
Drug–Drug Interaction Study to Evaluate the Pharmacokinetics, Safety, and Tolerability of Ipatasertib in Combination with Darolutamide in Patients with Advanced Prostate Cancer
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
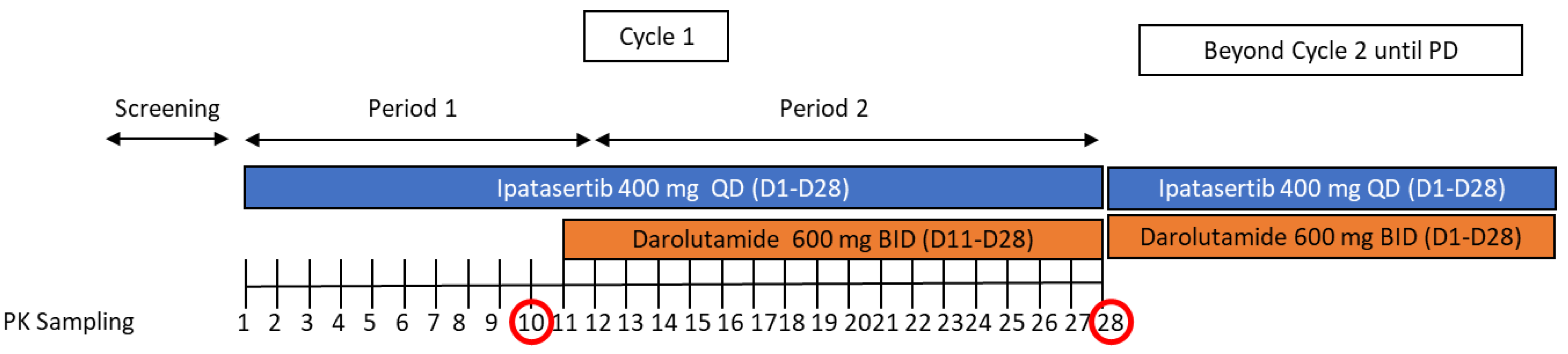
2.1. Study Design and Treatment
2.2. Safety Plan
2.3. Pharmacokinetic Analysis
2.4. Statistical Analysis
3. Results
3.1. Subjects
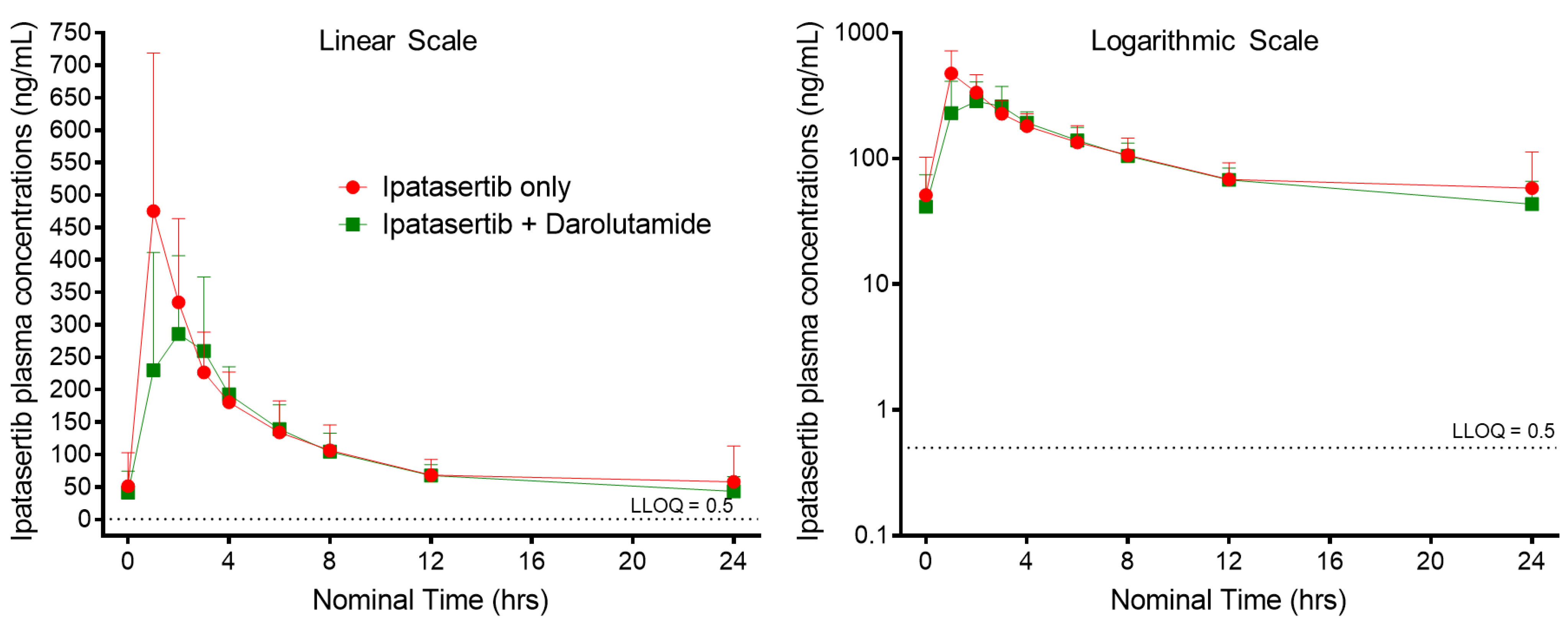
3.2. Ipatasertib Pharmacokinetics
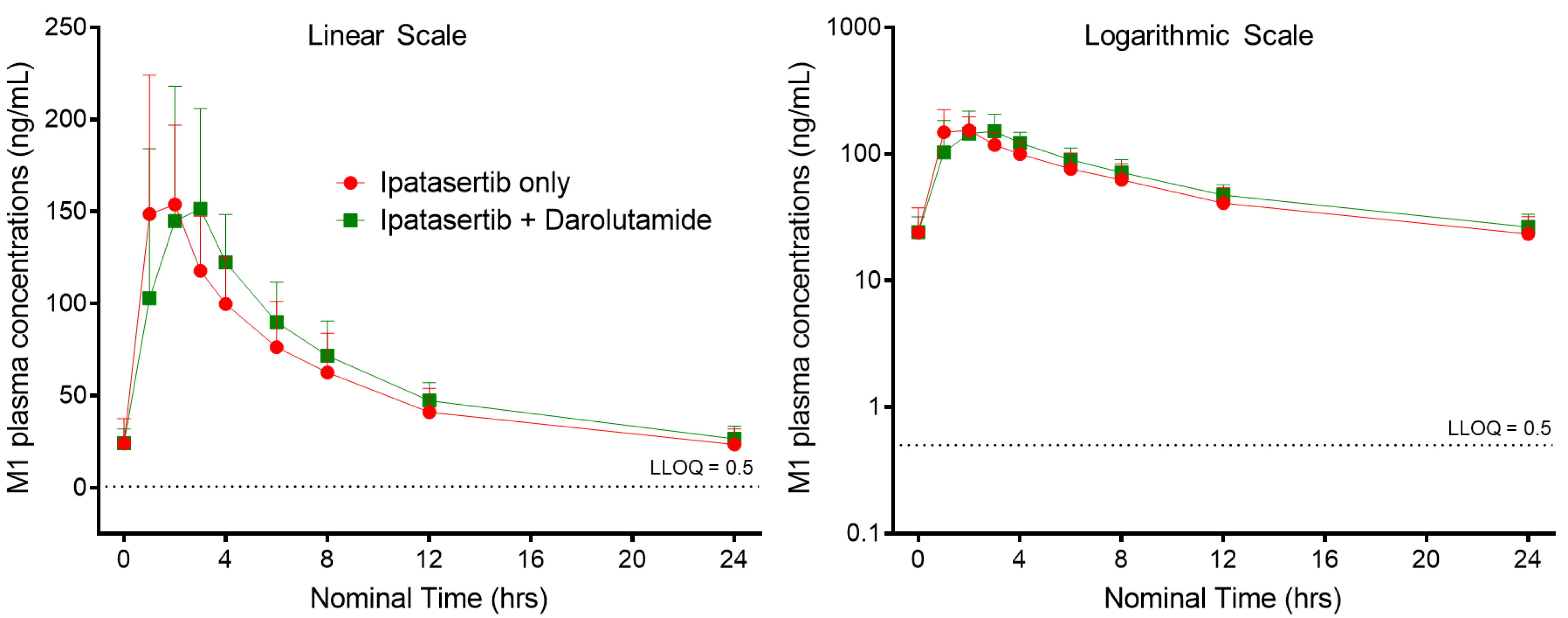
3.3. M1 (G-037720) Pharmacokinetics
3.4. Darolutamide and Keto-Darolutamide Pharmacokinetics
3.5. Safety Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adelaiye-Ogala, R.; Gryder, B.E.; Nguyen, Y.T.M.; Alilin, A.N.; Grayson, A.R.; Bajwa, W.; Jansson, K.H.; Beshiri, M.L.; Agarwal, S.; Rodriguez-Nieves, J.A.; et al. Targeting the PI3K/AKT Pathway Overcomes Enzalutamide Resistance by Inhibiting Induction of the Glucocorticoid Receptor. Mol. Cancer Ther. 2020, 19, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Saura, C.; Roda, D.; Roselló, S.; Oliveira, M.; Macarulla, T.; Pérez-Fidalgo, J.A.; Morales-Barrera, R.; Sanchis-García, J.M.; Musib, L.; Budha, N.; et