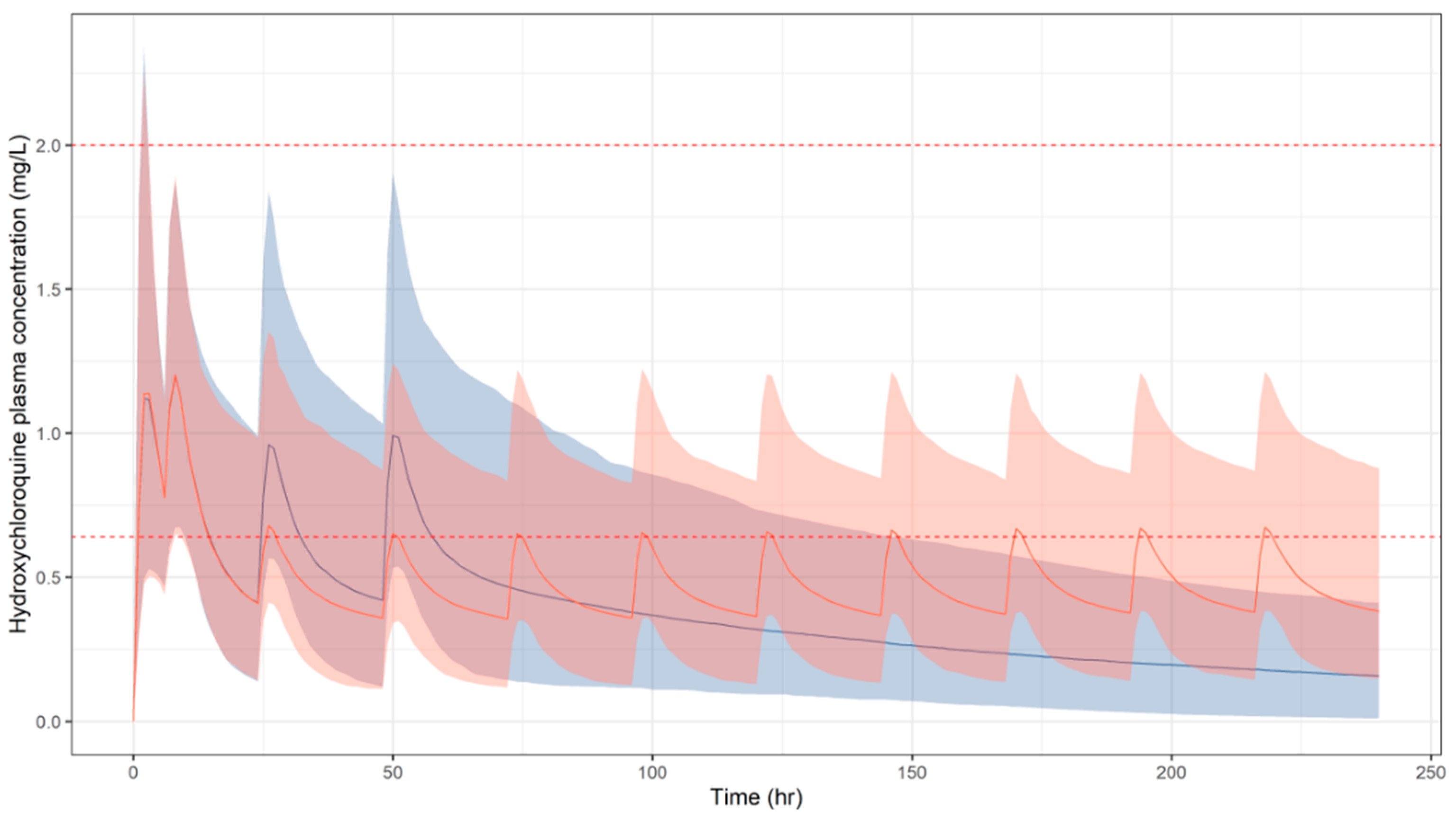
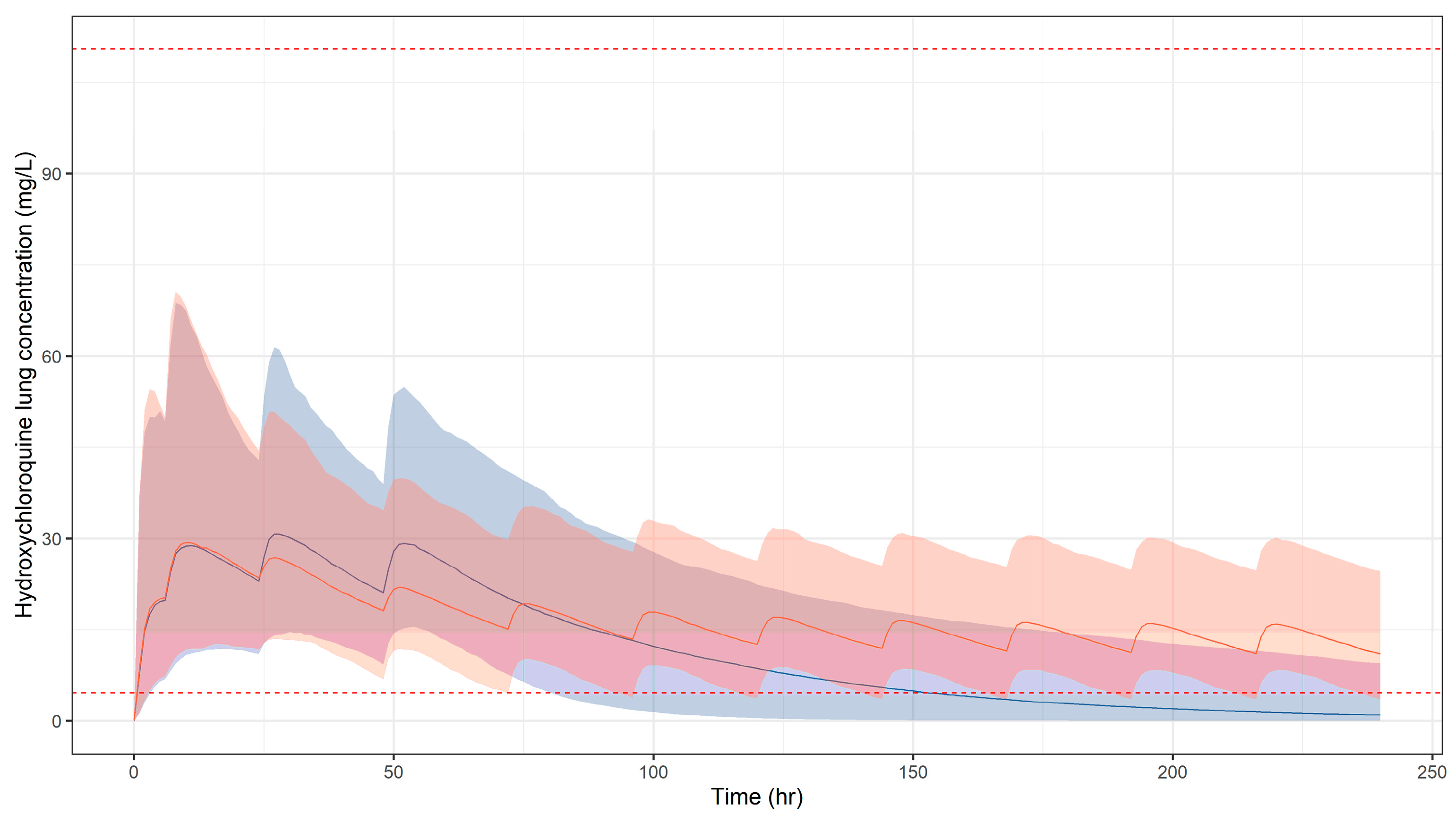
3.1. Predicted Pharmacokinetic Profiles in Paediatric Patients
3.2. Simulation Scenarios for Selected Doses and Dosing Regimens
4. Discussion
5. Limitations and Clinical Implications
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Rayner, C.R.; Smith, P.F.; Andes, D.; Andrews, K.; Derendorf, H.; Friberg, L.E.; Hanna, D.; Lepak, A.; Mills, E.; Polasek, T.M.; et al. Model-informed drug development for anti-infectives: State of the art and future. Clin. Pharmacol. Ther. 2021, 109, 867–891. [Google Scholar] [CrossRef]
- Danhof, M.; de Lange, E.; Della Pasqua, O.E.; Ploeger, B.A.; Voskuyl, R.A. Mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling in translational drug research. Trends Pharmacol. Sci. 2008, 29, 186–191. [Google Scholar] [CrossRef]
- Dong, Y.; Mo, X.; Hu, Y.; Qi, X.; Jiang, F.; Jiang, Z.; Tong, S. Epidemiology of COVID-19 among children in China. Pediatrics 2020, 145, e20200702. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, P.; Curtis, N. Coronavirus infections in children including COVID-19: An overview of the epidemiology, clinical features, diagnosis, treatment and prevention options in children. Pediatr. Infect. Dis. J. 2020, 39, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Götzinger, F.; Santiago-García, B.; Noguera-Julián, A.; Lanaspa, M.; Lancella, L.; Carducci, F.I.C.; Gabrovska, N.; Velizarova, S.; Prunk, P.; Osterman, V.; et al. COVID-19 in children and adolescents in Europe: A multinational, multicentre cohort study. Lancet Child Adolesc. Health 2020, 4, 653–661. [Google Scholar] [CrossRef]
- Riphagen, S.; Gomez, X.; Gonzalez-Martinez, C.; Wilkinson, N.; Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 2020, 395, 1607–1608. [Google Scholar] [CrossRef]
- Flood, S.M.; Osborne, C.M.; Martin, B.; Christopher Derderian, S.; Stenson, E.; Grebenhoff, J.A. Severe SARS-CoV-2 infection in a pediatric patient requiring extracorporeal membrane oxygenation. Case Rep. Pediatr. 2020, 8885022. [Google Scholar] [CrossRef]
- Bixler, D.; Miller, A.D.; Mattison, C.P.; Taylor, B.; Komatsu, K.; Pompa, X.P.; Moon, S.; Karmarkar, E.; Liu, C.Y.; Openshaw, J.J.; et al. SARS-CoV-2-associated deaths among persons aged <21 years—United States, 12 February–31 July 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1324–1329. [Google Scholar]
- Kalil, A.C. Treating COVID-19—Off-label drug use, compassionate use, and randomized clinical trials during pandemics. JAMA 2020, 323, 1897. [Google Scholar] [CrossRef] [PubMed]
- Chiotos, K.; Hayes, M.; Kimberlin, D.W.; Jones, S.B.; James, S.H.; Pinninti, S.G.; Yarbrough, A.; Abzug, M.J.; MacBrayne, C.E.; Soma, V.L.; et al. Multicenter initial guidance on use of antivirals for children with COVID-19/SARS-CoV-2. J. Pediatric Infect. Dis. Soc. 2020, 9, 701–715. [Google Scholar] [CrossRef]
