

Radiolabeling, Quality Control and In Vivo Imaging of Multimodal Targeted Nanomedicines

 ,
,  , , , ,
, , , ,
Abstract
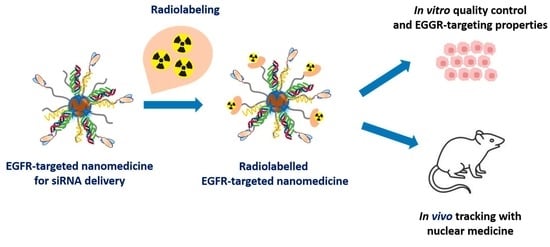
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
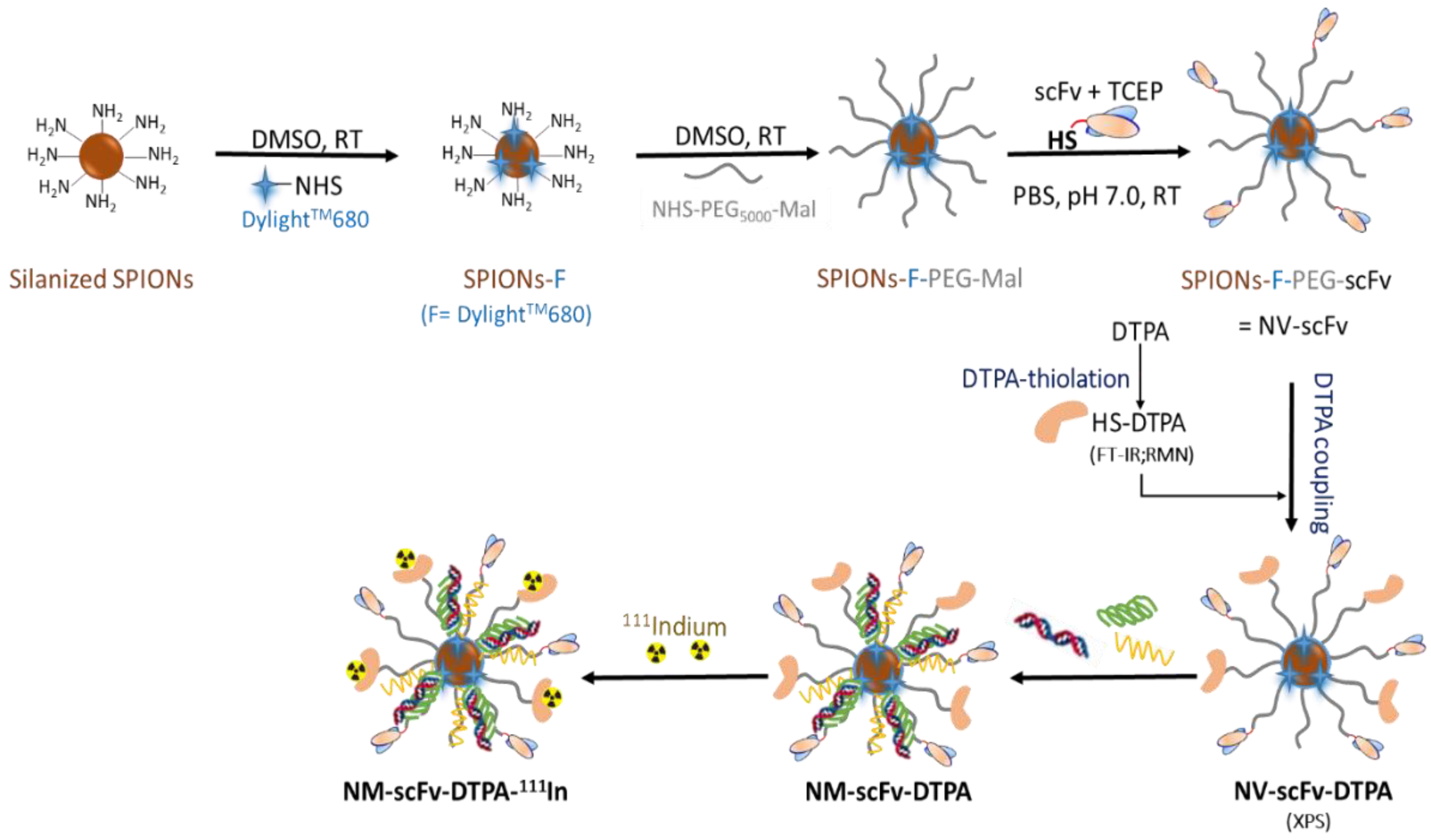
2.3. Preparation of DTPA-Derivatized EGFR-Targeted Nanomedicine (NM-scFv-DTPA)
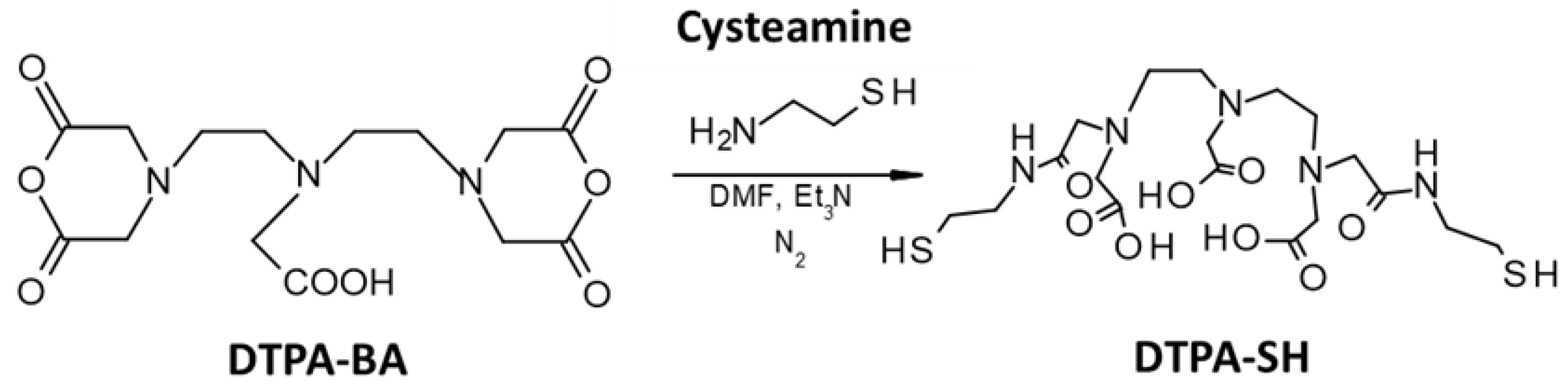
2.3.1. Thiolation of DTPA-BA
- (a)
- Synthesis of the thiolated DTPA (DTPA-SH)
- (b)
- Characterization of the thiolated DTPA
2.3.2. Preparation and Characterization of NV-scFv and DTPA-Derivatization
- (a)
- Preparation of NV-scFv
- (b)
- DTPA-derivatization of the NV-scFv (NV-scFv-DTPA)
- (c)
- Characterizations of NV-scFv-DTPA
2.3.3. Preparation and Characterization of EGFR-Targeted Nanomedicine (NM-scFv-DTPA)
2.4. In Vitro Evaluation of the Impact of DTPA-Coupling on NM-scFv Active Targeting Properties
2.4.1. Functionality Test of Grafted Antibody Fragments on NM-scFv-DTPA
2.4.2. Transfection Assay
2.5. Physico-Chemical Properties and siRNA Protection Capacity of NM-scFv-DTPA-115In3+
2.5.1. Preparation and Physico-Chemical Characterizations of NM-scFv-DTPA-115In
2.5.2. siRNA Protection Capacity of NM-scFv-DTPA-115In
2.6. Radiolabeling Efficiency for Determination of the Suitable DTPA/Fe for In Vivo SPECT-CT Imaging
2.7. Small-Animal In Vivo Experiments
2.7.1. Radiolabeling of NM-scFv-DTPA with 111InCl3
2.7.2. Stability Control of Optimized NM-scFv-DTPA-111In in In Vivo Conditions
2.7.3. Small-Animal SPECT-CT Imaging
3. Results and Discussion
3.1. Synthesis and Characterization of NV-scFv-DTPA
3.1.1. Synthesis and Characterization of DTPA-SH
3.1.2. DTPA Functionalization of NV-scFv
XPS Characterization
Physico-Chemical Properties Characterization
3.2. Selection of Appropriate DTPA/Fe Ratio for Efficient Radiolabeling of NM-scFv-DTPA
3.2.1. Formulation of NM-scFv-DTPA
3.2.2. Complexation of NM-scFv-DTPA with Non-Radioactive 115Indium
3.2.3. Radiolabeling Yield of NM-scFv-DTPA by 111In3+
