
Combination of DNA Vaccine and Immune Checkpoint Blockades Improves the Immune Response in an Orthotopic Unresectable Glioblastoma Model

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
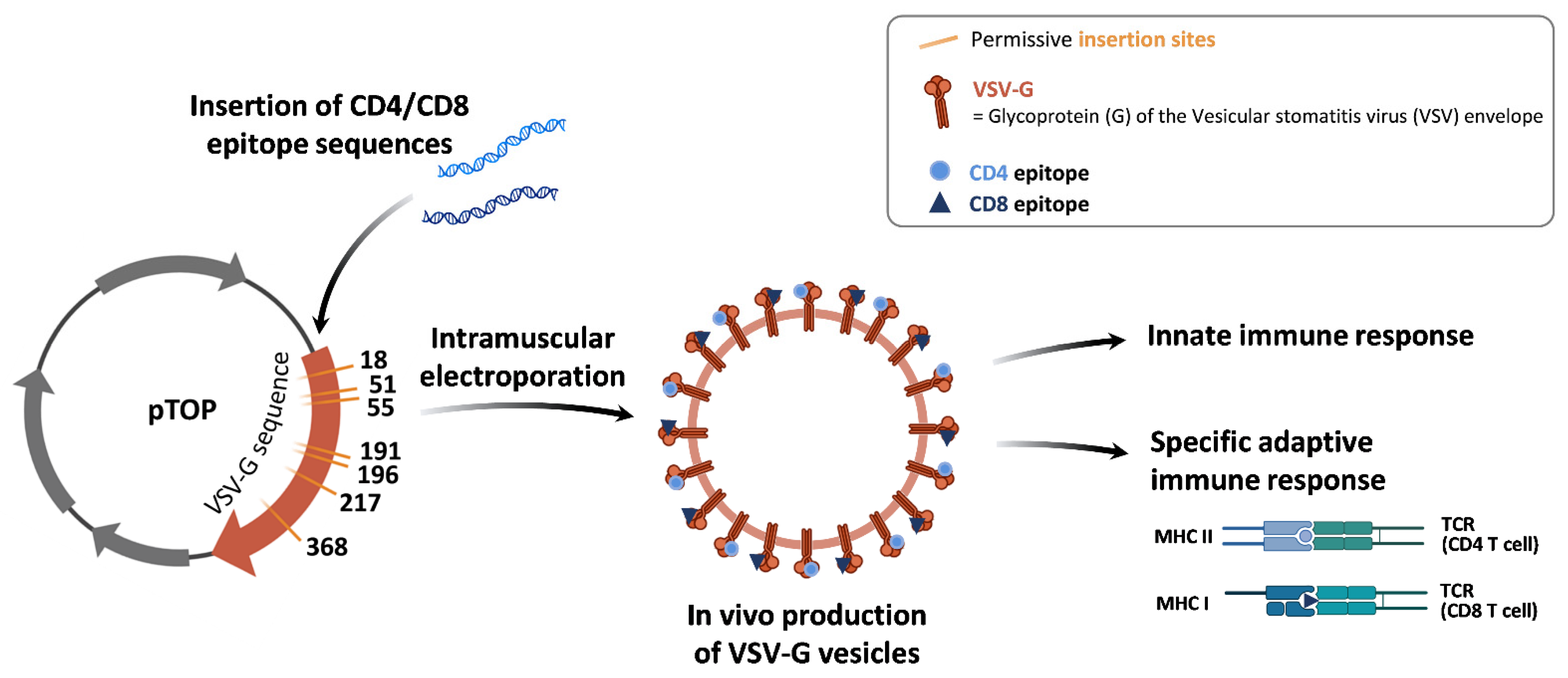
2.1. Plasmids
2.2. GL261 Cell Line
2.3. In Vivo Experiments
2.3.1. GL261 Mouse Models
2.3.2. Magnetic Resonance Imaging
2.3.3. Immunization
2.4. IFN-γ Enzyme-Linked Immunospot Assay
2.5. Flow Cytometry Analysis of the Immune Populations in Brains and Spleens
2.6. Statistics
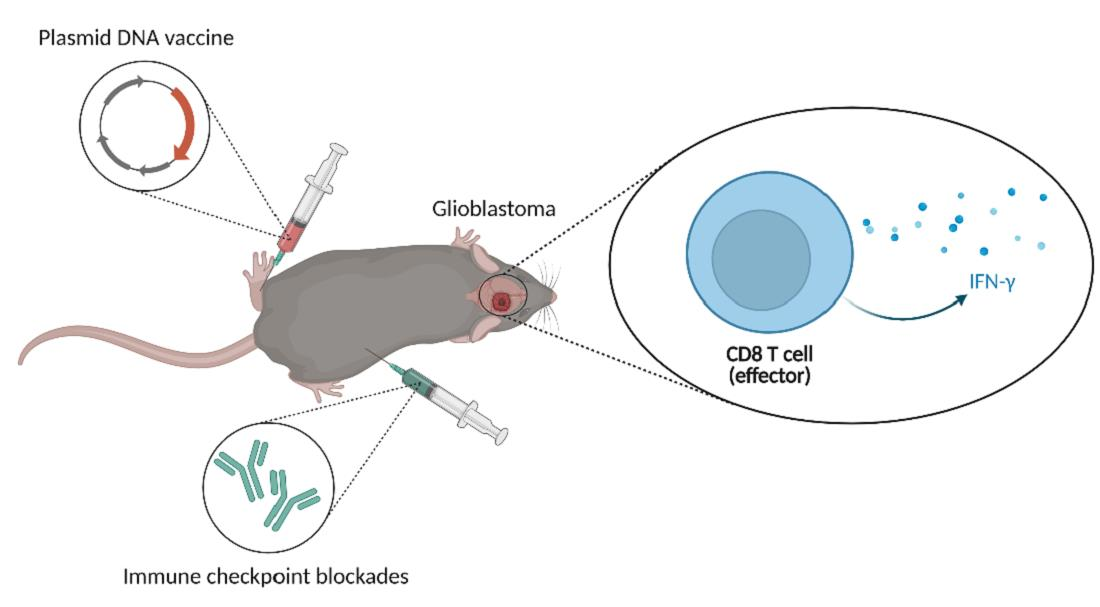
3. Results
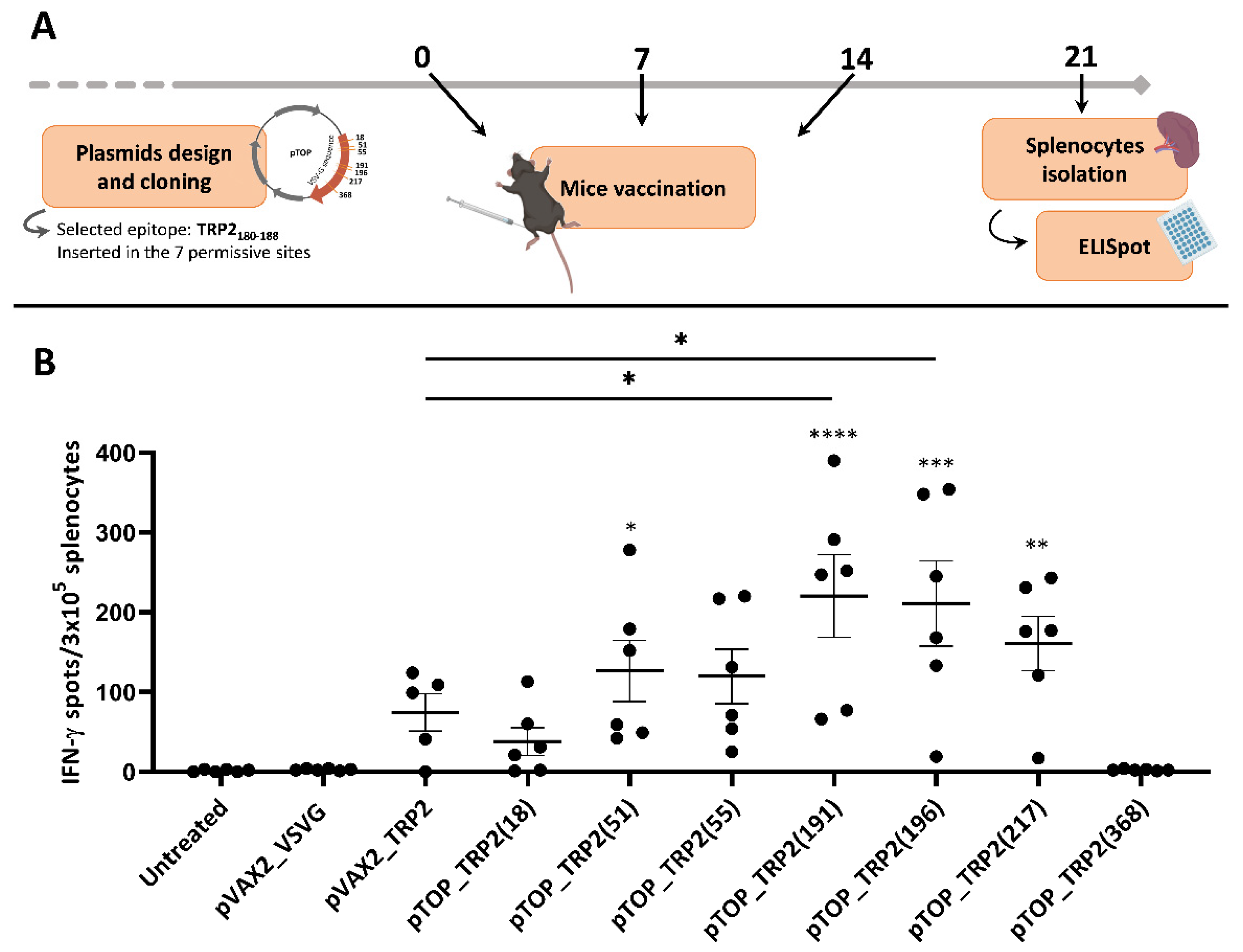
3.1. The Insertion Position of TRP2180–188 in VSV-G Influences the Immune Response
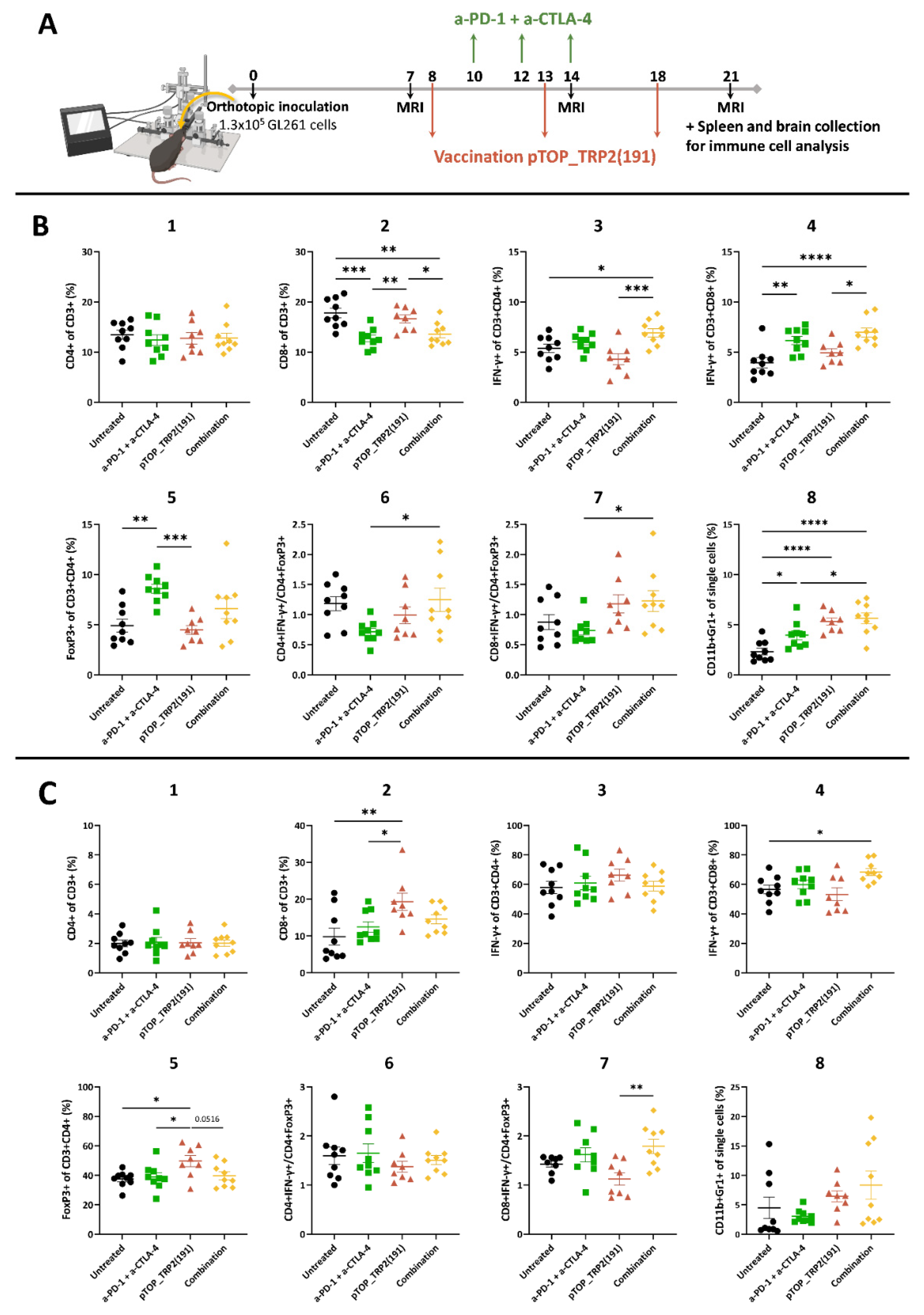
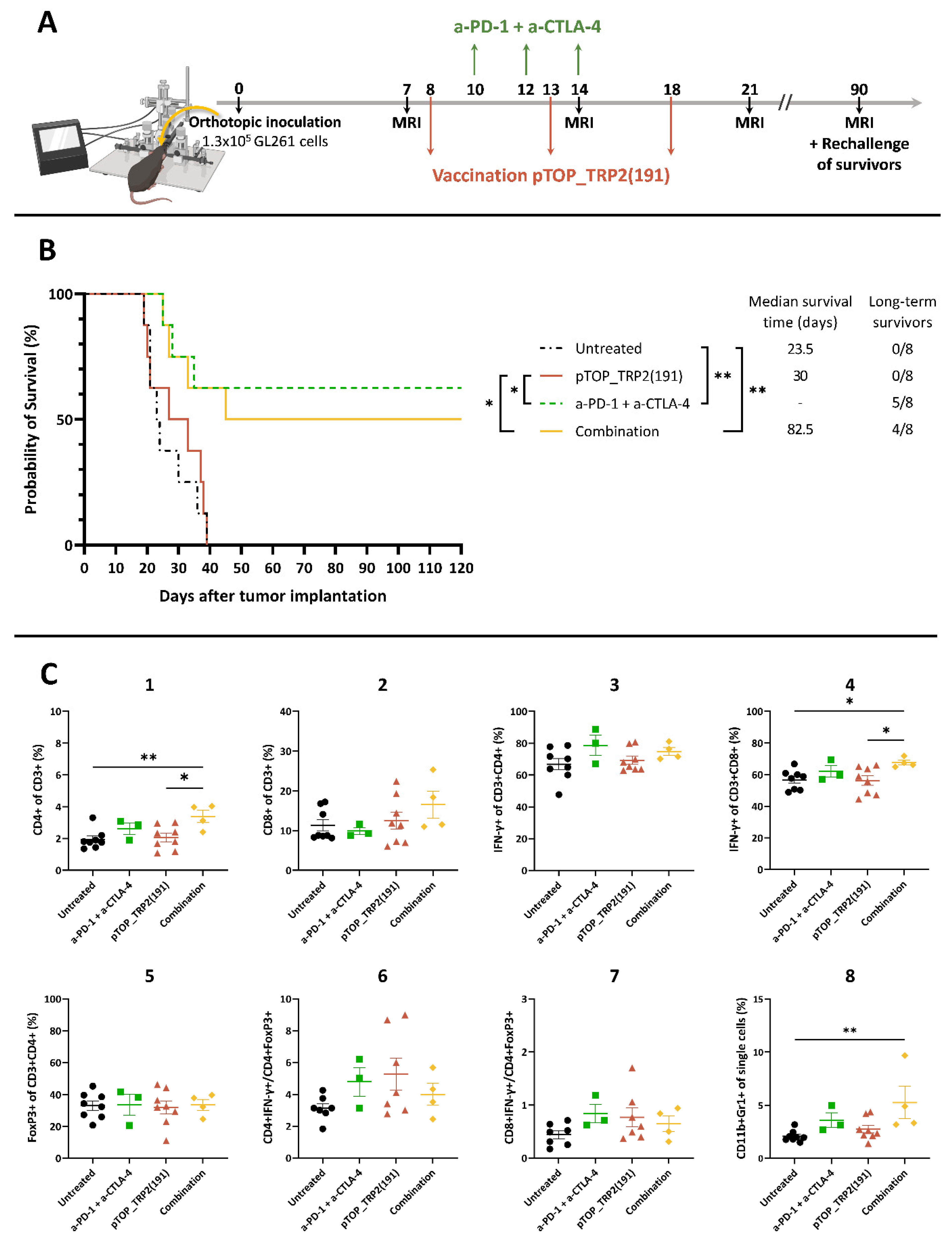
3.2. Combination Therapy Induces a Potent T Cell Response in the Spleen and Increases the Activation of CD8 T Cells in the Brain
3.3. Dual ICBs and Combination Therapy Increase Survival of GBM-Bearing Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.-C.; Pentheroudakis, G. High-Grade Glioma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2014, 25, iii93–iii101. [Google Scholar] [CrossRef] [PubMed]
- Lukas, R.V.; Wainwright, D.A.; Ladomersky, E.; Sachdev, S.; Sonabend, A.M.; Stupp, R. Newly Diagnosed Glioblastoma: A Review on Clinical Management. Oncol. Williston Park N 2019, 33, 91–100. [Google Scholar]
- Lesueur, P.; Lequesne, J.; Grellard, J.-M.; Dugué, A.; Coquan, E.; Brachet, P.-E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa Study of Concomitant Radiotherapy with Olaparib and Temozolomide in Unresectable or Partially Resectable Glioblastoma: OLA-TMZ-RTE-01 Trial Protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef] [PubMed]