- Clout, A.; Pasqua, O.D.; Hanna, M.G.; Orlu, M.; Pitceathly, R.D.S. Drug repurposing in neurological diseases: An integrated approach to reduce trial and error. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1270–1275. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020, 71, 732–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M.; et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Pizzorno, A.; Padey, B.; Dubios, J.; Julien, T.; Traversier, A.; Duliere, V.; Brun, P.; Lina, B.; Rosa-Calatrava, M.; Terrier, O. In vitro evaluation of antiviral activity of single and combined repurposable drugs against SARS-CoV-2. Antivir. Res. 2020, 181, 104878. [Google Scholar] [CrossRef] [PubMed]
- Pruijssers, A.J.; George, A.S.; Schafer, A.; Leist, S.R.; Gralinksi, L.E.; Dinnon III, K.H.; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D.; et al. Remdesivir inhibits SARS-CoV-2 in human lung cells and chimeric SARS-CoV expressing the SARS-CoV-2 RNA polymerase in mice. Cell Rep. 2020, 32, 107940. [Google Scholar] [CrossRef]
- Savarino, A.; Di Trani, L.; Donatelli, I.; Cauda, R.; Cassone, A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69. [Google Scholar] [CrossRef]
- Hashem, A.M.; Alghamdi, B.S.; Algaissi, A.A.; Alshehri, F.S.; Bukhari, A.; Alfaleh, M.; Memish, Z.A. Therapeutic use of chloroquine and hydroxychloroquine in COVID-19 and other viral infections: A narrative review. Travel Med. Infect. Dis. 2020, 35, 101735. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Chen, X.; Geiger, J.D. Role of endolysosomes in severe acute respiratory syndrome Coronavirus-2 infection and Coronavirus disease 2019 pathogenesis: Implications for potential treatments. Front. Pharmacol. 2020, 11, 1739. [Google Scholar] [CrossRef] [PubMed]
- Dehrmann, F.M.; Coetzer, T.; Pike, R.; Dennison, C. Mature Cathepsin L Is Substantially active in the ionic milieu of the extracellular medium. Arch. Biochem. Biophys. 1995, 324, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef] [Green Version]
- McChesney, E.W. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am. J. Med. 1983, 75, 11–18. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Humeniuk, R.; Mathias, A.; Cao, H.; Osinusi, A.; Shen, G.; Chng, E.; Ling, J.; Vu, A.; German, P. Safety, tolerability, and pharmacokinetics of remdesivir, an antiviral for treatment of COVID-19, in healthy subjects. Clin. Transl. Sci. 2020, 13, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Central Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, S.C.J.; Kebriaei, R.; Dresser, L.D. Dresser, Remdesivir: Review of pharmacology, pre-clinical data, and emerging clinical experience for COVID-19 Pharmacotherapy. J. Hum. Pharm. Drug Ther. 2020, 40, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Bahar, F.G.; Ohura, K.; Ogihara, T.; Imai, T. Species difference of esterase expression and hydrolase activity in plasma. J. Pharm. Sci. 2012, 101, 3979–3988. [Google Scholar] [CrossRef]