3.3. In Vitro Potency of Optimized NM-scFv-DTPA for siRNA Active Delivery into EGFR-Positive Cancer Cells
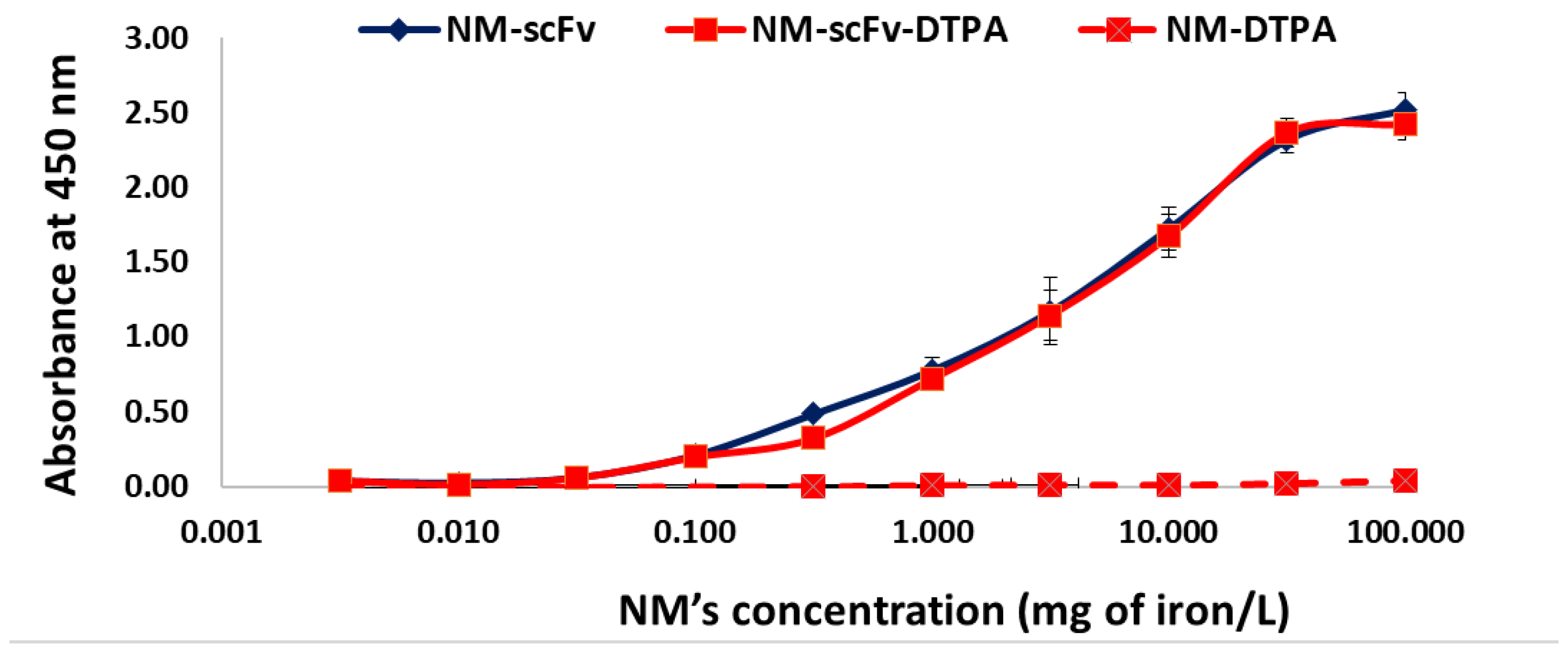
3.3.1. In Vitro Functionality towards EGFR with the ELISA Experiment
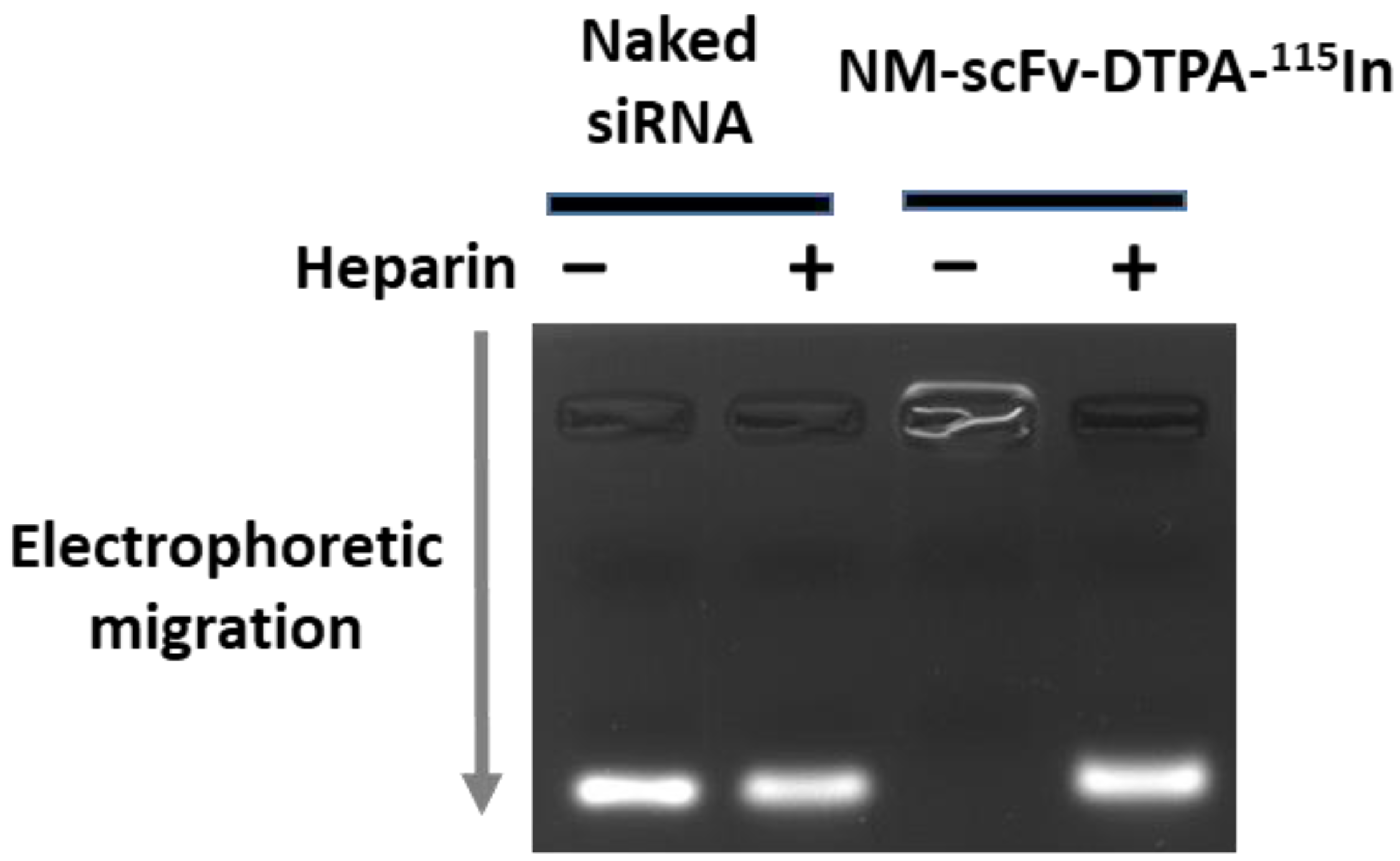
3.3.2. siRNA Protection Capacity of Optimized NM-scFv-DTPA-115In
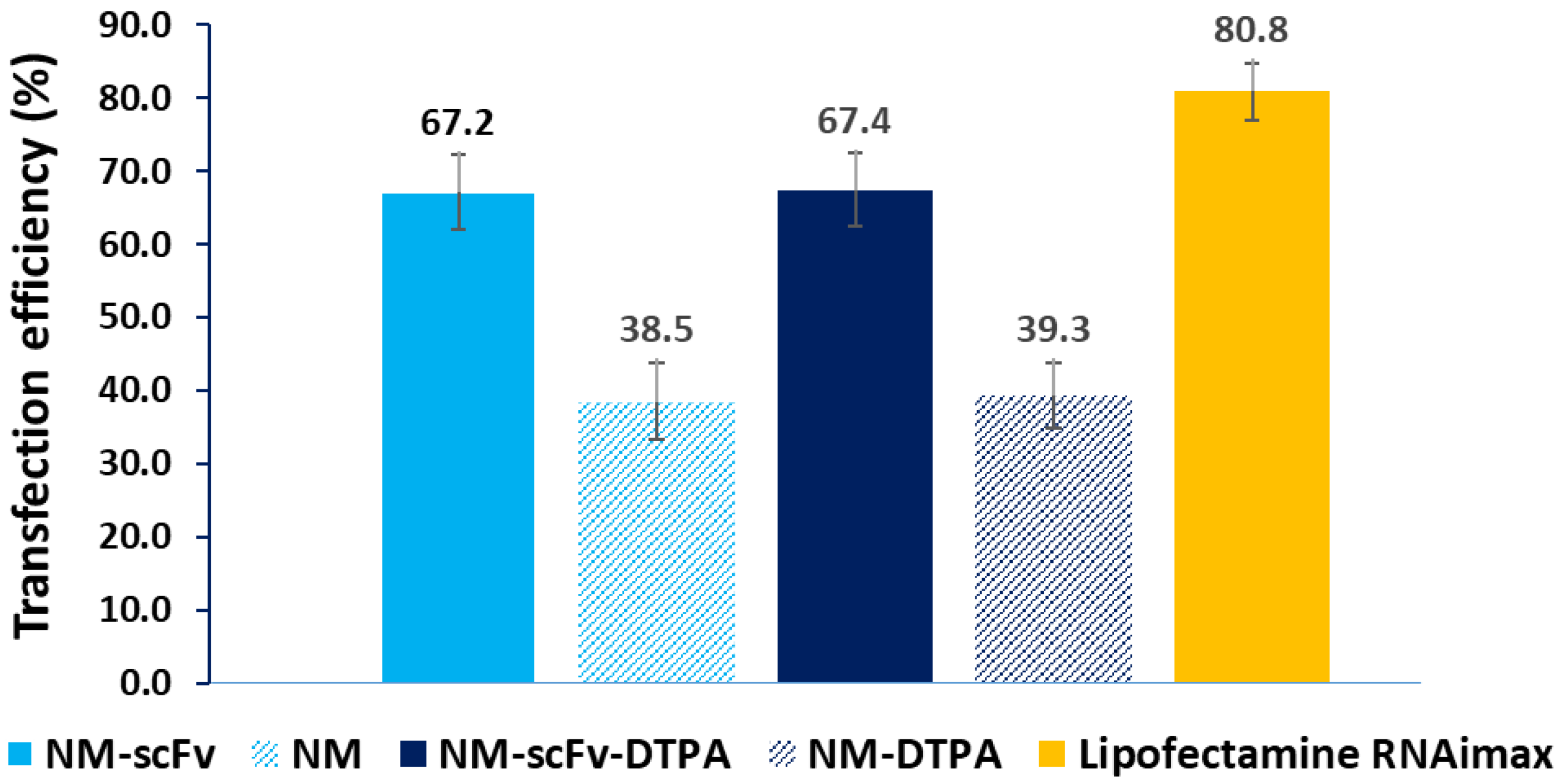
3.3.3. In Vitro Active siRNA Delivery into EGFR-Positive Non-Small Lung Cancer Cells
3.4. Stability Control of the Optimized Nanomedicine in In Vivo Conditions
3.4.1. Radiolabeling of the Optimized NM-scFv-DTPA with 111In
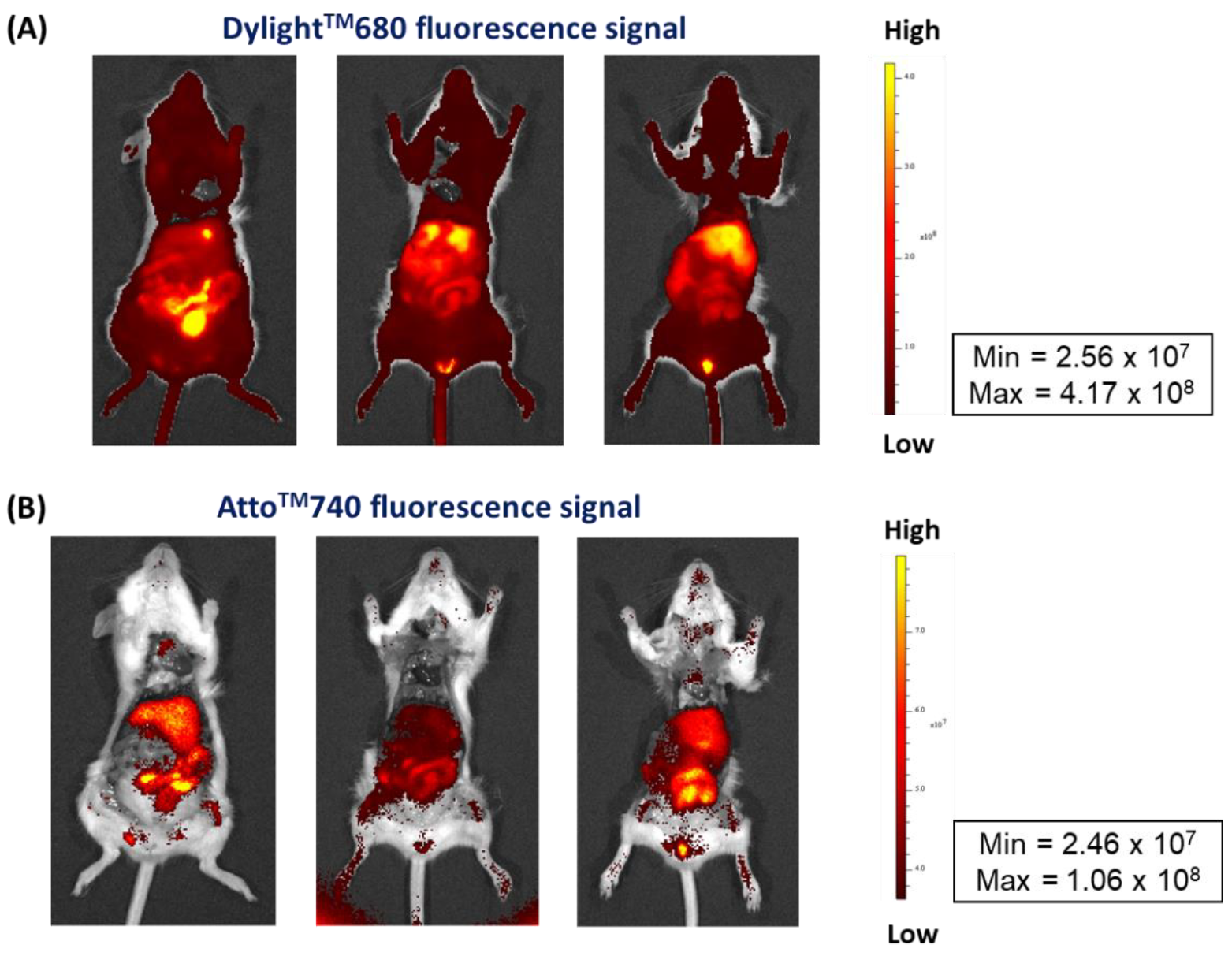
3.4.2. Stability Control of Radiolabeled NM-scFv in In Vivo Conditions Using NIFR Imaging
3.5. 3D In Vivo Tracking with SPECT-CT Imaging
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pawar, A.; Prabhu, P. Nanosoldiers: A Promising Strategy to Combat Triple Negative Breast Cancer. Biomed. Pharmacother. 2019, 110, 319–341. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active Targeting Schemes for Nanoparticle Systems in Cancer Therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Subhan, M.A.; Yalamarty, S.S.K.; Filipczak, N.; Parveen, F.; Torchilin, V.P. Recent Advances in Tumor Targeting via EPR Effect for Cancer Treatment. J. Pers. Med. 2021, 11, 571. [Google Scholar] [CrossRef] [PubMed]
- Villela Zumaya, A.L.; Mincheva, R.; Raquez, J.-M.; Hassouna, F. Nanocluster-Based Drug Delivery and Theranostic Systems: Towards Cancer Therapy. Polymers 2022, 14, 1188. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Aime, S.; Hennink, W.E.; Storm, G.; Kiessling, F. Theranostic Nanomedicine. Acc. Chem. Res. 2011, 44, 1029–1038. [Google Scholar] [CrossRef]
- Indoria, S.; Singh, V.; Hsieh, M.-F. Recent Advances in Theranostic Polymeric Nanoparticles for Cancer Treatment: A Review. Int. J. Pharm. 2020, 582, 119314. [Google Scholar] [CrossRef]
