
Contribution of CYP2D6 Functional Activity to Oxycodone Efficacy in Pain Management: Genetic Polymorphisms, Phenoconversion, and Tissue-Selective Metabolism




Abstract
:1. Introduction
2. Pharmacokinetic Considerations
2.1. CYP2D6 Activities, Polymorphisms, and Expression (GRADE High Quality ++++)
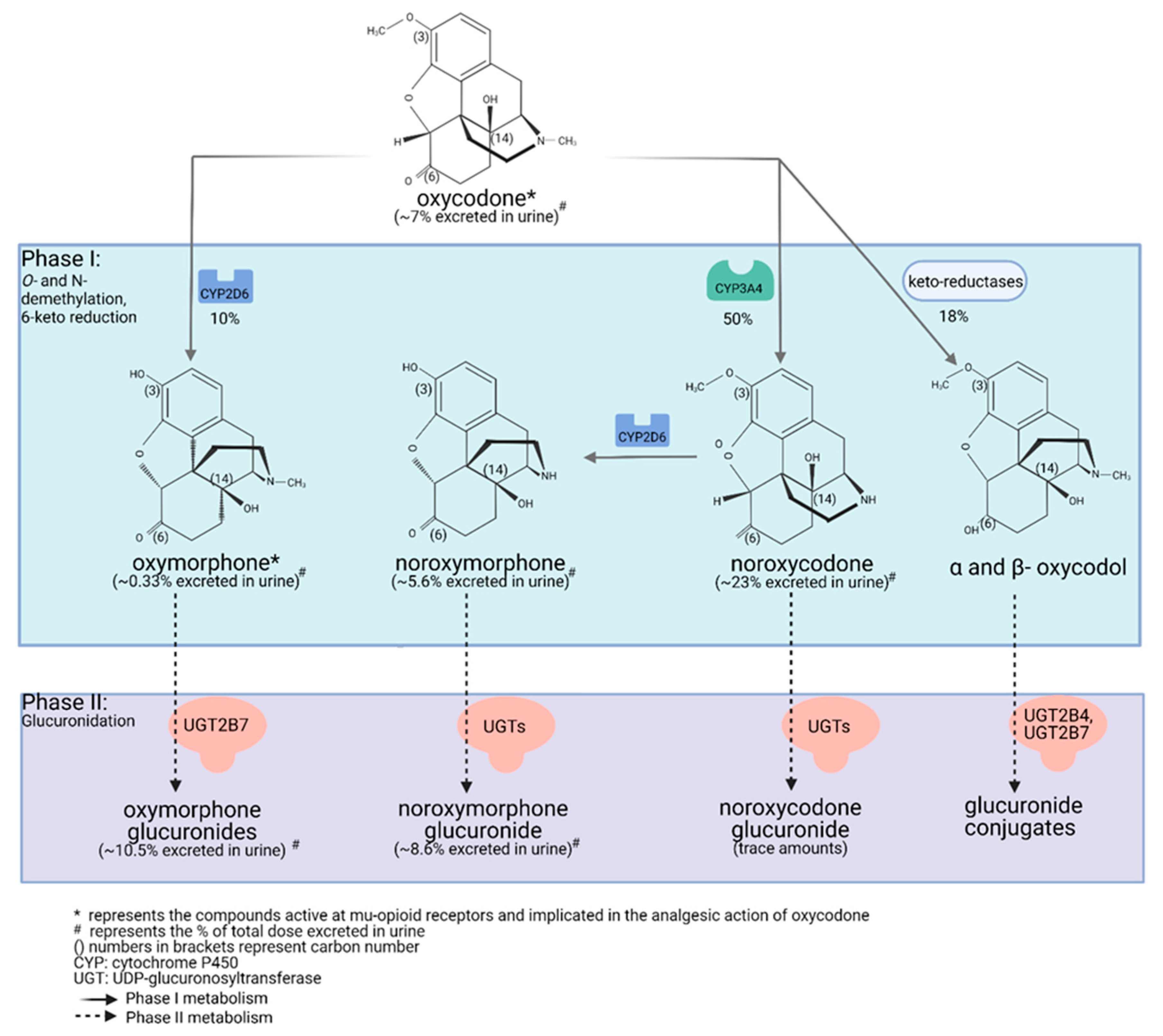
2.2. Oxycodone Metabolism in Humans (PK and PGx; GRADE High Quality ++++)
2.3. Oxycodone Distribution and Protein Binding (PK; GRADE Low Quality ++−−)
2.4. Oxymorphone Pharmacokinetics Following Direct Oxymorphone Administration (PK; GRADE Moderate Quality +++−)
3. Pharmacodynamic Considerations
3.1. Interaction of Oxycodone and Its Metabolites with Opioid Receptors (PD; GRADE Moderate Quality +++−)
3.2. Unlikely Formation of Active 6-Glucuronide and Other Glucuronide Metabolites (GRADE Moderate Quality +++−)
3.3. Opioid μ-Receptor (OPMR1) and Catechol-O-Methyl Transferase (COMT) Genetic Polymorphisms and Response to Opioid Drugs (PD and PGx; GRADE Low Quality ++−−)
4. Pharmacokinetic, Pharmacogenomic, and Pharmacodynamic Considerations
4.1. Pharmacogenetic Studies Relating Oxymorphone Formation to Pain Management by Oxycodone (PK, PD, and PGx; GRADE High Quality +)
4.2. Drug–Drug Interaction Studies Modulating CYP2D6 Activity to Assess Oxymorphone Contribution to Oxycodone Efficacy (PK, PD, and PGx Plus DDIs; GRADE Moderate Quality +++−)
4.3. Drug–Drug Interactions Studies Modulating CYP3A4 Activity to Assess Oxymorphone Contribution to Oxycodone Efficacy (PK-DDI and PD; GRADE Low Quality ++−−)
5. Other Covariables
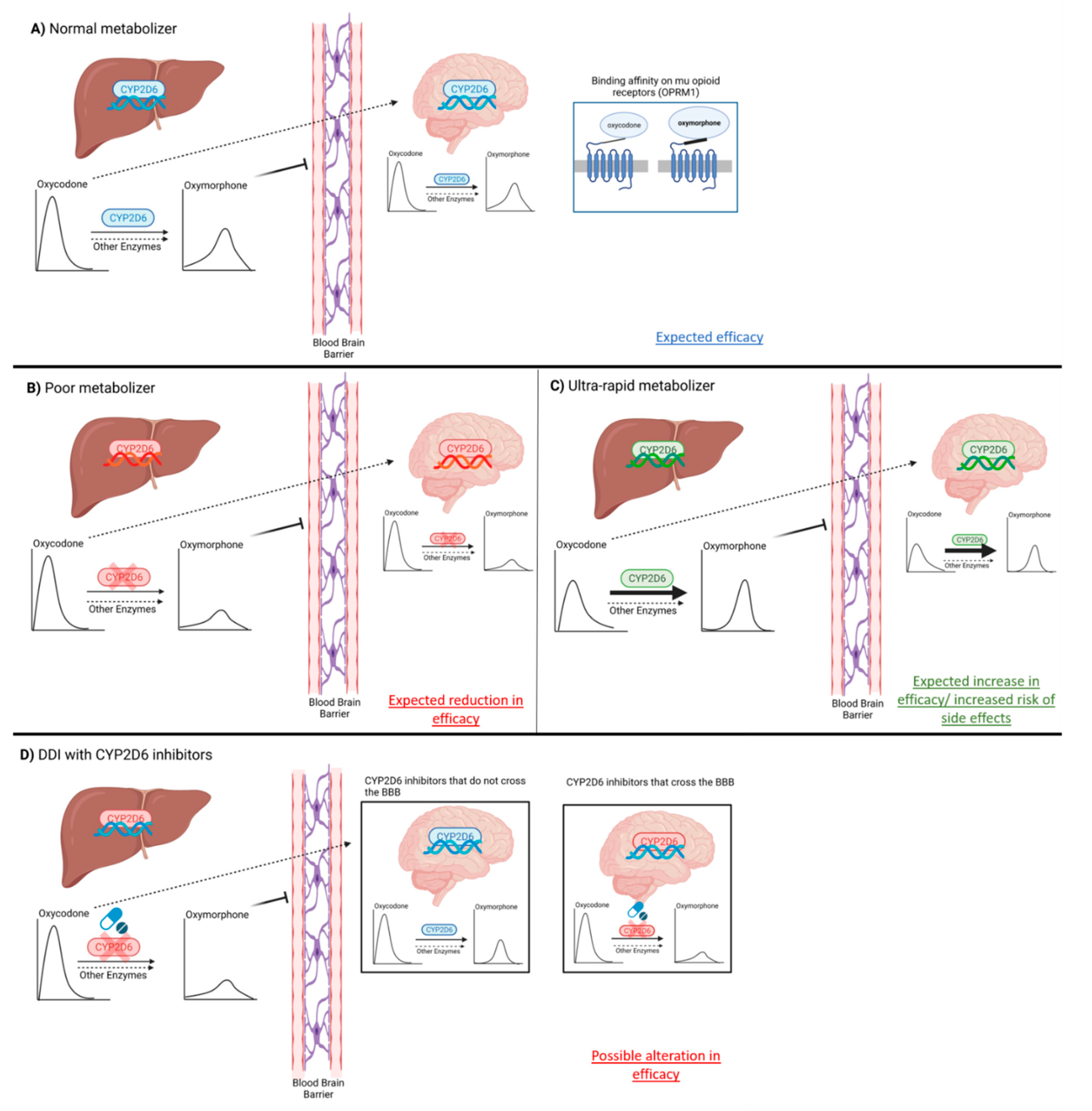
5.1. Phenoconversion as a Confounding Factor (GRADE Low Quality ++−−)
5.2. Liver vs. Brain CYP2D6 Activity (GRADE Low Quality ++−−)
6. Conclusions
- (1)
- (2)
- Pharmacogenomic and drug–drug interaction studies conducted in humans also confirmed the role of CYP2D6 in the formation of oxymorphone. Grade: high quality ++++;
- (3)
- Plasma concentrations of oxymorphone observed following oral administration of oxycodone, although in the low nanogram level, are of the same magnitude as those measured when oxymorphone is directly administered, and they have been shown to be efficacious for pain management in humans. This suggests that oxymorphone mediates most of the analgesic efficacy of oxycodone. Grade: high quality ++++;
- (4)
- Binding studies at the μ-opioid receptor level have clearly established the much greater agonist affinity of oxymorphone vs. oxycodone [44]. Grade: high quality ++++;
- (5)
- The estimated brain concentration ratio of oxycodone vs. oxymorphone, considering protein binding and brain distribution (tissue level) and weighted for the relative potency of both compounds as agonists on the μ-opioid receptor, suggests that oxycodone and oxymorphone contribute equally to analgesic effects. Grade: low quality ++−−;
- (6)