- Kaptein, S.J.F.; Jacobs, S.; Langendries, L.; Seldeslachts, L.; ter Horst, S.; Liesenborghs, L.; Hens, B.; Vergote, V.; Heylen, E.; Barthelemy, K.; et al. Favipiravir at high doses has potent antiviral activity in SARS-CoV-2−infected hamsters, whereas hydroxychloroquine lacks activity. Proc. Nat. Acad. Sci. USA 2020, 117, 26955–26965. [Google Scholar] [CrossRef]
- Rudraraju, A.V.; Amoyaw, P.N.A.; Hubin, T.J.; Khan, M.O.F. Determination of log P values of new cyclen based antimalarial drug leads using RP-HPLC. Die Pharm. 2014, 69, 655–662. [Google Scholar]
- Munster, T.; Gibbs, J.P.; Shen, D.; Baethge, B.A.; Botstein, G.R.; Caldwell, J.; Dietz, F.; Ettlinger, R.; Golden, H.E.; Lindsley, H.; et al. Hydroxychloroquine concentration-response relationships in patients with rheumatoid arthritis. Arthritis Rheum. 2002, 46, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Browning, D.J. Pharmacology of Chloroquine and Hydroxychloroquine. Hydroxychloroquine Chloroquine Retin. 2014, 35–63. [Google Scholar] [CrossRef]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) mediated by the viral polymerase and the proofreading exoribonuclease. MBio 2018, 9, e00221-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruijssers, A.J.; Denison, M. Nucleoside analogues for the treatment of coronavirus infections. Curr. Opin. Virol. 2019, 35, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Shannon, A.; Le, N.T.-T.; Selisko, B.; Eydoux, C.; Alvarez, K.; Guillemont, J.-C.; Decroly, E.; Peersen, O.; Ferron, F.; Canard, B. Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 RdRp and nsp14 Exonuclease active-sites. Antivir. Res. 2020, 178, 104793. [Google Scholar] [CrossRef] [PubMed]
- Green, N.; Ott, R.D.; Isaacs, R.J.; Fang, H. Cell-based assays to identify inhibitors of viral disease. Expert Opin. Drug Discov. 2008, 3, 671–676. [Google Scholar] [CrossRef] [Green Version]
- Yan, V.C.; Muller, F.L. Advantages of the parent nucleoside GS-441524 over remdesivir for COVID-19 treatment. ACS Med. Chem. Lett. 2020, 11, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA), Human Medicines Division. Summary on Compassionate Use. Remdesivir Gilead. Product No. EMEA/H/K/5622/CU. 3 April 2020. Available online: https://www.ema.europa.eu/en/documents/other/summary-compassionate-use-remdesivir-gilead_en.pdf (accessed on 24 April 2020).