- Yabroff, K.R.; Harlan, L.; Zeruto, C.; Abrams, J.; Mann, B. Patterns of Care and Survival for Patients with Glioblastoma Multiforme Diagnosed during 2006. Neuro-oncology 2012, 14, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, F.; Whiteside, G.; Perry, C. Ipilimumab: First Global Approval. Drugs 2011, 71, 1093–1104. [Google Scholar] [CrossRef]
- Hazarika, M.; Chuk, M.K.; Theoret, M.R.; Mushti, S.; He, K.; Weis, S.L.; Putman, A.H.; Helms, W.S.; Cao, X.; Li, H.; et al. U.S. FDA Approval Summary: Nivolumab for Treatment of Unresectable or Metastatic Melanoma Following Progression on Ipilimumab. Clin. Cancer Res. 2017, 23, 3484–3488. [Google Scholar] [CrossRef] [Green Version]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic Cancer Vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]
- Bausart, M.; Préat, V.; Malfanti, A. Immunotherapy for Glioblastoma: The Promise of Combination Strategies. J. Exp. Clin. Cancer Res. 2022, 41, 35. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs. Bevacizumab in Patients with Recurrent Glioblastoma: The Checkmate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with Temozolomide for Patients with Newly Diagnosed, EGFRvIII-Expressing Glioblastoma (ACT IV): A Randomised, Double-Blind, International Phase 3 Trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First Results on Survival from a Large Phase 3 Clinical Trial of an