- As the μ-opioid receptor is on the extracellular side of the neuron membrane in the synapse, the extracellular concentrations of oxycodone and oxymorphone could be more relevant than brain tissue levels. Unfortunately, these data are not available at the moment. Grade: very low quality +−−−;
- (7)
- Brain CYP2D6 activity, capable of converting oxycodone into oxymorphone, could explain the lack of association between oxymorphone plasma levels and efficacy observed in some drug–drug interactions and clinical studies. Grade: low quality ++−−;
- (8)
- The lack of oxycodone activity in genetically characterized CYP2D6 PMs, who functionally lack CYP2D6 activity not only in their liver but also in their brain, strongly supports the concept that oxycodone activity is mediated by oxymorphone. Grade: high quality ++++;
- (9)
- Clinically, the coadministration of other CYP2D6 substrates and inhibitors, and their capacity to blunt oxycodone efficacy, could be related to both their capacity to inhibit liver CYP2D6 activity and also to their capacity to cross the blood–brain barrier, penetrate CNS, and reach neurons and inhibit brain CYP2D6 activity, thus limiting local oxymorphone formation from oxycodone. Grade: low quality ++−−;
- (10)
- Finally, mutations on OPMR1 leading to less functional μ-opioid receptors and their role in the inter-individual response to active opioid products is too often overlooked. Grade: low quality ++−−.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- National Institute on Drug Abuse. Overdose Death Rates. Available online: https://www.drugabuse.gov/drug-topics/trends-statistics/overdose-death-rates (accessed on 22 February 2021).
- Kane, S. Oxycodone, ClinCalc DrugStats Database, Version. Available online: https://clincalc.com/DrugStats/Drugs/Oxycodone (accessed on 22 February 2021).
- Rui, P.; Santo, L.; Ashman, J.J. Trends in opioids prescribed at discharge from emergency departments among adults: United States 2006–2017. Nat. Health Stat. Rep. 2020, 135, 1–12. Available online: https://www.cdc.gov/nchs/data/nhsr/nhsr135-508 (accessed on 22 February 2021).
- Crews, K.R.; Monte, A.A.; Huddart, R.; Caudle, K.E.; Kharasch, E.D.; Gaedigk, A.; Dunnenberger, H.M.; Leeder, J.S.; Callaghan, J.T.; Samer, C.F.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for CYP2D6, OPRM1, and COMT Genotypes and Select Opioid Therapy. Clin. Pharmacol. Ther. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.R.; Kruhlak, N.L.; Kim, M.T.; Hawkins, E.G.; Stavitskaya, L. Predicting opioid receptor binding affinity of pharmacologically unclassified designer substances using molecular docking. PLoS ONE 2018, 13, e0197734. [Google Scholar] [CrossRef] [Green Version]
- Consult, E.-B.M. Quality of Evidence per GRADE Criteria. Available online: https://www.ebmconsult.com/articles/levels-of-evidence-and-recommendations (accessed on 5 April 2021).