- Yoo, J.; Park, C.; Yi, G.; Lee, D.; Koo, H. Active Targeting Strategies Using Biological Ligands for Nanoparticle Drug Delivery Systems. Cancers 2019, 11, 640. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Ehlerding, E.B.; Cai, W. Theranostic Nanoparticles. J. Nucl. Med. 2014, 55, 1919–1922. [Google Scholar] [CrossRef] [Green Version]
- Vinh Nguyen, P.; Allard-Vannier, E.; Chourpa, I.; Hervé-Aubert, K. Nanomedicines Functionalized with Anti-EGFR Ligands for Active Targeting in Cancer Therapy: Biological Strategy, Design and Quality Control. Int. J. Pharm. 2021, 605, 120795. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular Targeted Therapy: Treating Cancer with Specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Sun, W.; Chen, X.; Xie, C.; Wang, Y.; Lin, L.; Zhu, K.; Shuai, X. Co-Delivery of Doxorubicin and Anti-BCL-2 SiRNA by PH-Responsive Polymeric Vector to Overcome Drug Resistance in In Vitro and In Vivo HepG2 Hepatoma Model. Biomacromolecules 2018, 19, 2248–2256. [Google Scholar] [CrossRef]
- Nguyen, P.V.; Hervé-Aubert, K.; David, S.; Lautram, N.; Passirani, C.; Chourpa, I.; Aubrey, N.; Allard-Vannier, E. Targeted Nanomedicine with Anti-EGFR ScFv for SiRNA Delivery into Triple Negative Breast Cancer Cells. Eur. J. Pharm. Biopharm. 2020, 157, 74–84. [Google Scholar] [CrossRef]
- Ben Djemaa, S.; David, S.; Hervé-Aubert, K.; Falanga, A.; Galdiero, S.; Allard-Vannier, E.; Chourpa, I.; Munnier, E. Formulation and in Vitro Evaluation of a SiRNA Delivery Nanosystem Decorated with GH625 Peptide for Triple Negative Breast Cancer Theranosis. Eur. J. Pharm. Biopharm. 2018, 131, 99–108. [Google Scholar] [CrossRef]
- Ben Djemaa, S.; Munnier, E.; Chourpa, I.; Allard-Vannier, E.; David, S. Versatile Electrostatically Assembled Polymeric SiRNA Nanovectors: Can They Overcome the Limits of SiRNA Tumor Delivery? Int. J. Pharm. 2019, 567, 118432. [Google Scholar] [CrossRef]
- Wessels, J.T.; Busse, A.C.; Mahrt, J.; Dullin, C.; Grabbe, E.; Mueller, G.A. In Vivo Imaging in Experimental Preclinical Tumor Research—A Review. Cytometry 2007, 71A, 542–549. [Google Scholar] [CrossRef]