- Humeniuk, R.; Mathias, A.; Kirby, B.J.; Lutz, J.D.; Cao, H.; Osinusi, A.; Babusis, D.; Porter, D.; Wei, X.; Ling, J.; et al. Pharmacokinetic, pharmacodynamic, and drug-interaction profile of remdesivir, a SARS-CoV-2 replication inhibitor. Clin. Pharmacokinet. 2021, 60, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Tempestilli, M.; Caputi, P.; Avataneo, V.; Notari, S.; Forini, O.; Scorzolini, L.; Marchioni, L.; Bartoli, T.A.; Castilletti, C.; Lalle, E.; et al. Pharmacokinetics of remdesivir and GS-441524 in two critically ill patients who recovered from COVID-19. J. Antimicrob. Chemother. 2020, 75, 2977–2980. [Google Scholar] [CrossRef]
- Carmichael, S.J.; Charles, B.; Tett, S.E. Population pharmacokinetics of hydroxychloroquine in patients with rheumatoid arthritis. Ther. Drug Monit. 2003, 25, 671–681. [Google Scholar] [CrossRef]
- Höglund, R.; Moussavi, Y.; Ruengweerayut, R.; Cheomung, A.; Äbelö, A.; Na-Bangchang, K. Population pharmacokinetics of a three-day chloroquine treatment in patients with Plasmodium vivax infection on the Thai-Myanmar border. Malar. J. 2016, 15, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.-S.; Im, J.-S.; Cho, J.-Y.; Bae, K.-S.; Klein, T.A.; Yeom, J.-S.; Kim, T.-S.; Choi, J.-S.; Jang, I.-J.; Park, J.-W. Pharmacokinetics of hydroxychloroquine and its clinical implications in chemoprophylaxis against malaria caused by plasmodium vivax. Antimicrob. Agents Chemother. 2009, 53, 1468–1475. [Google Scholar] [CrossRef] [Green Version]
- Chhonker, Y.S.; Sleightholm, R.L.; Li, J.; Oupický, D.; Murry, D.J. Simultaneous quantitation of hydroxychloroquine and its metabolites in mouse blood and tissues using LC–ESI–MS/MS: An application for pharmacokinetic studies. J. Chromatogr. B 2018, 1072, 320–327. [Google Scholar] [CrossRef]
- Smit, C.; Peeters, M.Y.M.; van den Anker, J.N.; Knibbe, C.A.J.M. Chloroquine for SARS-CoV-2: Implications of its unique pharmacokinetic and safety properties. Clin. Pharm. 2020, 59, 659–669. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Graham, R.L.; Menachery, V.D.; Gralinski, L.E.; Case, J.B.; Leist, S.R.; Pyrc, K.; Feng, J.Y.; Trantcheva, I.; et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017, 9, 3653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boerma, E.C.; Bethlehem, C.; Stellingwerf, F.; De Lange, F.; Streng, K.W.; Koetsier, P.M.; Bootsma, I.T. Hemodynamic characteristics of mechanically ventilated COVID-19 patients: A cohort analysis. Crit. Care Res. Pr. 2021, 2021, 1–7. [Google Scholar] [CrossRef]
- National Center for Health Statistics. Centers for Disease Control and Prevention. Available online: https://wwwn.cdc.gov/nchs/nhanes/ (accessed on 1 September 2020).