- Wang, B.; Yang, L.-P.; Zhang, X.-Z.; Huang, S.-Q.; Bartlam, M.; Zhou, S.-F. New insights into the structural characteristics and functional relevance of the human cytochrome P450 2D6 enzyme. Drug Metab. Rev. 2009, 41, 573–643. [Google Scholar] [CrossRef]
- Snider, N.T.; Sikora, M.J.; Sridar, C.; Feuerstein, T.J.; Rae, J.M.; Hollenberg, P.F. The endocannabinoid anandamide is a substrate for the human polymorphic cytochrome P450 2D. J. Pharmacol. Exp. Ther. 2008, 327, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Bertilsson, L.; Dahl, M.-L.; Dalén, P.; Al-Shurbaji, A. Molecular genetics of CYP2D6: Clinical relevance with focus on psychotropic drugs. Br. J. Clin. Pharmacol. 2002, 53, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haufroid, V.; Hantson, P. CYP2D6 genetic polymorphisms and their relevance for poisoning due to amfetamines, opioid analgesics and antidepressants. Clin. Toxicol. 2015, 53, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.W.; Jiang, X.L.; Winter, J.C.; Yu, A.M. Psychedelic 5-methoxy-N,N-dimethyltryptamine: Metabolism, pharmacokinetics, drug interactions, and pharmacological actions. Curr. Drug. Metab. 2010, 11, 659–666. [Google Scholar] [CrossRef] [Green Version]
- The Pharmacogene Variation PharmVar Consortium. CYP2D6 cytochrome P450 Family 2 Subfamily D Member. Available online: https://www.pharmvar.org/gene/CYP2D6 (accessed on 16 September 2016).
- Maréchal, J.D.; Kemp, C.A.; Roberts, G.C.; Paine, M.J.; Wolf, C.R.; Sutcliffe, M.J. Insights into drug metabolism by cytochromes P450 from modelling studies of CYP2D6-drug interactions. Br. J. Pharm. 2008, 153, S82–S89. [Google Scholar] [CrossRef] [Green Version]
- Kapelyukh, Y.; Wolf, R. CYP2D6 substrates and drug metabolism. In CYP2D6: Genetics, Pharmacology and Clinical Relevance; Future Medicine Ltd.: London, UK, 2014; pp. 80–100. [Google Scholar]
- Anzenbacher, P.; Anzenbacherová, E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol. Life Sci. 2001, 58, 737–747. [Google Scholar] [CrossRef]
- Del Tredici, A.L.; Malhotra, A.; Dedek, M.; Espin, F.; Roach, D.; Zhu, G.D.; Voland, J.; Moreno, T.A. Frequency of CYP2D6 Alleles Including Structural Variants in the United States. Front. Pharm. 2018, 9, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byeon, J.Y.; Kim, Y.H.; Lee, C.M.; Kim, S.H.; Chae, W.K.; Jung, E.H.; Choi, C.I.; Jang, C.G.; Lee, S.Y.; Bae, J.W.; et al. CYP2D6 allele frequencies in Korean population, comparison with East Asian, Caucasian and African populations, and the comparison of metabolic activity of CYP2D6 genotypes. Arch. Pharm. Res. 2018, 41, 921–930. [Google Scholar] [CrossRef] [PubMed]
- LLerena, A.; Naranjo, M.E.; Rodrigues-Soares, F.; Penas, L.E.M.; Fariñas, H.; Tarazona-Santos, E. Interethnic variability of CYP2D6 alleles and of predicted and measured metabolic phenotypes across world populations. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Oscarson, M.; Hidestrand, M.; Johansson, I.; Ingelman-Sundberg, M. A Combination of Mutations in the CYP2D6*17 (CYP2D6Z) Allele Causes Alterations in Enzyme Function. Mol. Pharmacol. 1997, 52, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Grangeon, A.; Clermont, V.; Barama, A.; Gaudette, F.; Turgeon, J.; Michaud, V. Development and validation of an absolute protein assay for the simultaneous quantification of fourteen CYP450s in human microsomes by HPLC-MS/MS-based targeted proteomics. J. Pharm. Biomed. Anal. 2019, 173, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Clermont, V.; Grangeon, A.; Barama, A.; Turgeon, J.; Lallier, M.; Malaise, J.; Michaud, V. Activity and mRNA expression levels of selected cytochromes P450 in various sections of the human small intestine. Br. J. Clin. Pharm. 2019, 85, 1367–1377. [Google Scholar] [CrossRef]
- Fonne-Pfister, R.; Bargetzi, M.J.; Meyer, U.A. MPTP, the neurotoxin inducing Parkinson’s disease, is a potent competitive inhibitor of human and rat cytochrome P450 isozymes (P450bufI, P450db1) catalyzing debrisoquine 4-hydroxylation. Biochem. Biophys. Res. Commun. 1987, 148, 1144–1150. [Google Scholar] [CrossRef]
- Gilham, D.E.; Cairns, W.; Paine, M.J.; Modi, S.; Poulsom, R.; Roberts, G.C.; Wolf, C.R. Metabolism of MPTP by cytochrome P4502D6 and the demonstration of 2D6 mRNA in human foetal and adult brain by in situ hybridization. Xenobiotica 1997, 27, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Miksys, S.L.; Tyndale, R.F. Drug-metabolizing cytochrome P450s in the brain. J. Psychiatry Neurosci. 2002, 27, 406–415. [Google Scholar]
- McFadyen, M.C.; Melvin, W.T.; Murray, G.I. Cytochrome P450 in normal human brain and brain tumours. Biochem. Soc. Trans. 1997, 25, S577. [Google Scholar] [CrossRef] [Green Version]
- Mann, A.; Miksys, S.; Lee, A.; Mash, D.C.; Tyndale, R.F. Induction of the drug metabolizing enzyme CYP2D in monkey brain by chronic nicotine treatment. Neuropharmacology 2008, 55, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Siegle, I.; Fritz, P.; Eckhardt, K.; Zanger, U.M.; Eichelbaum, M. Cellular localization and regional distribution of CYP2D6 mRNA and protein expression in human brain. Pharmacogenetics 2001, 11, 237–245. [Google Scholar] [CrossRef]
- Dutheil, F.; Dauchy, S.; Diry, M.; Sazdovitch, V.; Cloarec, O.; Mellottée, L.; Bièche, I.; Ingelman-Sundberg, M.; Flinois, J.P.; de Waziers, I.; et al. Xenobiotic-metabolizing enzymes and transporters in the normal human brain: Regional and cellular mapping as a basis for putative roles in cerebral function. Drug Metab. Dispos. 2009, 37, 1528–1538. [Google Scholar] [CrossRef] [Green Version]