Autologous Dendritic Cell Vaccine in Newly Diagnosed Glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatoum, A.; Mohammed, R.; Zakieh, O. The Unique Invasiveness of Glioblastoma and Possible Drug Targets on Extracellular Matrix. Cancer Manag. Res. 2019, 11, 1843–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karmur, B.S.; Philteos, J.; Abbasian, A.; Zacharia, B.E.; Lipsman, N.; Levin, V.; Grossman, S.; Mansouri, A. Blood-Brain Barrier Disruption in Neuro-Oncology: Strategies, Failures, and Challenges to Overcome. Front. Oncol. 2020, 10, 563840. [Google Scholar] [CrossRef] [PubMed]
- Pombo Antunes, A.R.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the Glioblastoma Immune Microenvironment as Basis for the Development of New Immunotherapeutic Strategies. eLife 2020, 9, e52176. [Google Scholar] [CrossRef]
- Garg, A.D.; Vandenberk, L.; Van Woensel, M.; Belmans, J.; Schaaf, M.; Boon, L.; De Vleeschouwer, S.; Agostinis, P. Preclinical Efficacy of Immune-Checkpoint Monotherapy Does Not Recapitulate Corresponding Biomarkers-Based Clinical Predictions in Glioblastoma. OncoImmunology 2017, 6, e1295903. [Google Scholar] [CrossRef] [Green Version]
- Skaga, E.; Kulesskiy, E.; Fayzullin, A.; Sandberg, C.J.; Potdar, S.; Kyttälä, A.; Langmoen, I.A.; Laakso, A.; Gaál-Paavola, E.; Perola, M.; et al. Intertumoral Heterogeneity in Patient-Specific Drug Sensitivities in Treatment-Naïve Glioblastoma. BMC Cancer 2019, 19, 628. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.; Wang, Y.; Wang, Q.; Fang, S.; Shan, X.; Wang, J.; Jiang, T. Intratumor Heterogeneity, Microenvironment, and Mechanisms of Drug Resistance in Glioma Recurrence and Evolution. Front. Med. 2021, 15, 551–561. [Google Scholar] [CrossRef]