- Liu, M.; Anderson, R.; Lan, X.; Conti, P.S.; Chen, K. Recent Advances in the Development of Nanoparticles for Multimodality Imaging and Therapy of Cancer. Med. Res. Rev. 2020, 40, 909–930. [Google Scholar] [CrossRef]
- Jin, K.-T.; Yao, J.-Y.; Ying, X.-J.; Lin, Y.; Chen, Y.-F. Nanomedicine and Early Cancer Diagnosis: Molecular Imaging Using Fluorescence Nanoparticles. CTMC 2020, 20, 2737–2761. [Google Scholar] [CrossRef]
- Anani, T.; Rahmati, S.; Sultana, N.; David, A.E. MRI-Traceable Theranostic Nanoparticles for Targeted Cancer Treatment. Theranostics 2021, 11, 579–601. [Google Scholar] [CrossRef]
- Frangioni, J. In Vivo Near-Infrared Fluorescence Imaging. Curr. Opin. Chem. Biol. 2003, 7, 626–634. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Li, Z.; Chen, K.; Hsu, A.R.; Xu, C.; Xie, J.; Sun, S.; Chen, X. PET/MRI Dual-Modality Tumor Imaging Using Arginine-Glycine-Aspartic (RGD)-Conjugated Radiolabeled Iron Oxide Nanoparticles. J. Nucl. Med. 2008, 49, 1371–1379. [Google Scholar] [CrossRef]
- Leary, J.; Key, J. Nanoparticles for Multimodal in Vivo Imaging in Nanomedicine. IJN 2014, 2014, 711–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crișan, G.; Moldovean-Cioroianu, N.S.; Timaru, D.-G.; Andrieș, G.; Căinap, C.; Chiș, V. Radiopharmaceuticals for PET and SPECT Imaging: A Literature Review over the Last Decade. IJMS 2022, 23, 5023. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Nabi, H.; Doerr, R.J. Clinical Applications of Indium-111-Labeled Monoclonal Antibody Imaging in Colorectal Cancer Patients. Semin. Nucl. Med. 1993, 23, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Psimadas, D.; Georgoulias, P.; Valotassiou, V.; Loudos, G. Molecular Nanomedicine Towards Cancer: 111In-Labeled Nanoparticles. J. Pharm. Sci. 2012, 101, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Jani, A.B.; Liauw, S.L.; Blend, M.J. The Role of Indium-111 Radioimmunoscintigraphy in Post-Radical Retropubic Prostatectomy Management of Prostate Cancer Patients. Clin. Med. Res. 2007, 5, 123–131. [Google Scholar] [CrossRef] [Green Version]