- Hiroi, T.; Imaoka, S.; Funae, Y. Dopamine formation from tyramine by CYP2D. Biochem. Biophys. Res. Commun. 1998, 249, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Bromek, E.; Haduch, A.; Daniel, W.A. The ability of cytochrome P450 2D isoforms to synthesize dopamine in the brain: An in vitro study. Eur. J. Pharm. 2010, 626, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.M.; Idle, J.R.; Byrd, L.G.; Krausz, K.W.; Küpfer, A.; Gonzalez, F.J. Regeneration of serotonin from 5-methoxytryptamine by polymorphic human CYP2D. Pharmacogenetics 2003, 13, 173–181. [Google Scholar] [CrossRef]
- Yu, A.M.; Idle, J.R.; Gonzalez, F.J. Polymorphic cytochrome P450 2D6: Humanized mouse model and endogenous substrates. Drug Metab. Rev. 2004, 36, 243–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peñas-Lledó, E.M.; Llerena, A. CYP2D6 variation, behaviour and psychopathology: Implications for pharmacogenomics-guided clinical trials. Br. J. Clin. Pharm. 2014, 77, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Lalovic, B.; Phillips, B.; Risler, L.L.; Howald, W.; Shen, D.D. Quantitative Contribution of Cyp2d6 and Cyp3a to Oxycodone Metabolism in Human Liver and Intestinal Microsomes. Drug Metab. Dispos. 2004, 32, 447–454. [Google Scholar] [CrossRef] [Green Version]
- Cone, E.J.; Darwin, W.D.; Buchwald, W.F.; Gorodetzky, C.W. Oxymorphone metabolism and urinary excretion in human, rat, guinea pig, rabbit, and dog. Drug Metab. Dispos. 1983, 11, 446–450. [Google Scholar]
- Rasmussen, I. Identification of Cytochrome P450 Isoforms Involved ín the Metabolísm oÍ Oxycodone. Master’s Thesis, Universify of Oslo, Oslo, Norway, December 2000. [Google Scholar]
- Romand, S.; Spaggiari, D.; Marsousi, N.; Samer, C.; Desmeules, J.; Daali, Y.; Rudaz, S. Characterization of oxycodone in vitro metabolism by human cytochromes P450 and UDP-glucuronosyltransferases. J. Pharm. Biomed. Anal. 2017, 144, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Fritz, A.; Busch, D.; Lapczuk, J.; Ostrowski, M.; Drozdzik, M.; Oswald, S. Expression of clinically relevant drug-metabolizing enzymes along the human intestine and their correlation to drug transporters and nuclear receptors: An intra-subject analysis. Basic Clin. Pharm. Toxicol. 2019, 124, 245–255. [Google Scholar] [CrossRef]
- Pöyhiä, R.; Olkkola, K.T.; Seppälä, T.; Kalso, E. The pharmacokinetics of oxycodone after intravenous injection in adults. Br. J. Clin. Pharm. 1991, 32, 516–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korjamo, T.; Tolonen, A.; Ranta, V.-P.; Turpeinen, M.; Kokki, H. Metabolism of Oxycodone in Human Hepatocytes from Different Age Groups and Prediction of Hepatic Plasma Clearance. Front. Pharm. 2012, 2, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pöyhiä, R.; Seppälä, T.; Olkkola, K.T.; Kalso, E. The pharmacokinetics and metabolism of oxycodone after intramuscular and oral administration to healthy subjects. Br. J. Clin. Pharm. 1992, 33, 617–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baselt, R.C.; Stewart, C.B. Determination of Oxycodone and a Major Metabolite in Urine by Electron-Capture GLC. J. Anal. Toxicol. 1978, 2, 107–109. [Google Scholar] [CrossRef]
- Weinstein, S.H.; Gaylord, J.C. Determination of oxycodone in plasma and identification of a major metabolite. J. Pharm. Sci. 1979, 68, 527–528. [Google Scholar] [CrossRef]
- Lalovic, B.; Kharasch, E.; Hoffer, C.; Risler, L.; Liu-Chen, L.-Y.; Shen, D.D. Pharmacokinetics and pharmacodynamics of oral oxycodone in healthy human subjects: Role of circulating active metabolites. Clin. Pharmacol. Ther. 2006, 79, 461–479. [Google Scholar] [CrossRef]
- Nieminen, T.H.; Hagelberg, N.M.; Saari, T.I.; Pertovaara, A.; Neuvonen, M.; Laine, K.; Neuvonen, P.; Olkkola, K.T. Rifampin Greatly Reduces the Plasma Concentrations of Intravenous and Oral Oxycodone. Anesthesiology 2009, 110, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Sirhan-Daneau, A.M.V.; Manzini, C.; Schwab, R.; Demers, A.; Roy, I.; Lafrenchi, P.; Chauny, J.M.; St-Onge, M.; Gaudette, F.; Belanger, F.; et al. Role of CYP2D6 in the pharmacokinetics and pharmacodynamics of oxycodone in healthy volunteers during co-treatment with placebo or quinidine. Can. J. Clin. Pharm. 2010, 17, 1. [Google Scholar]
- Sirhan-Daneau, A.M.V.; St-Onge, M.; Turgeon, J. Pharmacokinetics of oxycodone in extensive and poor metabolizers of CYP2D6 during co-treatment with placebo or quinidine. In Proceedings of the European ISSX Meeting, Lisbon, Portugal, 17–20 May 2009. [Google Scholar]
- Samer, C.F.; Daali, Y.; Wagner, M.; Hopfgartner, G.; Eap, C.; Rebsamen, M.C.; Rossier, M.; Hochstrasser, D.; Dayer, P.; Desmeules, J.A. The effects of CYP2D6 and CYP3A activities on the pharmacokinetics of immediate release oxycodone. Br. J. Pharmacol. 2010, 160, 907–918. [Google Scholar] [CrossRef]