- Karunajeewa, H.A.; Ilett, K.F.; Mueller, I.; Siba, P.; Law, I.; Page-Sharp, M.; Lin, E.; Lammey, J.; Batty, K.T.; Davis, T.M.E. Pharmacokinetics and efficacy of piperaquine and chloroquine in melanesian children with uncomplicated malaria. Antimicrob. Agents Chemother. 2008, 52, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Zahr, N.; Urien, S.; Funck-Brentano, C.; Vantomme, H.; Garcelon, N.; Melki, I.; Boistault, M.; Boyer, O.; Bader-Meunier, B. Evaluation of hydroxychloroquine blood concentrations and effects in childhood-onset systemic lupus erythematosus. Pharmaceuticals 2021, 14, 273. [Google Scholar] [CrossRef]
- Ursing, J.; Eksborg, S.; Rombo, L.; Bergqvist, Y.; Blessborn, D.; Rodrigues, A.; Kofoed, P.-E. Chloroquine is grossly underdosed in young children with malaria: Implications for drug resistance. PLoS ONE 2014, 9, e86801. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, E.; Cong, Y.; Dewald, L.E.; Dyall, J.; Yu, S.; Hart, B.J.; Zhou, H.; Gross, R.; Logue, J.; Cai, Y.; et al. Testing therapeutics in cell-based assays: Factors that influence the apparent potency of drugs. PLoS ONE 2018, 13, e0194880. [Google Scholar] [CrossRef] [Green Version]
- Frisk-Holmberg, M.; Bergkvist, Y.; Domeij-Nyberg, B.; Hellström, L.; Jansson, F. Chloroquine serum concentration and side effects: Evidence for dose-dependent kinetics. Clin. Pharmacol. Ther. 1979, 25, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Jordan, P.; Brookes, J.G.; Nikolic, G.; Le Couteur, D. Hydroxychloroquine overdose: Toxicokinetics and management. J. Toxicol. Clin. Toxicol. 1999, 37, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Durcan, L.; Clarke, W.A.; Magder, L.S.; Petri, M. Hydroxychloroquine blood levels in systemic lupus erythematosus: Clarifying dosing controversies and improving adherence. J. Rheumatol. 2015, 42, 2092–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez-Echevarría, A.; Perez-Martinez, A.; del Valle, L.G.; Ara, M.F.; Melendo, S.; Ruiz de Valbuena, M.; Vazquez-Martinez, J.L.; Morales-Martinez, A.; Remesal, A.; de Valbuena, M.R.; et al. Compassionate use of remdesivir in children with COVID-19. Eur. J. Pediatrics 2020, 180, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Reckers, A.; Wu, A.H.B.; Ong, C.M.; Gandhi, M.; Metcalfe, J.; Gerona, R. A combined assay for quantifying remdesivir and its metabolite, along with dexamethasone, in serum. J. Antimicrob. Chemother. 2021, 76, 1865–1873. [Google Scholar] [CrossRef] [PubMed]
- De Sena, L.W.P.; Mello, A.G.N.C.; Ferreira, M.V.D.; de Ataide, M.A.; Dias, R.M.; Vieira, J.L.F. Doses of chloroquine in the treatment of malaria by Plasmodium vivax in patients between 2 and 14 years of age from the Brazilian Amazon basin. Malar. J. 2019, 18, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borba, M.G.S.; Val, F.F.A.; Sampaio, V.S. Effect of high vs. low doses of chloroquine diphosphate as adjunctive therapy for patients hospitalized with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection: A randomized clinical trial. JAMA Netw. Open 2020, 3, e208857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellanti, F.; Della Pasqua, O. Modelling and simulation as research tools in paediatric drug development. Eur. J. Clin. Pharmacol. 2011, 67, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Cella, M.; De Vries, F.G.; Burger, D.; Danhof, M.; Della Pasqua, O. A model-based approach to dose selection in early pediatric development. Clin. Pharmacol. Ther. 2010, 87, 294–302. [Google Scholar] [CrossRef]