- De Gooyer, J.M.; Elekonawo, F.M.K.; Bos, D.L.; van der Post, R.S.; Pèlegrin, A.; Framery, B.; Cailler, F.; Vahrmeijer, A.L.; de Wilt, J.H.W.; Rijpkema, M. Multimodal CEA-Targeted Image-Guided Colorectal Cancer Surgery Using 111In-Labeled SGM-101. Clin. Cancer Res. 2020, 26, 5934–5942. [Google Scholar] [CrossRef]
- Bombardieri, E.; Ambrosini, V.; Aktolun, C.; Baum, R.P.; Bishof-Delaloye, A.; Del Vecchio, S.; Maffioli, L.; Mortelmans, L.; Oyen, W.; Pepe, G.; et al. 111In-Pentetreotide Scintigraphy: Procedure Guidelines for Tumour Imaging. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1441–1448. [Google Scholar] [CrossRef]
- Wangler, B.; Schirrmacher, R.; Bartenstein, P.; Wangler, C. Chelating Agents and Their Use in Radiopharmaceutical Sciences. MRMC 2011, 11, 968–983. [Google Scholar] [CrossRef]
- Alric, C.; Taleb, J.; Le Duc, G.; Mandon, C.; Billotey, C.; Le Meur-Herland, A.; Brochard, T.; Vocanson, F.; Janier, M.; Perriat, P.; et al. Gadolinium Chelate Coated Gold Nanoparticles as Contrast Agents for Both X-Ray Computed Tomography and Magnetic Resonance Imaging. J. Am. Chem. Soc. 2008, 130, 5908–5915. [Google Scholar] [CrossRef]
- Pijeira, M.S.O.; Viltres, H.; Kozempel, J.; Sakmár, M.; Vlk, M.; İlem-Özdemir, D.; Ekinci, M.; Srinivasan, S.; Rajabzadeh, A.R.; Ricci-Junior, E.; et al. Radiolabeled Nanomaterials for Biomedical Applications: Radiopharmacy in the Era of Nanotechnology. EJNMMI Radiopharm. Chem. 2022, 7, 8. [Google Scholar] [CrossRef]
- Grenier, L.; Beyler, M.; Platas-Iglesias, C.; Closson, T.; Gómez, D.E.; Seferos, D.S.; Liu, P.; Ornatsky, O.I.; Baranov, V.; Tripier, R. Highly Stable and Inert Complexation of Indium(III) by Reinforced Cyclam Dipicolinate and a Bifunctional Derivative for Bead Encoding in Mass Cytometry. Chem. Eur. J. 2019, 25, 15387–15400. [Google Scholar] [CrossRef]
- Psimadas, D.; Bouziotis, P.; Georgoulias, P.; Valotassiou, V.; Tsotakos, T.; Loudos, G. Radiolabeling Approaches of Nanoparticles with 99m Tc: RADIOLABELING OF NANOPARTICLES WITH 99M TC. Contrast Media Mol. Imaging 2013, 8, 333–339. [Google Scholar] [CrossRef]