- Andreassen, T.N.; Eftedal, I.; Klepstad, P.; Davies, A.; Bjordal, K.; Lundström, S.; Kaasa, S.; Dale, O. Do CYP2D6 genotypes reflect oxycodone requirements for cancer patients treated for cancer pain? A cross-sectional multicentre study. Eur. J. Clin. Pharmacol. 2011, 68, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Stamer, U.M.; Zhang, L.; Book, M.; Lehmann, L.E.; Stuber, F.; Musshoff, F. CYP2D6 Genotype Dependent Oxycodone Metabolism in Postoperative Patients. PLoS ONE 2013, 8, e60239. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, M.; Takenoshita-Nakaya, S.; Takeba, Y.; Nishimura, Y.; Oda, M.; Watanabe, M.; Ohta, Y.; Kobayashi, S.; Ohtsubo, T.; Kobayashi, S.; et al. Evaluation of CYP2D6 Protein Expression and Activity in the Small Intestine to Determine Its Metabolic Capability in the Japanese Population. Biol. Pharm. Bull. 2017, 40, 1344–1351. [Google Scholar] [CrossRef] [Green Version]
- Paine, M.F.; Hart, H.L.; Ludington, S.S.; Haining, R.L.; Rettie, A.E.; Zeldin, D.C. The Human Intestinal Cytochrome P450 “Pie”. Drug Metab. Dispos. 2006, 34, 880–886. [Google Scholar] [CrossRef]
- Li, A.P.; Ho, M.D.; Alam, N.; Mitchell, W.; Wong, S.; Yan, Z.; Kenny, J.R.; Hop, C.E.C.A. Inter-individual and inter-regional variations in enteric drug metabolizing enzyme activities: Results with cryopreserved human intestinal mucosal epithelia (CHIM) from the small intestines of 14 donors. Pharmacol. Res. Perspect. 2020, 8, e00645. [Google Scholar] [CrossRef] [PubMed]
- Gaudette, F.; Sirhan-Daneau, A.; St-Onge, M.; Turgeon, J.; Michaud, V. Development of a sensitive method for the determination of oxycodone and its major metabolites noroxycodone and oxymorphone in human plasma by liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2016, 1008, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Heiskanen, T.; Olkkola, K.T.; Kalso, E. Effects of blocking CYP2D6 on the pharmacokinetics and pharmacodynamics of oxycodone*. Clin. Pharmacol. Ther. 1998, 64, 603–611. [Google Scholar] [CrossRef]
- Lemberg, K.; Heiskanen, T.; Neuvonen, M.; Kontinen, V.; Neuvonen, P.; Dahl, M.-L.; Kalso, E. Does co-administration of paroxetine change oxycodone analgesia: An interaction study in chronic pain patients. Scand. J. Pain 2010, 1, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Hagelberg, N.M.; Nieminen, T.H.; Saari, T.I.; Neuvonen, M.; Neuvonen, P.J.; Laine, K.; Olkkola, K.T. Voriconazole drastically increases exposure to oral oxycodone. Eur. J. Clin. Pharmacol. 2008, 65, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saari, T.I.; Grönlund, J.; Hagelberg, N.M.; Neuvonen, M.; Laine, K.; Neuvonen, P.; Olkkola, K.T. Effects of itraconazole on the pharmacokinetics and pharmacodynamics of intravenously and orally administered oxycodone. Eur. J. Clin. Pharmacol. 2010, 66, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Grönlund, J.; Saari, T.; Hagelberg, N.; Martikainen, I.K.; Neuvonen, P.J.; Olkkola, K.T.; Laine, K. Effect of Telithromycin on the Pharmacokinetics and Pharmacodynamics of Oral Oxycodone. J. Clin. Pharmacol. 2010, 50, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Boström, E.; Hammarlund-Udenaes, M.; Simonsson, U. Blood–Brain Barrier Transport Helps to Explain Discrepancies in In Vivo Potency between Oxycodone and Morphine. Anesthesiology 2008, 108, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boström, E.; Simonsson, U.S.H.; Hammarlund-Udenaes, M. In Vivo Blood-Brain Barrier Transport of Oxycodone in the Rat: Indications for Active Influx and Implications for Pharmacokinetics/Pharmacodynamics. Drug Metab. Dispos. 2006, 34, 1624–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okura, T.; Higuchi, K.; Deguchi, Y. The Blood-Brain Barrier Transport Mechanism Controlling Analgesic Effects of Opioid Drugs in CNS. Yakugaku Zasshi 2015, 135, 697–702. [Google Scholar] [CrossRef] [Green Version]
- Sadiq, M.W.; Boström, E.; Keizer, R.; Bjorkman, S.; Hammarlund-Udenaes, M. Oxymorphone Active Uptake at the Blood–Brain Barrier and Population Modeling of its Pharmacokinetic–Pharmacodynamic Relationship. J. Pharm. Sci. 2013, 102, 3320–3331. [Google Scholar] [CrossRef]
- Samer, C.F.; Daali, Y.; Wagner, M.; Hopfgartner, G.; Eap, C.; Rebsamen, M.C.; Rossier, M.; Hochstrasser, D.; Dayer, P.; Desmeules, J.A. Genetic polymorphisms and drug interactions modulating CYP2D6 and CYP3A activities have a major effect on oxycodone analgesic efficacy and safety. Br. J. Pharmacol. 2010, 160, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Coffman, B.L.; King, C.D.; Rios, G.R.; Tephly, T.R. The glucuronidation of opioids, other xenobiotics, and androgens by human UGT2B7Y(268) and UGT2B7H(268). Drug Metab. Dispos. 1998, 26, 73–77. [Google Scholar]
- Adams, M.P.; Ahdieh, H. Single- and Multiple-Dose Pharmacokinetic and Dose-Proportionality Study of Oxymorphone Immediate-Release Tablets. Drugs 2005, 6, 91–99. [Google Scholar] [CrossRef]
- Adams, M.P.; Ahdieh, H. Pharmacokinetics and dose-proportionality of oxymorphone extended release and its metabolites: Results of a randomized crossover study. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2004, 24, 468–476. [Google Scholar] [CrossRef]
- Lemberg, K.K.; Korpi, E.R.; Siiskonen, A.O.; Yli-Kauhaluoma, J.T.; Kontinen, V.K.; Viljakka, K.M.; Kalso, E.A. Oxycodone’s Mechanism of Action and Potency Differences after Spinal and Systemic Routes of Administration. Anesthesiology 2007, 106, 1064–1065. [Google Scholar] [CrossRef] [Green Version]
- Monory, K.; Greiner, E.; Sartania, N.; Sallai, L.; Pouille, Y.; Schmidhammer, H.; Hanoune, J.; Borsodi, A. Opioid binding profiles of new hydrazone, oxime, carbazone and semicarbazone derivatives of 14-alkoxymorphinans. Life Sci. 1999, 64, 2011–2020. [Google Scholar] [CrossRef]