al. A First-in-Human Phase I Study of the ATP-Competitive AKT Inhibitor Ipatasertib Demonstrates Robust and Safe Targeting of AKT in Patients with Solid Tumors. Cancer Discov. 2017, 7, 102–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sane, R.S.; Cheung, K.W.K.; Cho, E.; Liederer, B.M.; Hanover, J.; Malhi, V.; Plise, E.; Wong, S.; Musib, L. Evaluation of Ipatasertib Interactions with Itraconazole and Coproporphyrin I and III in a Single Drug Interaction Study in Healthy Subjects. J. Pharmacol. Exp. Ther. 2021, 378, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Sutaria, D.S.; Agarwal, P.; Huang, K.C.; Miles, D.; Rotmensch, J.; Hinton, H.; Gallo, J.D.; Rasuo, G.; Sane, R. Mitigating Ipatasertib Induced Glucose Increase through Dose and Meal Timing Modifications. Clin Transl Sci. in press. 2022. [Google Scholar]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): A multicentre, randomised, double-blind, phase 3 trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef]
- Isakoff, S.; Tabernero, J.; Molife, L.; Soria, J.-C.; Cervantes, A.; Vogelzang, N.; Patel, M.; Hussain, M.; Baron, A.; Argilés, G.; et al. Antitumor activity of ipatasertib combined with chemotherapy: Results from a phase Ib study in solid tumors. Ann. Oncol. 2020, 31, 626–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, R.; Poland, B.; Wada, R.; Liu, Q.; Musib, L.; Maslyar, D.; Cho, E.; Yu, W.; Ma, H.; Jin, J.Y.; et al. Exposure–Response-Based Product Profile–Driven Clinical Utility Index for Ipatasertib Dose Selection in Prostate Cancer. CPT Pharmacometrics Syst. Pharmacol. 2019, 8, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Smith, M.R.; Tombal, B. Clinical Development of Darolutamide: A Novel Androgen Receptor Antagonist for the Treatment of Prostate Cancer. Clin. Genitourin. Cancer 2018, 16, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212099Orig1s000lbl.pdf (accessed on 8 September 2022).
- Available online: https://www.ema.europa.eu/en/documents/product-information/nubeqa-epar-product-information_en.pdf (accessed on 8 September 2022).
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212099Orig1s000MultidisciplineR.pdf (accessed on 8 September 2022).