- Grossman, S.A.; Ye, X.; Piantadosi, S.; Desideri, S.; Nabors, L.B.; Rosenfeld, M.; Fisher, J.; NABTT CNS Consortium. Survival of Patients with Newly Diagnosed Glioblastoma Treated with Radiation and Temozolomide in Research Studies in the United States. Clin. Cancer Res. 2010, 16, 2443–2449. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.; Bastiancich, C.; Bausart, M.; Ligot, S.; Lambricht, L.; Vanvarenberg, K.; Ucakar, B.; Gallez, B.; Préat, V.; Vandermeulen, G. New Generation of DNA-Based Immunotherapy Induces a Potent Immune Response and Increases the Survival in Different Tumor Models. J. Immunother. Cancer 2021, 9, e001243. [Google Scholar] [CrossRef]
- Lambricht, L.; Lopes, A.; Kos, S.; Sersa, G.; Préat, V.; Vandermeulen, G. Clinical Potential of Electroporation for Gene Therapy and DNA Vaccine Delivery. Expert Opin. Drug Deliv. 2016, 13, 295–310. [Google Scholar] [CrossRef]
- Vandermeulen, G.; Vanvarenberg, K.; De Beuckelaer, A.; De Koker, S.; Lambricht, L.; Uyttenhove, C.; Reschner, A.; Vanderplasschen, A.; Grooten, J.; Préat, V. The Site of Administration Influences Both the Type and the Magnitude of the Immune Response Induced by DNA Vaccine Electroporation. Vaccine 2015, 33, 3179–3185. [Google Scholar] [CrossRef] [PubMed]