- Thompson, C.M.; Wojno, H.; Greiner, E.; May, E.L.; Rice, K.C.; Selley, D.E. Activation of G-Proteins by Morphine and Codeine Congeners: Insights to the Relevance of O- and N-Demethylated Metabolites at μ- and δ-Opioid Receptors. J. Pharmacol. Exp. Ther. 2003, 308, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Lemberg, K.K.; Kontinen, V.K.; Siiskonen, A.O.; Viljakka, K.M.; Yli-Kauhaluoma, J.T.; Korpi, E.R.; Kalso, E.A. Antinociception by Spinal and Systemic Oxycodone: Why Does the Route Make a Difference? Anesthesiology 2006, 105, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.R.; Irvine, R.J.; Somogyi, A.A.; Bochner, F. Mu receptor binding of some commonly used opioids and their metabolites. Life Sci. 1991, 48, 2165–2171. [Google Scholar] [CrossRef]
- Wang, P.; Stone, J.A.; Chen, K.H.; Gross, S.F.; Haller, C.A.; Wu, A.H. Incomplete Recovery of Prescription Opioids in Urine using Enzymatic Hydrolysis of Glucuronide Metabolites. J. Anal. Toxicol. 2006, 30, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, A.E.; Sato, H.; Nielsen, L.M.; Staahl, C.; Droney, J.; Gretton, S.; Branford, R.; Drewes, A.; Arendt-Nielsen, L.; Riley, J.; et al. The genetic influences on oxycodone response characteristics in human experimental pain. Fundam. Clin. Pharmacol. 2015, 29, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Taqi, M.M.; Faisal, M.; Zaman, H. OPRM1 A118G Polymorphisms and Its Role in Opioid Addiction: Implication on Severity and Treatment Approaches. Pharm. Pers. Med. 2019, 12, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Yan, T.; Gong, S.; Deng, H.; Zhang, G.; Wang, J. Opioid receptor mu 1 (OPRM1) A118G polymorphism (rs1799971) and postoperative nausea and vomiting. Am. J. Transl. Res. 2018, 10, 2764–2780. [Google Scholar]
- Yu, Z.; Wen, L.; Shen, X.; Zhang, H. Effects of the OPRM1 A118G Polymorphism (rs1799971) on Opioid Analgesia in Cancer Pain. Clin. J. Pain 2019, 35, 77–86. [Google Scholar] [CrossRef]
- Gong, X.-D.; Wang, J.-Y.; Liu, F.; Yuan, H.-H.; Zhang, W.-Y.; Guo, Y.-H.; Jiang, B. Gene Polymorphisms of OPRM1 A118G and ABCB1 C3435T May Influence Opioid Requirements in Chinese Patients with Cancer Pain. Asian Pac. J. Cancer Prev. 2013, 14, 2937–2943. [Google Scholar] [CrossRef] [Green Version]
- Lötsch, J.; von Hentig, N.; Freynhagen, R.; Griessinger, N.; Zimmermann, M.; Doehring, A.; Rohrbacher, M.; Sittl, R.; Geisslinger, G. Cross-sectional analysis of the influence of currently known pharmacogenetic modulators on opioid therapy in outpatient pain centers. Pharm. Genom. 2009, 19, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Zwisler, S.T.; Enggaard, T.P.; Noehr-Jensen, L.; Mikkelsen, S.; Verstuyft, C.; Becquemont, L.; Sindrup, S.; Brosen, K. The antinociceptive effect and adverse drug reactions of oxycodone in human experimental pain in relation to genetic variations in the OPRM1 and ABCB1 genes. Fundam. Clin. Pharmacol. 2009, 24, 517–524. [Google Scholar] [CrossRef]
- Cajanus, K.; Kaunisto, M.; Tallgren, M.; Jokela, R.; Kalso, E. How Much Oxycodone Is Needed for Adequate Analgesia After Breast Cancer Surgery: Effect of the OPRM1 118A>G Polymorphism. J. Pain 2014, 15, 1248–1256. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.C.; Park, J.-Y.; Myung, S.-K.; Ahn, H.; Fukuda, K.-I.; Liao, Q. OPRM1 A118G Gene Variant and Postoperative Opioid Requirement. Anesthesiology 2014, 121, 825–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.-Y.; Xu, X.-Q.; Bao, Y.; He, J.; Shi, L.; Deng, J.-H.; Gao, X.-J.; Tang, H.-L.; Wang, Y.-M.; Lu, L. The impact of genetic variation on sensitivity to opioid analgesics in patients with postoperative pain: A systematic review and meta-analysis. Pain Physician 2015, 18, 131–152. [Google Scholar] [PubMed]
- Khalil, H.; Sereika, S.M.; Dai, F.; Alexander, S.; Conley, Y.; Gruen, G.; Meng, L.; Siska, P.; Tarkin, I.; Henker, R. OPRM1 and COMT Gene-Gene Interaction Is Associated with Postoperative Pain and Opioid Consumption After Orthopedic Trauma. Biol. Res. Nurs. 2016, 19, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Laugsand, E.A.; Fladvad, T.; Skorpen, F.; Maltoni, M.; Kaasa, S.; Fayers, P.; Klepstad, P. Clinical and genetic factors associated with nausea and vomiting in cancer patients receiving opioids. Eur. J. Cancer 2011, 47, 1682–1691. [Google Scholar] [CrossRef]
- Zwisler, S.T.; Enggaard, T.P.; Mikkelsen, S.; Verstuyft, C.; Becquemont, L.; Sindrup, S.H.; Brosen, K. Lack of Association of OPRM1 and ABCB1 Single-Nucleotide Polymorphisms to Oxycodone Response in Postoperative Pain. J. Clin. Pharmacol. 2012, 52, 234–242. [Google Scholar] [CrossRef]
- Rakvåg, T.T.; Klepstad, P.; Baar, C.; Kvam, T.-M.; Dale, O.; Kaasa, S.; Krokan, H.E.; Skorpen, F. The Val158Met polymorphism of the human catechol-O-methyltransferase (COMT) gene may influence morphine requirements in cancer pain patients. Pain 2005, 116, 73–78. [Google Scholar] [CrossRef]
- Fragoso, R.M.; Pereira, D.; Medeiros, R. Pain polymorphisms and opioids: An evidence based review. Mol. Med. Rep. 2018, 19, 1423–1434. [Google Scholar] [CrossRef] [Green Version]
- Tammimäki, A.; Männistö, P. Catechol-O-methyltransferase gene polymorphism and chronic human pain. Pharm. Genom. 2012, 22, 673–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, S.; Skorpen, F. Variation in theCOMTgene: Implications for pain perception and pain treatment. Pharmacogenomics 2009, 10, 669–684. [Google Scholar] [CrossRef]