- Pitkänen, L.; Striegel, A.M. Size-Exclusion Chromatography of Metal Nanoparticles and Quantum Dots. TrAC Trends Anal. Chem. 2016, 80, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Helbok, A.; Decristoforo, C.; Dobrozemsky, G.; Rangger, C.; Diederen, E.; Stark, B.; Prassl, R.; von Guggenberg, E. Radiolabeling of Lipid-Based Nanoparticles for Diagnostics and Therapeutic Applications: A Comparison Using Different Radiometals. J. Liposome Res. 2010, 20, 219–227. [Google Scholar] [CrossRef]
- Gill, M.R.; Menon, J.U.; Jarman, P.J.; Owen, J.; Skaripa-Koukelli, I.; Able, S.; Thomas, J.A.; Carlisle, R.; Vallis, K.A. 111 In-Labelled Polymeric Nanoparticles Incorporating a Ruthenium-Based Radiosensitizer for EGFR-Targeted Combination Therapy in Oesophageal Cancer Cells. Nanoscale 2018, 10, 10596–10608. [Google Scholar] [CrossRef] [Green Version]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal Growth Factor Receptor (EGFR) in Lung Cancer: An Overview and Update. J. Thorac. Dis 2010, 2, 48–51. [Google Scholar]
- Rosenkranz, A.A.; Slastnikova, T.A. Epidermal Growth Factor Receptor: Key to Selective Intracellular Delivery. Biochem. Mosc. 2020, 85, 967–993. [Google Scholar] [CrossRef]
- Rho, J.K.; Choi, Y.J.; Choi, Y.R.; Kim, S.Y.; Choi, S.J.; Choi, C.-M.; Na, I.I.; Lee, J.C. The Effect of Acquired Cisplatin Resistance on Sensitivity to EGFR Tyrosine Kinase Inhibitors in EGFR Mutant Lung Cancer Cells. Oncol. Res. 2011, 19, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Behlke, M.A. Progress towards in Vivo Use of SiRNAs. Mol. Ther. 2006, 13, 644–670. [Google Scholar] [CrossRef]