- Taggart, D.; Andreou, T.; Scott, K.J.; Williams, J.; Rippaus, N.; Brownlie, R.J.; Ilett, E.J.; Salmond, R.J.; Melcher, A.; Lorger, M. Anti–PD-1/Anti–CTLA-4 Efficacy in Melanoma Brain Metastases Depends on Extracranial Disease and Augmentation of CD8 T Cell Trafficking. Proc. Natl. Acad. Sci. USA 2018, 115, E1540–E1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayyar, N.; Singh, M.; Stocking, J.; Brehm, M.; Brastianos, P. 62. Presence of Extracranial Tumors Influences Response to Immune Checkpoint Inhibitors in a Pre-Clinical Model of Melanoma Brain Metastasis. Neuro-Oncol. Adv. 2020, 2, ii13. [Google Scholar] [CrossRef]
- Vigneron, N.; Van den Eynde, B.J. Proteasome Subtypes and the Processing of Tumor Antigens: Increasing Antigenic Diversity. Curr. Opin. Immunol. 2012, 24, 84–91. [Google Scholar] [CrossRef]
- Howles, G.P.; Bing, K.F.; Qi, Y.; Rosenzweig, S.J.; Nightingale, K.R.; Johnson, G.A. Contrast-Enhanced In Vivo Magnetic Resonance Microscopy of the Mouse Brain Enabled by Noninvasive Opening of the Blood-Brain Barrier with Ultrasound. Magn. Reson. Med. 2010, 64, 995–1004. [Google Scholar] [CrossRef] [Green Version]
- Moreno, H.; Hua, F.; Brown, T.; Small, S. Longitudinal Mapping of Mouse Cerebral Blood Volume with MRI. NMR Biomed. 2006, 19, 535–543. [Google Scholar] [CrossRef]
- McKelvey, K.J.; Hudson, A.L.; Prasanna Kumar, R.; Wilmott, J.S.; Attrill, G.H.; Long, G.V.; Scolyer, R.A.; Clarke, S.J.; Wheeler, H.R.; Diakos, C.I.; et al. Temporal and Spatial Modulation of the Tumor and Systemic Immune Response in the Murine Gl261 Glioma Model. PLoS ONE 2020, 15, e0226444. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.E.; Hu, J.C.; Athanasiou, K.A. Ammonium–Chloride–Potassium Lysing Buffer Treatment of Fully Differentiated Cells Increases Cell Purity and Resulting Neotissue Functional Properties. Tissue Eng. Part C Methods 2016, 22, 895–903. [Google Scholar] [CrossRef] [Green Version]