- Zwisler, S.T.; Enggaard, T.P.; Noehr-Jensen, L.; Pedersen, R.S.; Mikkelsen, S.; Nielsen, F.; Brosen, K.; Sindrup, S.H. The Hypoalgesic Effect of Oxycodone in Human Experimental Pain Models in Relation to the CYP2D6 Oxidation Polymorphism. Basic Clin. Pharmacol. Toxicol. 2009, 104, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Susce, M.T.; Murray-Carmichael, E.; de Leon, J. Response to hydrocodone, codeine and oxycodone in a CYP2D6 poor metabolizer. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2006, 30, 1356–1358. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.; Mobley, E.; Wang, Z. Complicated Pain Management in a CYP450 2D6 Poor Metabolizer. Pain Pract. 2007, 7, 352–356. [Google Scholar] [CrossRef] [PubMed]
- de Leon, J.; Dinsmore, L.; Wedlund, P. Adverse Drug Reactions to Oxycodone and Hydrocodone in CYP2D6 Ultrarapid Metabolizers. J. Clin. Psychopharmacol. 2003, 23, 420–421. [Google Scholar] [CrossRef]
- Tyndale, R.F.; Droll, K.P.; Sellers, E.M. Genetically deficient CYP2D6 metabolism provides protection against oral opiate dependence. Pharmacogenetics 1997, 7, 375–379. [Google Scholar] [CrossRef]
- Kummer, O.; Hammann, F.; Moser, C.; Schaller, O.; Drewe, J.; Krähenbühl, S. Effect of the inhibition of CYP3A4 or CYP2D6 on the pharmacokinetics and pharmacodynamics of oxycodone. Eur. J. Clin. Pharmacol. 2011, 67, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Zwisler, S.T.; Enggaard, T.P.; Mikkelsen, S.; Brosen, K.; Sindrup, S.H. Impact of the CYP2D6 genotype on post-operative intravenous oxycodone analgesia. Acta Anaesthesiol. Scand. 2010, 54, 232–240. [Google Scholar] [CrossRef]
- Grönlund, J.; Saari, T.I.; Hagelberg, N.M.; Neuvonen, P.J.; Olkkola, K.T.; Laine, K. Exposure to oral oxycodone is increased by concomitant inhibition of CYP2D6 and 3A4 pathways, but not by inhibition of CYP2D6 alone. Br. J. Clin. Pharm. 2010, 70, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Homma, M.; Momo, K.; Okoshi, Y.; Wada, T.; Hara, A.; Chiba, S.; Kohda, Y. Effects of voriconazole co-administration on oxycodone-induced adverse events: A case in the retrospective survey. Eur. J. Clin. Pharm. 2011, 67, 859–861. [Google Scholar] [CrossRef]
- Grönlund, J.; Saari, T.I.; Hagelberg, N.M.; Neuvonen, P.J.; Laine, K.; Olkkola, K.T. Effect of inhibition of cytochrome P450 enzymes 2D6 and 3A4 on the pharmacokinetics of intravenous oxycodone: A randomized, three-phase, crossover, placebo-controlled study. Clin. Drug Investig. 2011, 31, 143–153. [Google Scholar] [CrossRef]
- Darakjian, L.; Deodhar, M.; Turgeon, J.; Michaud, V. Chronic Inflammatory Status Observed in Patients with Type 2 Diabetes Induces Modulation of Cytochrome P450 Expression and Activity. Int. J. Mol. Sci. 2021, 22, 4967. [Google Scholar] [CrossRef] [PubMed]
- Deodhar, M.; Al Rihani, S.B.; Arwood, M.J.; Darakjian, L.; Dow, P.; Turgeon, J.; Michaud, V. Mechanisms of CYP450 Inhibition: Understanding Drug-Drug Interactions Due to Mechanism-Based Inhibition in Clinical Practice. Pharmaceutics 2020, 12, 846. [Google Scholar] [CrossRef]
- Gravel, S.; Chiasson, J.L.; Turgeon, J.; Grangeon, A.; Michaud, V. Modulation of CYP450 Activities in Patients with Type 2 Diabetes. Clin. Pharmacol. Ther. 2019, 106, 1280–1289. [Google Scholar] [CrossRef]
- Magliocco, G.; Matthey, A.; Bararpour, N.; Joye, T.; Gloor, Y.; Desmeules, J.; Thomas, A.; Daali, Y. Metabolomics Reveals Five Endogenous Biomarkers in Human Urine And Plasma To Predict Cyp2d6 Activity. Authorea 2021. [Google Scholar] [CrossRef]
- Grangeon, A.; Gravel, S.; Gaudette, F.; Turgeon, J.; Michaud, V. Highly sensitive LC-MS/MS methods for the determination of seven human CYP450 activities using small oral doses of probe-drugs in human. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1040, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Gravel, S.; Chiasson, J.L.; Gaudette, F.; Turgeon, J.; Michaud, V. Use of 4β-Hydroxycholesterol Plasma Concentrations as an Endogenous Biomarker of CYP3A Activity: Clinical Validation in Individuals with Type 2 Diabetes. Clin Pharm. 2019, 106, 831–840. [Google Scholar] [CrossRef]
- Michaud, V.; Bikmetov, R.; Smith, M.K.; Dow, P.; Darakjian, L.; Deodhar, M.; Cicali, B.; Bain, K.; Turgeon, J. An Observational Study of Drug Interactions Involving CYP2D6 among Opioid Users: Impact on Healthcare Costs. J. Pers. Med. 2021, submitted. [Google Scholar]
- Jannetto, P.J.; Wong, S.H.; Gock, S.B.; Laleli-Sahin, E.; Schur, B.C.; Jentzen, J.M. Pharmacogenomics as Molecular Autopsy for Postmortem Forensic Toxicology: Genotyping Cytochrome P450 2D6 for Oxycodone Cases. J. Anal. Toxicol. 2002, 26, 438–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funae, Y.; Kishimoto, W.; Cho, T.; Niwa, T.; Hiroi, T. CYP2D in the Brain. Drug Metab. Pharmacokinet. 2003, 18, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Syvänen, S.; Schenke, M.; Berg, D.-J.V.D.; Voskuyl, R.A.; De Lange, E.C. Alteration in P-glycoprotein Functionality Affects Intrabrain Distribution of Quinidine More Than Brain Entry—A Study in Rats Subjected to Status Epilepticus by Kainate. AAPS J. 2012, 14, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, R.J.; Kemp, P.M.; Johnson, R.D. Distribution of Paroxetine in Postmortem Fluids and Tissues. 2015. Available online: https://www.faa.gov/data_research/research/med_humanfacs/oamtechreports/2010s/media/201511.pdf (accessed on 5 June 2020).