- Landen, C.N.; Chavez-Reyes, A.; Bucana, C.; Schmandt, R.; Deavers, M.T.; Lopez-Berestein, G.; Sood, A.K. Therapeutic EphA2 Gene Targeting In Vivo Using Neutral Liposomal Small Interfering RNA Delivery. Cancer Res. 2005, 65, 6910–6918. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.; Szigyártó, I.C.; Gyurkó, I.; Dóczi, R.; Horváth, I.; Máthé, D.; Szigeti, K. Radiolabeling and Quantitative In Vivo SPECT/CT Imaging Study of Liposomes Using the Novel Iminothiolane- 99m Tc-Tricarbonyl Complex. Contrast Media Mol. Imaging 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.; Tang, Q.; Yin, D.; Tang, C.; He, E.; Zou, L.; Peng, Q. The Protein Corona and Its Effects on Nanoparticle-Based Drug Delivery Systems. Acta Biomater. 2021, 129, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Iochmann, S.; Lerondel, S.; Bléchet, C.; Lavergne, M.; Pesnel, S.; Sobilo, J.; Heuzé-Vourc’h, N.; Le Pape, A.; Reverdiau, P. Monitoring of Tumour Progression Using Bioluminescence Imaging and Computed Tomography Scanning in a Nude Mouse Orthotopic Model of Human Small Cell Lung Cancer. Lung Cancer 2012, 77, 70–76. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
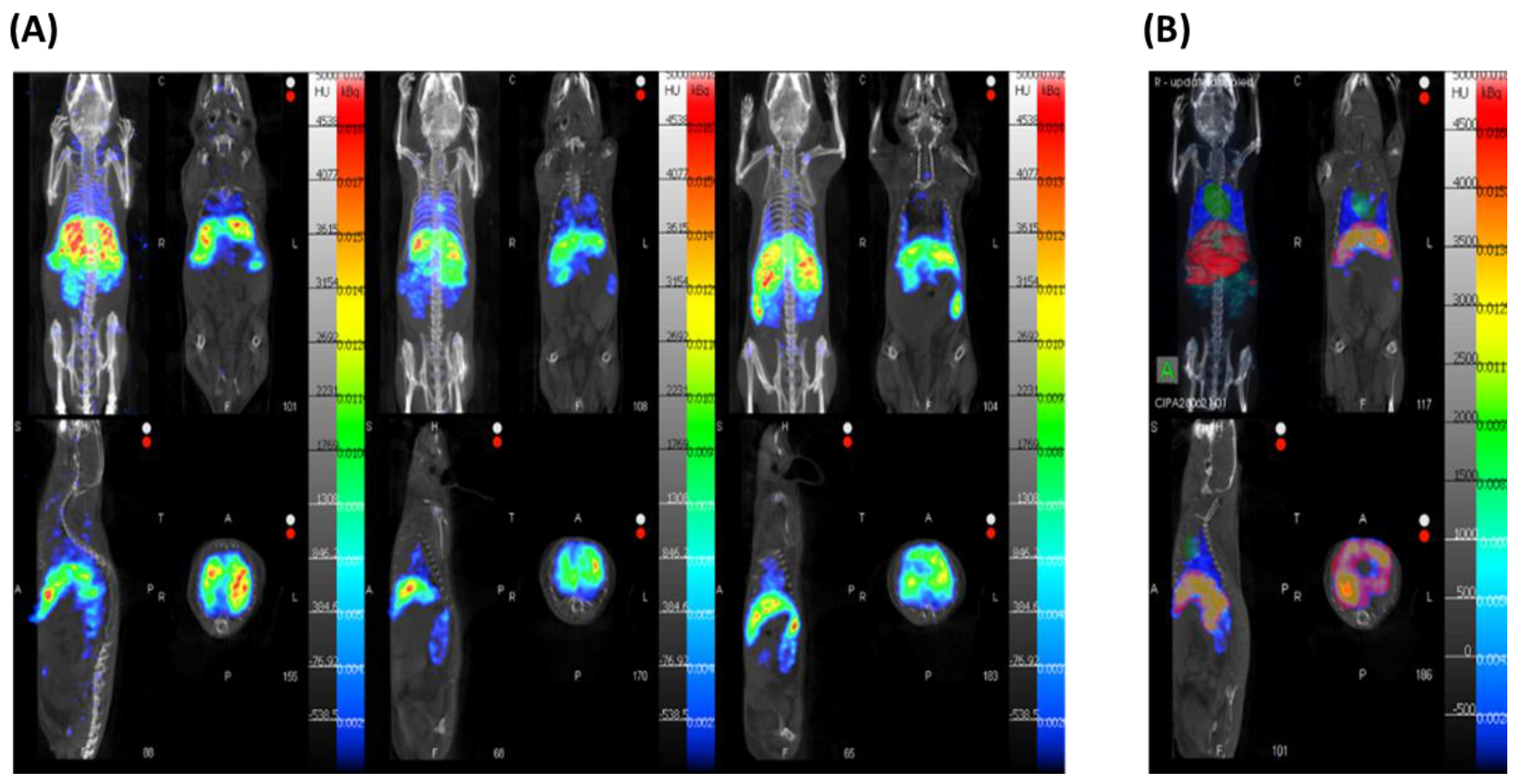{kind=link}
| Batches | C | O | N | Fe | Na | Cl | P | K | Ca | Si | N/Fe |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NV-scFv | 42.7 | 38.1 | 4.2 | 4.1 | 4.5 | 1.6 | 2.7 | 0.5 | - | 1.6 | 1.0 |
| NV-scFv-DTPA0.6 | 43.0 | 38.2 | 3.4 | 1.9 | 5.5 | 1.5 | 4.5 | 0.4 | 0.7 | 0.9 | 1.8 |
| Batch | DTPA/Fe Ratio | DH (nm) | PDi | ζ (mV) | Number of scFv/NV |
|---|---|---|---|---|---|
| NV-scFv | 0 | 75.4 ± 2.0 | 0.17 ± 0.01 | −3.5 ± 1.0 | 26 ± 3 |
| NV | 78.8 ± 0.2 | 0.16 ± 0.01 | −4.9 ± 1.1 | 0 | |
| NV-scFv-DTPA0.03 | 0.03 | 85.0 ± 2.1 | 0.18 ± 0.01 | −3.3 ± 0.3 | 24 ± 2 |
| NV-scFv-DTPA0.3 | 0.3 | 75.6 ± 1.3 | 0.16 ± 0.01 | −2.9 ± 0.6 | 28 ± 2 |
| NV-scFv-DTPA0.6 | 0.6 | 79.9 ± 1.5 | 0.16 ± 0.01 | −4.4 ± 0.9 | 24 ± 2 |
| Batch | DTPA/Fe Ratio | DH (nm) | PDi | ζ (mV) |
|---|---|---|---|---|
| NM | 0 | 99.0 ± 2.5 | 0.27 ± 0.01 | +15.3 ± 1.3 |
| NM-scFv | 0 | 100.0 ± 3.5 | 0.24 ± 0.03 | +21.9 ± 4.4 |
| NM-scFv-DTPA0.03 | 0.03 | 92.4 ± 5.0 | 0.21 ± 0.01 | +18.5 ± 2.6 |
| NM-scFv-DTPA0.3 | 0.3 | 91.0 ± 4.7 | 0.23 ± 0.01 | +12.5 ± 3.4 |