- Blaszczyk-Thurin, M.; Shen, C.T.; Ertl, H.C. A DNA Vaccine Expressing Tyrosinase-Related Protein-2 Induces T-Cell-Mediated Protection against Mouse Glioblastoma. Cancer Gene Ther. 2003, 10, 678–688. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Fitzgerald, E.B.; Southwood, S.; Sette, A.; Rosenberg, S.A.; Kawakami, Y. Identification of a Shared HLA-A*0201-Restricted T-Cell Epitope from the Melanoma Antigen Tyrosinase-Related Protein 2 (TRP2). Cancer Res. 1998, 58, 4895–4901. [Google Scholar]
- Ammayappan, A.; Peng, K.-W.; Russell, S.J. Characteristics of Oncolytic Vesicular Stomatitis Virus Displaying Tumor-Targeting Ligands. J. Virol. 2013, 87, 13543–13555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toes, R.E.M.; Nussbaum, A.K.; Degermann, S.; Schirle, M.; Emmerich, N.P.N.; Kraft, M.; Laplace, C.; Zwinderman, A.; Dick, T.P.; Müller, J.; et al. Discrete Cleavage Motifs of Constitutive and Immunoproteasomes Revealed by Quantitative Analysis of Cleavage Products. J. Exp. Med. 2001, 194, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritzell, S.; Sandén, E.; Eberstål, S.; Visse, E.; Darabi, A.; Siesjö, P. Intratumoral Temozolomide Synergizes with Immunotherapy in a T Cell-Dependent Fashion. Cancer Immunol. Immunother. 