- Bellanti, F.; van Wijk, R.C.; Danhof, M.; Della Pasqua, O. Integration of PKPD relationships into benefit-risk analysis. Br. J. Clin. Pharmacol. 2015, 80, 979–991. [Google Scholar] [CrossRef] [Green Version]
- Venisse, N.; Peytavin, G.; Bouchet, S.; Gagnieu, M.-C.; Garraffo, R.; Guilhaumou, R.; Solas, C. Concerns about pharmacokinetic (PK) and pharmacokinetic-pharmacodynamic (PK-PD) studies in the new therapeutic area of COVID-19 infection. Antivir. Res. 2020, 181, 104866. [Google Scholar] [CrossRef]
- Laaksonen, A.-L.; Koskiahde, V.; Juva, K. Dosage of antimalarial drugs for children with juvenile rheumatoid arthritis and systemic lupus erythematosus: A clinical study with determination of serum concentrations of chloroquine and hydroxychloroquine. Scand. J. Rheumatol. 1974, 3, 103–108. [Google Scholar] [CrossRef]
- Mzayek, F.; Deng, H.; Mather, F.J.; Wasilevich, E.C.; Liu, H.; Hadi, C.M.; Chansolme, D.H.; Murphy, H.A.; Melek, B.H.; Tenaglia, A.N.; et al. Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clin. Trials 2007, 2, e6. [Google Scholar] [CrossRef]
- Aljayyoussi, G.; Rajoli, R.K.R.; Pertinez, H.; Pennington, S.H.; Hong, W.D.; O’Neil, P.M.; Owen, A.; Ward, S.A.; Biahini, G.A. Modelling of systemic versus pulmonary chloroquine exposure in man for COVID-19 dose selection. Medrxiv 2020. [Google Scholar] [CrossRef]
- Maharaj, A.R.; Wu, H.; Hornik, C.P.; Balevic, S.J.; Hornik, C.D.; Smith, P.B.; Gonzalez, D.; Zimmerman, K.O.; Benjamin, D.K.; Cohen-Wolkowiez, M.; et al. Simulated assessment of pharmacokinetically guided dosing for investigational treatments of pediatric patients with Coronavirus disease 2019. JAMA Pediatr. 2020, 174, e202422. [Google Scholar] [CrossRef]
- Chorin, E.; Dai, M.; Shulman, E.; Wadhwani, L.; Bar-Cohen, R.; Barbhaiya, C.; Aizer, A.; Holmes, D.; Bernstein, S.; Spinelli, M.; et al. The QT interval in patients with COVID-19 treated with hydroxychloroquine and azithromycin. Nat. Med. 2020, 26, 808–809. [Google Scholar] [CrossRef] [PubMed]
- Perinel, S.; Launay, M.; Botelho-Nevers, E.; Diconne, E.; Louf-Durier, A.; Lachand, R.; Murgier, M.; Page, D.; Vermesch, R.; Thierry, G.; et al. Towards optimization of hydroxychloroquine dosing in intensive care unit COVID-19 patients. Clin. Infect. Dis. 2020, 71, 2227–2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahévas, M.; Tran, V.-T.; Roumier, M.; Chabrol, A.; Paule, R.; Guillaud, C.; Fois, E.; Lepeule, R.; Szwebel, T.-A.; Lescure, X.; et al. Clinical efficacy of hydroxychloroquine in patients with COVID-19 pneumonia who require oxygen: Observational comparative study using routine care data. BMJ 2020, 369, m1844. [Google Scholar] [CrossRef]
- Boulware, D.R.; Pullen, M.F.; Bangdiwala, A.S.; Pastick, K.A.; Lofgren, S.M.; Okafor, E.C.; Skipper, C.P.; Nascene, A.A.; Nicol, M.R.; Abassi, M.; et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for COVID-19. N. Engl. J. Med. 2020, 383, 517–525. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the treatment of COVID-19—final report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, P.G.; Du, P.R.; Zhao, P.J.; Jin, P.Y.; Fu, P.S.; Gao, P.L.; Cheng, P.Z.; Lu, P.Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Clewell, H. Quantitative in vitro–in vivo extrapolations: Essential issues. Toxicol. Lett. 2011, 205, S16. [Google Scholar] [CrossRef]
- Jermain, B.; Hanafin, P.O.; Cao, Y.; Lifschitz, A.; Lanusse, C.; Rao, G.G. Development of a minimal physiologically-based pharmacokinetic model to simulate lung exposure in humans following oral administration of ivermectin for COVID-19 drug repurposing. J. Pharm. Sci. 2020, 109, 3574–3578. [Google Scholar] [CrossRef]