| NM-scFv-DTPA0.6 | 0.6 | 145.9 ± 3.9 | 0.27 ± 0.01 | +6.79 ± 0.93 |
| Batch | DTPA/Fe Molar Ratio | DH (nm) | PDi | ζ (mV) |
|---|---|---|---|---|
| NM-scFv-DTPA0.03-115In | 0.03 | 103.0 ± 9.1 | 0.27 ± 0.01 | +0.7 ± 0.3 |
| NM-scFv-DTPA0.3-115In | 0.3 | 113.2 ± 0.5 | 0.24 ± 0.01 | +2.0 ± 1.8 |
| NM-scFv-DTPA0.6-115In | 0.6 | 206.1 ± 10.4 | 0.21 ± 0.02 | +0.3 ± 0.1 |
| Batch | Before Filtration | After Membrane Filtration | Filtration Yield | ||
|---|---|---|---|---|---|
| DH (nm) | PDi | DH (nm) | PDi | ||
| NM-scFv-DTPA0.03-115In | 65.9 ± 3.3 (70%) 720.4 ± 17.5 (30%) | 0.42 ± 0.01 | 77.6 ± 3.5 (100%) | 0.24 ± 0.01 | 81.5% |
| NM-scFv-DTPA0.3-115In | 82.4 ± 2.6 (70%) 1851 ± 326.1 (30%) | 0.53 ± 0.01 | 85.7 ± 0.6 (100%) | 0.23 ± 0.01 | 77.8% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, P.-V.; Allard-Vannier, E.; Aubrey, N.; Labrugère-Sarroste, C.; Chourpa, I.; Sobilo, J.; Le Pape, A.; Hervé-Aubert, K. Radiolabeling, Quality Control and In Vivo Imaging of Multimodal Targeted Nanomedicines. Pharmaceutics 2022, 14, 2679. https://doi.org/10.3390/pharmaceutics14122679
Nguyen P-V, Allard-Vannier E, Aubrey N, Labrugère-Sarroste C, Chourpa I, Sobilo J, Le Pape A, Hervé-Aubert K. Radiolabeling, Quality Control and In Vivo Imaging of Multimodal Targeted Nanomedicines. Pharmaceutics. 2022; 14(12):2679. https://doi.org/10.3390/pharmaceutics14122679
Chicago/Turabian StyleNguyen, Phuoc-Vinh, Emilie Allard-Vannier, Nicolas Aubrey, Christine Labrugère-Sarroste, Igor Chourpa, Julien Sobilo, Alain Le Pape, and Katel Hervé-Aubert. 2022. "Radiolabeling, Quality Control and In Vivo Imaging of Multimodal Targeted Nanomedicines" Pharmaceutics 14, no. 12: 2679. https://doi.org/10.3390/pharmaceutics14122679
APA StyleNguyen, P. -V., Allard-Vannier, E., Aubrey, N., Labrugère-Sarroste, C., Chourpa, I., Sobilo, J., Le Pape, A., & Hervé-Aubert, K. (2022). Radiolabeling, Quality Control and In Vivo Imaging of Multimodal Targeted Nanomedicines. Pharmaceutics, 14(12), 2679. https://doi.org/10.3390/pharmaceutics14122679