- Masand, P.S.; Narasimhan, M.; Patkar, A.A. Paroxetine for Somatic Pain Associated with Physical Illness. Prim. Care Companion J. Clin. Psychiatry 2006, 8, 122–130. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| CYP2D6 Activity | Oxycodone/Oxymorphone Concentration Ratio in Plasma | Oxycodone/Oxymorphone Free-Drug Concentration Ratio in Plasma | Oxycodone/Oxymorphone Free-Drug Concentration Ratio in the Brain * | Oxycodone/Oxymorphone Relative Contribution to μ-Opioid Receptor-Binding Considering the Free-Drug Concentration Ratio in the Brain ** |
|---|---|---|---|---|
| UM CYP2D6 | 32:1 | 19:1 | 57:1 | 0.6:1 |
| NM CYP2D6 | 43:1 | 26:1 | 78:1 | 0.8:1 |
| PM CYP2D6 | 300:1 | 180:1 | 540:1 | 5.4:1 |
| With potent CYP2D6 inhibitors | 110:1 | 66:1 | 198:1 | 2:1 |
| With potent CYP3A4 inhibitors | 56:1 to 21:1 | 34:1 to 13:1 | 102:1 to 39:1 | 1:1 to 0.4:1 |
| Studies | Study Design | PK, PD, and/or PGx Measures | Outcomes | Conclusion |
|---|---|---|---|---|
| CYP2D6 polymorphisms: Effect on oxycodone pharmacokinetics and pharmacodynamics | ||||
| Only PGx measurements | ||||
| Zwisler et al. (2009) [91] | 33 healthy volunteers were administered 10 mg oral OXY or a placebo in a crossover design |
|
| OXM may be important for anti-nociception mediated by OXY |
| Zwisler et al. (2010) [97] | 270 patients undergoing thyroid surgery or hysterectomy received IV OXY and morphine (rescue medication). The study was open label; however, CYP2D6 genotypes were double-blinded |
|
| OXM may not be important for anti-nociception based on genotype grouping Caveats: Phenotyping was not conducted, *5 allele was not analyzed |
| Samer et al. (2010) [64] | 10 healthy volunteers were administered oral OXY (0.2 mg/kg) with or without the CYP3A4 inhibitor ketoconazole and CYP2D6 inhibitor quinidine in a randomized crossover design |
|
| |
| Andreassen et al. (2012) [49] | 2294 patients with malignant pain from multiple centers who had CYP2D6 PGx data available were selected for analyses; OXY was administered by SC, orally, or IV |
|
| OXM may not be important for analgesia Caveats: No phenotyping, limited sample sizes, no phenoconversion consideration for concomitant CYP2D6 and 3A4 medicine |
| No PGx; only DDI measurement | ||||
| Kummer et al. (2011) [96] | 14 healthy volunteers, all NMs based on genotyping, were administered 0.2 mg/kg oral OXY with or without ketoconazole and paroxetine |
|
| OXM formation may be essential for OXY mediated analgesia |
| PGx and DDI measurements | ||||
| Heiskanen et al. (1998) [55] | 10 healthy volunteers were administered 20 mg oral OXY either with a placebo or quinidine |
|
| OXM may not be responsible for psychomotor effects Caveats: No pain tests were carried out to determine analgesic effects |
| Hagelberg et al. (2009) [57] | 12 healthy volunteers were given oral OXY and co-administered either a placebo or voriconazole (CYP3A4 inhibitor) |
|
| OXM does not seem to mediate analgesia Caveats: There was no sub-analysis of CYP2D6 PGx, since PM would not have been affected by CYP3A4 inhibition |
| Lemberg et al. (2010) [56] | 20 patients with pain were administered oral OXY and instructed to take morphine for breakthrough pain. After a stable dose was achieved, patients were instructed to take either paroxetine or a placebo. The change in the morphine dose requirement for breakthrough pain was measured |
|
| OXM may not be important for pain control Caveats: No phenotyping; no poor metabolizers were found in the patient population (the group that shows the highest inefficacy) |
| Saari et al. (2010) [58] | 10 healthy volunteers were given a placebo or itraconazole followed by IV or oral OXY in a four-way crossover design |
|
| OXY and OXM may both be important for drug effect; OXY-mediated analgesic effect depends on the test used to measure it Caveats: PGx sub-analysis was not conducted due to the small sample size in each group; there were no PMs in the study |
| Gronlund et al. (2010) [98] | 11 healthy subjects were administered a placebo or oral OXY, alone or in combination with paroxetine (CYP2D6 inhibitor) or with paroxetine plus itraconazole (CYP2D6 and CYP3A4 inhibitors) |
|
| OXM may be important for mediating some effects of OXY based on the worsened deterioration performance ratings for paroxetine alone Caveats: No PGx sub-analysis was conducted due to the small sample size in each group |
| COMT polymorphisms: Effect on opioid/oxycodone efficacy | ||||
| Rakvag et al. (2005) [87] | 207 Caucasian patients undergoing morphine treatment for cancer pain were selected and COMT Val/Met polymorphism was analyzed |
|
| Val/Met polymorphism in COMT may be responsible for morphine PK; however, it does not seem to influence PD measurements |
| Laugsand et al. (2011) [85] | 1579 cancer patients receiving opioids reported intensity of nausea and vomiting |
|
| COMT polymorphisms did not seem to affect opioid-mediated adverse reactions |
| OPRM1 polymorphism: Effect on opioid/oxycodone efficacy | ||||
| Lostch et al. (2009) [79] | Data were collected from 352 Caucasian patients with chronic pain treated with opioids for a minimum of 1 month |
|
| OPRM1G allele carriers may have lower efficacy of opioids compared to homozygous AA carriers Caveats: Though a significant association was found, statistical significance failed in a subsequent ANOVA test |
| Zwisler et al. (2010) [80] | Oral OXY efficacy was tested in 33 healthy volunteers using pain tests |
|
| OPRM1 and ABCB1 variants may contribute to a differential response to OXY |
| Zwisler et al. (2012) [86] | OXY-mediated analgesia was analyzed in 268 postoperative patients (OXY administered via IV) |
|
| OPRM1 and ABCB1 genotypes may not predict variable OXY response |
| Gong et al. (2013) [78] | Patients with cancer pain who were taking opioids (including OXY) were recruited |
|
| OPRM1A118G polymorphism may be important for opioid activity |
| Cajanus et al. (2014) [81] | Association between the OPRM1 A118G polymorphism and OXY-mediated analgesia was studied in 1000 women undergoing surgery for breast cancer (OXY administered via IV) |
|
| |
| Hwang et al. (2014) [82] | 18 articles, totaling 4607 participants, were reviewed in a meta-analysis to determine whether there is an association between OPRM1 A118G polymorphism and response to opioids |
|
| OPRM1 polymorphism may be a determinant of opioid efficacy |
| Olesen et al. (2015) [74] | Meta-analyses from three previously published studies of Caucasian healthy volunteers to determine association with OPRM1 variants and pain tests after OXY, morphine, and/or CR665 administration |
|
| Choice of pain test in healthy volunteer studies can introduce confounding factors in comparing different studies |
| Ren et al. (2015) [83] | A systematic review–meta-analysis approach was applied to determine association between opioid efficacy and genetic polymorphisms |
|
| OPRM1A118G polymorphism may be important for opioid efficacy |
| Khalil et al. (2017) [84] | 153 postoperative pain patients receiving opioids were recruited |
|
| OPRM1 may be important in variable response to opioids |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deodhar, M.; Turgeon, J.; Michaud, V. Contribution of CYP2D6 Functional Activity to Oxycodone Efficacy in Pain Management: Genetic Polymorphisms, Phenoconversion, and Tissue-Selective Metabolism. Pharmaceutics 2021, 13, 1466. https://doi.org/10.3390/pharmaceutics13091466
Deodhar M, Turgeon J, Michaud V. Contribution of CYP2D6 Functional Activity to Oxycodone Efficacy in Pain Management: Genetic Polymorphisms, Phenoconversion, and Tissue-Selective Metabolism. Pharmaceutics. 2021; 13(9):1466. https://doi.org/10.3390/pharmaceutics13091466
Chicago/Turabian StyleDeodhar, Malavika, Jacques Turgeon, and Veronique Michaud. 2021. "Contribution of CYP2D6 Functional Activity to Oxycodone Efficacy in Pain Management: Genetic Polymorphisms, Phenoconversion, and Tissue-Selective Metabolism" Pharmaceutics 13, no. 9: 1466. https://doi.org/10.3390/pharmaceutics13091466
APA StyleDeodhar, M., Turgeon, J., & Michaud, V. (2021). Contribution of CYP2D6 Functional Activity to Oxycodone Efficacy in Pain Management: Genetic Polymorphisms, Phenoconversion, and Tissue-Selective Metabolism. Pharmaceutics, 13(9), 1466. https://doi.org/10.3390/pharmaceutics13091466