- Xiao, F.; Fofana, I.; Thumann, C.; Mailly, L.; Alles, R.; Robinet, E.; Meyer, N.; Schaeffer, M.; Habersetzer, F.; Doffoël, M.; et al. Synergy of entry inhibitors with direct-acting antivirals uncovers novel combinations for prevention and treatment of hepatitis C. Gut 2014, 64, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Cremades, M.; Solans, B.P.; Hughes, E.; Ernest, J.P.; Wallender, E.; Aweeka, F.; Luetkemeyer, A.F.; Savic, R.M. Optimizing hydroxychloroquine dosing for patients with COVID-19: An integrative modeling approach for effective drug repurposing. Clin. Pharmacol. Ther. 2020, 108, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.; Li, J.; Parvangada, A.; Perry, J.; Cihlar, T.; Mo, H.; Porter, D.; Svarovskia, E. Genetic conservation of SARS-CoV-2 RNA replication complex in globally circulating isolates and recently emerged variants from humans and minks suggests minimal pre-existing resistance to remdesivir. Antivir. Res. 2021, 188, 105033. [Google Scholar] [CrossRef] [PubMed]
- Cheng, V.; Abdul-Aziz, M.-H.; Roberts, J.A.; Shekar, K. Optimising drug dosing in patients receiving extracorporeal membrane oxygenation. J. Thorac. Dis. 2018, 10, S629–S641. [Google Scholar] [CrossRef] [PubMed]
- Havers, F.; Flannery, B.; Clippard, J.R.; Gaglani, M.; Zimmerman, R.; Jackson, L.A.; Petrie, J.G.; McLean, H.Q.; Nowalk, M.P.; Jackson, M.L.; et al. Use of influenza antiviral medications among outpatients at high risk for influenza-associated complications during the 2013–2014 influenza season. Clin. Infect. Dis. 2015, 60, 1677–1680. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Wong, G.; Audet, J.; Bello, A.; Fernando, L.; Alimonti, J.B.; Fausther-Bovendo, H.; Wei, H.; Aviles, J.; Hiatt, E.; et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 2014, 514, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Wölfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virologic assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, B.N.; Feldmann, F.; Schwarz, B.; Meade-White, K.; Porter, D.P.; Schulz, J.; van Doremalen, N.; Leighton, I.; Yinda, C.K.; Perez-Perez, L.; et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 2020, 585, 273–276. [Google Scholar] [CrossRef]
- de Wit, E.; Feldmann, F.; Cronin, J.; Jordan, R.; Okumura, A.; Thomas, T.; Scott, D.; Cihlar, T.; Feldmann, H. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl. Acad. Sci. USA 2020, 117, 6771–6776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schäfer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O.; et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First Case of 2019 Novel Coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Andrés-Esteban, E.M.; Quintana-Diaz, M.; Ramírez-Cervantes, K.L.; Benayas-Peña, I.; Silva-Obregón, A.; Magallón-Botaya, R.; Santolalla-Arnedo, I.; Juárez-Vela, R.; Gea-Caballero, V. Outcomes of hospitalized patients with COVID-19 according to level of frailty. PeerJ 2021, 9, e11260. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Mechanisms Associated with Antiviral Activity | Derived PKPD Parameters (mg/L) | Reported IC50 (Value or Range) (mg/L) | Reference |
|---|---|---|---|---|
| Chloroquine | CQ has been considered as a broad-spectrum antiviral drug. Although the exact mechanisms are not fully understood, CQ shows antiviral and immunomodulatory properties by altering the acidic conditions necessary for virus-to-cell fusion and entry into host cells. CQ is thought to disrupt this mechanism through the de-acidification of endosomal/lysosomal organelles, which orchestrate enzyme cleavage of the SARS-CoV-2 spike glycoprotein. Post entry, CQ is also reported to de-acidify the Golgi apparatus, which can affect post-translational processes, such as proteolysis and glycosylation, preventing escape of the virus from the host cell. Additionally, binding of the S-protein through angiotensin-converting enzyme 2 is downregulated by CQ. | IC95 = 6.87–145.25 | 0.36–7.64 | [12,14] |