- Zurth, C.; Koskinen, M.; Fricke, R.; Prien, O.; Korjamo, T.; Graudenz, K.; Denner, K.; Bairlein, M.; von Bühler, C.-J.; Wilkinson, G.; et al. Drug–Drug Interaction Potential of Darolutamide: In Vitro and Clinical Studies. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 747–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shore, N.; Zurth, C.; Fricke, R.; Gieschen, H.; Graudenz, K.; Koskinen, M.; Ploeger, B.; Moss, J.; Prien, O.; Borghesi, G.; et al. Evaluation of Clinically Relevant Drug–Drug Interactions and Population Pharmacokinetics of Darolutamide in Patients with Nonmetastatic Castration-Resistant Prostate Cancer: Results of Pre-Specified and Post Hoc Analyses of the Phase III ARAMIS Trial. Target. Oncol. 2019, 14, 527–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornio, A.; Filppula, A.M.; Niemi, M.; Backman, J.T. Clinical Studies on Drug–Drug Interactions Involving Metabolism and Transport: Methodology, Pitfalls, and Interpretation. Clin. Pharmacol. Ther. 2019, 105, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Malhi, V.; Budha, N.; Sane, R.; Huang, J.; Liederer, B.; Meng, R.; Patel, P.; Deng, Y.; Cervantes, A.; Tabernero, J.; et al. Single- and multiple-dose pharmacokinetics, potential for CYP3A inhibition, and food effect in patients with cancer and healthy subjects receiving ipatasertib. Cancer Chemother. Pharmacol. 2021, 88, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; Chen, Y.; Musib, L.; Jin, J.Y.; Cheung, K.W.K.; Yoshida, K.; Sane, R. Assessment of cytochrome P450 3A4-mediated drug–drug interactions for ipatasertib using a fit-for-purpose physiologically based pharmacokinetic model. Cancer Chemother. Pharmacol. 2022, 89, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, J.A.; de Vries, M.; Krauwinkel, W.; Ohtsu, Y.; Noukens, J.; van der Walt, J.-S.; Mol, R.; Mordenti, J.; Ouatas, T. Pharmacokinetic Drug Interaction Studies with Enzalutamide. Clin. Pharmacokinet. 2015, 54, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Parameter | Category/Statistic | No of Subjects a (%) |
|---|---|---|
| Sex, n (%) | Male | 15 (100%) |
| Indication | Metastatic Castration-Resistant Prostate Cancer | 15 (100%) |
| Non-Metastatic Castration-Resistant Prostate Cancer | 0 | |
| Race | White | 15 (100%) |
| Black of African American | 0 | |
| Asian | 0 | |
| American Indian or Alaska Native | 0 | |
| Native Hawaiian or other Pacific Islander | 0 | |
| Other | 0 | |
| Ethnicity | Hispanic or Latino | 0 |
| Not Hispanic or Latino | 15 (100%) | |
| Age (y) | Median (range) | 69.0 (56–79) |
| Weight (kg) | Mean (range) | 92.55 (73.0–123.0) |
| Height (cm) | Mean (range) | 173.0 (161.0–189.0) |
| BMI (kg/m2) | Mean (range) | 31.01 (23.8–40.6) |
| ECOG | 0 | 10 (66.7%) |
| 1 | 5 (33.3%) |
| PK Parameter | Ipatasertib (n = 14) | Ipatasertib with Darolutamide (n = 13) * | |
|---|---|---|---|
| Ipatasertib | AUC0–tau,ss (h·ng/mL) | 2665 (31.2) | 2399 (19.6) |
| AUC0–t,ss (h·ng/mL) | 2666 (31.1) | 2399 (19.6) | |
| Cmax,ss (ng/mL) | 462 (51.0) | 355 (39.8) | |
| Tmax,ss (h) | 1.04 (1.00, 6.10) | 2.00 (1.00, 3.00) | |
| M1 (G-037720) | AUC0–tau,ss (h·ng/mL) | 1329 (27.6) | 1475 (21.7) |
| AUC0–t (h·ng/mL) | 1329 (27.6) | 1475 (21.7) | |
| Cmax,ss (ng/mL) | 169 (36.1) | 177 (41.1) | |
| Tmax,ss (h) | 1.10 (1.00, 4.10) | 2.00 (1.00, 4.00) |
| Test | Reference | Geometric LS Mean Ratio (Test/Ref) | |||||