2013, 62, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Jahan, N.; Talat, H.; Alonso, A.; Saha, D.; Curry, W.T. Triple Combination Immunotherapy with GVAX, Anti-PD-1 Monoclonal Antibody, and Agonist Anti-OX40 Monoclonal Antibody Is Highly Effective against Murine Intracranial Glioma. OncoImmunology 2019, 8, e1577108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genoud, V.; Marinari, E.; Nikolaev, S.I.; Castle, J.C.; Bukur, V.; Dietrich, P.-Y.; Okada, H.; Walker, P.R. Responsiveness to Anti-PD-1 and Anti-CTLA-4 Immune Checkpoint Blockade in SB28 and GL261 Mouse Glioma Models. OncoImmunology 2018, 7, e1501137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267.e5. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable Therapeutic Efficacy Utilizing Combinatorial Blockade against IDO, CTLA-4, and PD-L1 in Mice with Brain Tumors. Clin. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef] [Green Version]
- Haddad, A.F.; Young, J.S.; Amara, D.; Berger, M.S.; Raleigh, D.R.; Aghi, M.K.; Butowski, N.A. Mouse Models of Glioblastoma for the Evaluation of Novel Therapeutic Strategies. Neuro-Oncol. Adv. 2021, 3, vdab100. [Google Scholar] [CrossRef]
- Xu, M.; Yao, Y.; Hua, W.; Wu, Z.; Zhong, P.; Mao, Y.; Zhou, L.; Luo, F.; Chu, Y. Mouse Glioma Immunotherapy Mediated by A2B5 + GL261 Cell Lysate-Pulsed Dendritic Cells. J. Neurooncol. 2014, 116, 497–504. [Google Scholar] [CrossRef]
- Chamberlain, M.C.; Kim, B.T. Nivolumab for Patients with Recurrent Glioblastoma Progressing on Bevacizumab: A Retrospective Case Series. J. Neurooncol. 2017, 133, 561–569. [Google Scholar] [CrossRef]