| Hydroxy-chloroquine | HCQ, a derivative of CQ, is a safer compound than CQ in animals, and is considered to exert the same mechanisms of anti-viral activity as CQ upon SARS-CoV-2. | IC95 = 4.59–110.5 | 0.242–5.81 | [12,14] |
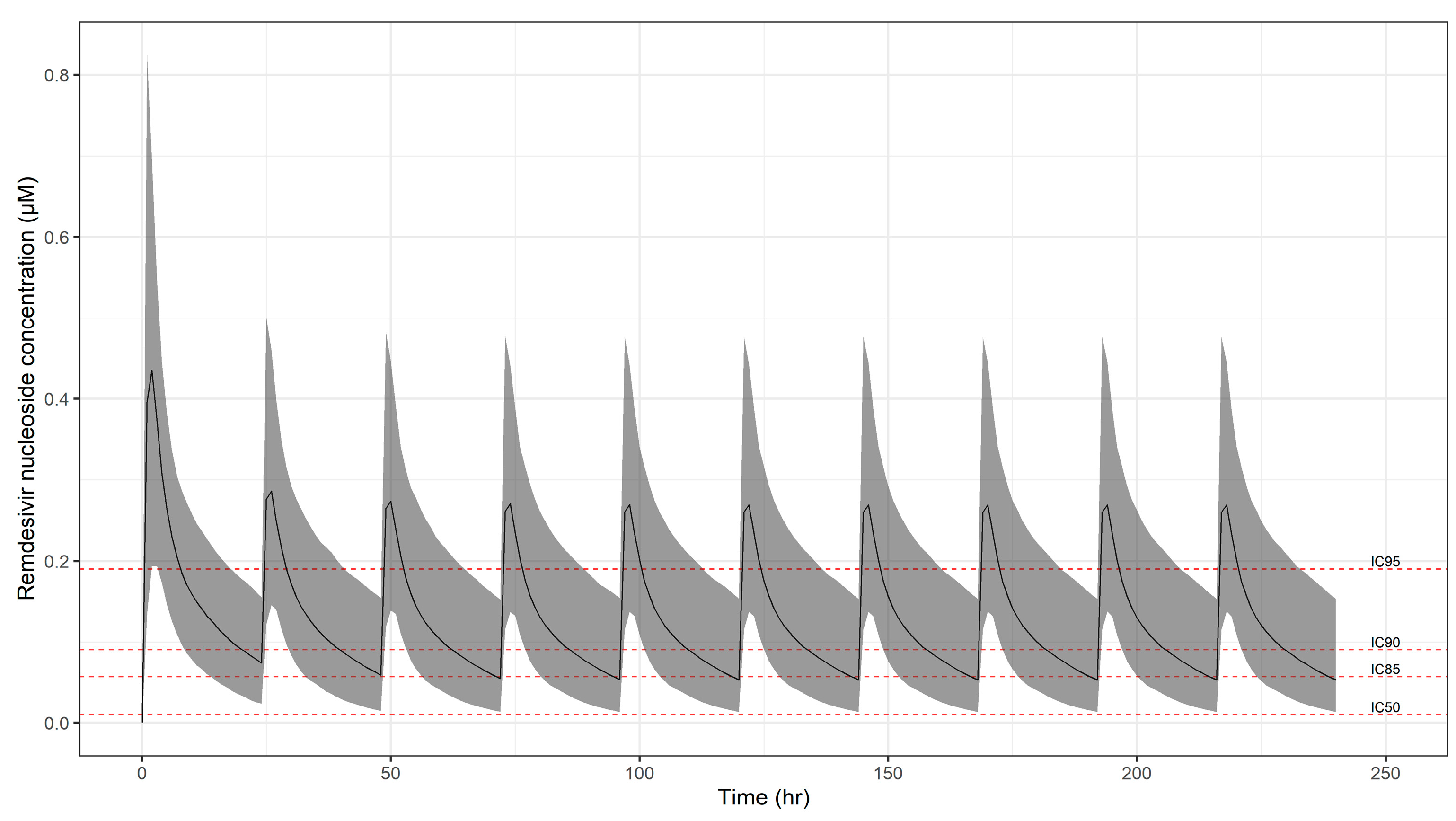
| Remdesivir | RDV’s antiviral activity, sterically interacting with the viral RNA-dependent RNA polymerase (RdRp) to induce delayed chain termination, has been shown in vitro against multiple coronaviruses. RDV also alters pan-CoV RdRp function by inhibiting viral replication even in settings with intact exonuclease proof-reading activity. Biochemical data from recombinant respiratory syncytial virus RdRp suggested the primary mechanism of action was through delayed chain termination. | IC85 = 0.034 IC90 = 0.054 IC95 = 0.114 | 0.006 | [16] |
| Drug | Classification | Dosing Regimen |
|---|---|---|
| CQ * | Standard dose | Loading: 10 mg/kg on day 1 †, 5 mg/kg 8 h later 5 mg/kg on days 2, 3 Total cumulative = 25 mg/kg over 3 days |
| High dose | Loading: 10 mg/kg b.i.d. on days 1 † and 2 5 mg/kg b.i.d. on day 3 Total cumulative = 50 mg/kg over 3 days | |
| HCQ * | Short course | 10 mg/kg + 5 mg/kg after 6, 24 and 48 h |
| Extended course | Loading: 10 mg/kg + 5 mg/kg after 6 h on day 1 † and 2.5 mg/kg at 24 h (day 2) 2.5 mg/kg q.d. thereafter for 8 days | |
| RDV | FDA approved doses | 5 mg/kg q.d. (infused over 30 min) on day 1 † and 2.5 mg/kg q.d. (infused over 30 min) for 9 subsequent days or 200 mg q.d. on day 1 † and 100 mg q.d. for 9 subsequent days ** |
| Drug | Regimen | Probability of Target Attainment (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| Best Case Target Concentration * | Worst Case Target Concentration a | |||||||
| Day 1 | Day 3 | Day 7 | Day 10 | Day 1 | Day 3 | Day 7 | ||
| CQ | Total 25 mg/kg | 100 | 100 | 100 | - | 0 | 0 | 0 |
| Total 50 mg/kg | 100 | 100 | 100 | - | 0 | 0 | 0 | |
| HCQ | Short dose | 100 | 99 | 41 | - | 0 | 0 | 0 |
| Extended dose | 100 | 97 | 96 | - | 0 | 0 | 0 | |
| RDV | 5 mg/kg + 2.5 mg/kg b or 200 mg + 100 mg | - | - | - | 8 ** | N/A | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romano, F.; D’Agate, S.; Pasqua, O.D. Model-Informed Repurposing of Medicines for SARS-CoV-2: Extrapolation of Antiviral Activity and Dose Rationale for Paediatric Patients. Pharmaceutics 2021, 13, 1299. https://doi.org/10.3390/pharmaceutics13081299
Romano F, D’Agate S, Pasqua OD. Model-Informed Repurposing of Medicines for SARS-CoV-2: Extrapolation of Antiviral Activity and Dose Rationale for Paediatric Patients. Pharmaceutics. 2021; 13(8):1299. https://doi.org/10.3390/pharmaceutics13081299
Chicago/Turabian StyleRomano, Federico, Salvatore D’Agate, and Oscar Della Pasqua. 2021. "Model-Informed Repurposing of Medicines for SARS-CoV-2: Extrapolation of Antiviral Activity and Dose Rationale for Paediatric Patients" Pharmaceutics 13, no. 8: 1299. https://doi.org/10.3390/pharmaceutics13081299
APA StyleRomano, F., D’Agate, S., & Pasqua, O. D. (2021). Model-Informed Repurposing of Medicines for SARS-CoV-2: Extrapolation of Antiviral Activity and Dose Rationale for Paediatric Patients. Pharmaceutics, 13(8), 1299. https://doi.org/10.3390/pharmaceutics13081299