|---|---|---|---|---|---|---|---|
| Analyte | PK Parameter | n | Geometric Mean | n | Geometric Mean | Estimate | 90% CI |
| Ipatasertib | AUC0–tau,ss (ng·h/mL) | 13 | 2399 | 13 | 2609 | 0.92 | (0.84, 1.01) |
| Cmax,ss (ng/mL) | 13 | 355 | 13 | 452 | 0.79 | (0.62, 0.99) | |
| M1 (G-037720) | AUC0–tau,ss (ng·h/mL) | 13 | 1475 | 13 | 1349 | 1.09 | (1.03, 1.16) |
| Cmax,ss (ng/mL) | 13 | 177 | 13 | 170 | 1.04 | (0.87, 1.25) | |
| PK Parameter | Ipatasertib with Darolutamide | Single-Agent Darolutamide from the Literature (11–13) | |
|---|---|---|---|
| Darolutamide (n = 14) | AUC0–tau,ss (h·µg/mL) | 51.0 (26.8) | - |
| AUC0–t,ss (h·µg/mL) | 51.7 (27.0) | 52.82 (33.9) | |
| Cmax,ss (µg/mL) | 5.43 (27.0) | 4.79 (30.9) | |
| Tmax,ss (h) | 4.23 (2.00, 8.25) | - | |
| Keto-darolutamide (n = 14) | AUC0–tau,ss (h·µg/mL) | 77.0 (32.7) | - |
| AUC0–t,ss (h·µg/mL) | 78.0 (33.5) | 82.8 (39.5) | |
| Cmax,ss (µg/mL) | 8.41 (28.6) | 9.4 (35.4) | |
| Tmax,ss (h) | 4.13 (1.00, 8.25) | - |
| AE Category | Ipatasertib (N = 14) n (%) | Ipatasertib with Darolutamide (N = 14) n (%) |
|---|---|---|
| Total number of patients with at least one AE | 2 (14.3) | 8 (57.1) |
| Total number of AE events | 5 | 39 |
| Total number of deaths | 0 | 0 |
| Total number of patients with at least one | ||
| Any treatment discontinuation | 0 | 1 (7.1) |
| AE leading to discontinuation of ipatasertib | 0 | 1 (7.1) |
| AE leading to dose reduction of ipatasertib | 0 | 1 (7.1) |
| AE leading to dose interruption of ipatasertib | 0 | 2 (14.3) |
| AE leading to discontinuation of darolutamide | 0 | 1 (7.1) |
| AE leading to dose reduction of darolutamide | 0 | 0 |
| AE leading to dose interruption of darolutamide | 0 | 2 (14.3) |
| Grade ≥ 3 AE | 0 | 2 (14.3) |
| Grade 5 AE | 0 | 0 |
| Serious AE | 0 | 2 (14.3) |
| AE related to any treatment | 1 (7.1) | 4 (28.6) |
| AE related to ipatasertib only | 1 (7.1) | 4 (28.6) |
| AE related to darolutamide only | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutaria, D.S.; Rasuo, G.; Harris, A.; Johnson, R.; Miles, D.; Gallo, J.D.; Sane, R. Drug–Drug Interaction Study to Evaluate the Pharmacokinetics, Safety, and Tolerability of Ipatasertib in Combination with Darolutamide in Patients with Advanced Prostate Cancer. Pharmaceutics 2022, 14, 2101. https://doi.org/10.3390/pharmaceutics14102101
Sutaria DS, Rasuo G, Harris A, Johnson R, Miles D, Gallo JD, Sane R. Drug–Drug Interaction Study to Evaluate the Pharmacokinetics, Safety, and Tolerability of Ipatasertib in Combination with Darolutamide in Patients with Advanced Prostate Cancer. Pharmaceutics. 2022; 14(10):2101. https://doi.org/10.3390/pharmaceutics14102101
Chicago/Turabian StyleSutaria, Dhruvitkumar S., Grozdana Rasuo, Adam Harris, Ryan Johnson, Dale Miles, Jorge Daniel Gallo, and Rucha Sane. 2022. "Drug–Drug Interaction Study to Evaluate the Pharmacokinetics, Safety, and Tolerability of Ipatasertib in Combination with Darolutamide in Patients with Advanced Prostate Cancer" Pharmaceutics 14, no. 10: 2101. https://doi.org/10.3390/pharmaceutics14102101
APA StyleSutaria, D. S., Rasuo, G., Harris, A., Johnson, R., Miles, D., Gallo, J. D., & Sane, R. (2022). Drug–Drug Interaction Study to Evaluate the Pharmacokinetics, Safety, and Tolerability of Ipatasertib in Combination with Darolutamide in Patients with Advanced Prostate Cancer. Pharmaceutics, 14(10), 2101. https://doi.org/10.3390/pharmaceutics14102101