- Fidanza, M.; Gupta, P.; Sayana, A.; Shanker, V.; Pahlke, S.-M.; Vu, B.; Krantz, F.; Azameera, A.; Wong, N.; Anne, N.; et al. Enhancing Proteasomal Processing Improves Survival for a Peptide Vaccine Used to Treat Glioblastoma. Sci. Transl. Med. 2021, 13, eaax4100. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bausart, M.; Vanvarenberg, K.; Ucakar, B.; Lopes, A.; Vandermeulen, G.; Malfanti, A.; Préat, V. Combination of DNA Vaccine and Immune Checkpoint Blockades Improves the Immune Response in an Orthotopic Unresectable Glioblastoma Model. Pharmaceutics 2022, 14, 1025. https://doi.org/10.3390/pharmaceutics14051025
Bausart M, Vanvarenberg K, Ucakar B, Lopes A, Vandermeulen G, Malfanti A, Préat V. Combination of DNA Vaccine and Immune Checkpoint Blockades Improves the Immune Response in an Orthotopic Unresectable Glioblastoma Model. Pharmaceutics. 2022; 14(5):1025. https://doi.org/10.3390/pharmaceutics14051025
Chicago/Turabian StyleBausart, Mathilde, Kevin Vanvarenberg, Bernard Ucakar, Alessandra Lopes, Gaëlle Vandermeulen, Alessio Malfanti, and Véronique Préat. 2022. "Combination of DNA Vaccine and Immune Checkpoint Blockades Improves the Immune Response in an Orthotopic Unresectable Glioblastoma Model" Pharmaceutics 14, no. 5: 1025. https://doi.org/10.3390/pharmaceutics14051025
APA StyleBausart, M., Vanvarenberg, K., Ucakar, B., Lopes, A., Vandermeulen, G., Malfanti, A., & Préat, V. (2022). Combination of DNA Vaccine and Immune Checkpoint Blockades Improves the Immune Response in an Orthotopic Unresectable Glioblastoma Model. Pharmaceutics, 14(5), 1025. https://doi.org/10.3390/pharmaceutics14051025