
Micro- and Nano-Systems Developed for Tolcapone in Parkinson’s Disease

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of TC-Loaded Microparticles
2.3. Preparation of TC-Loaded PLGA Nanoparticles
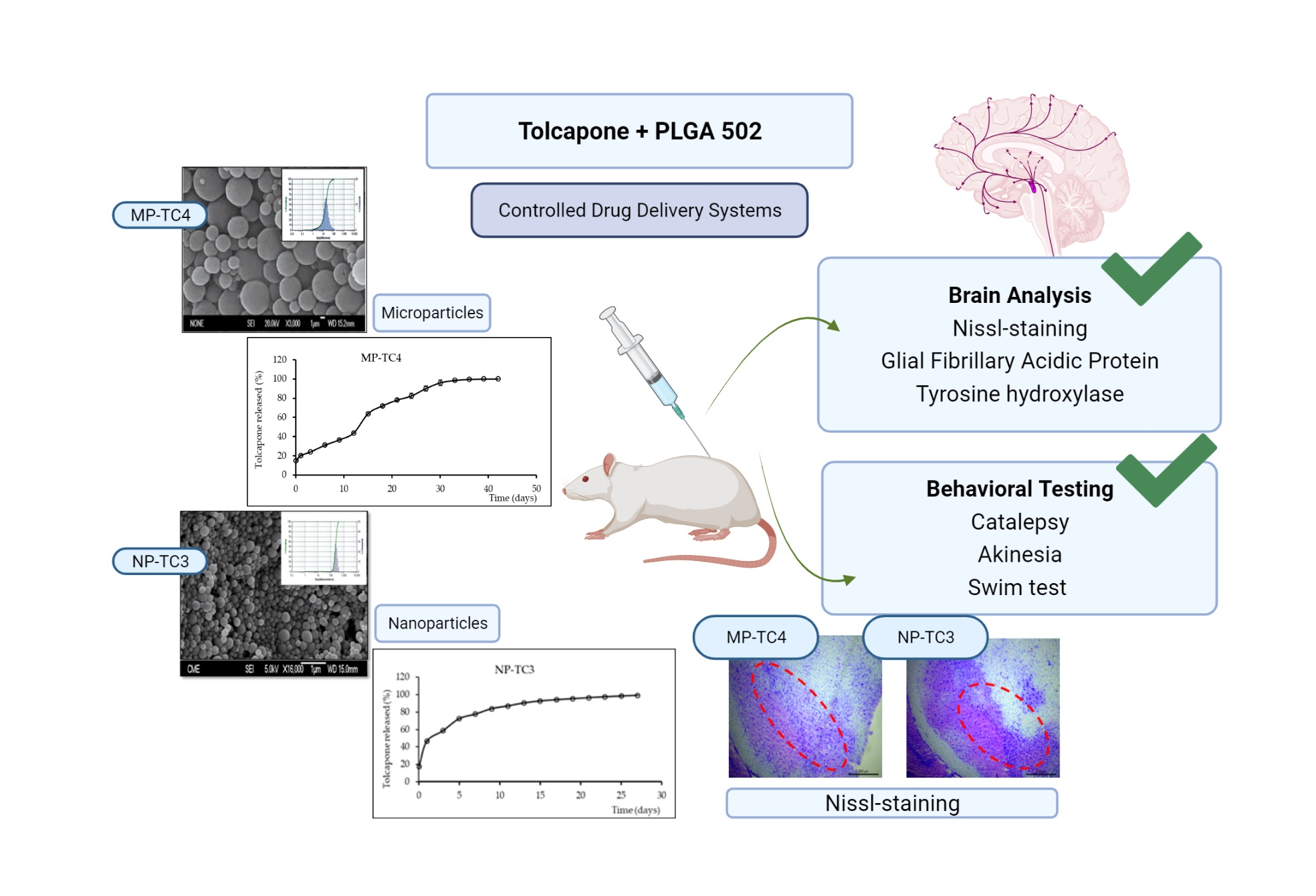
2.4. Characterization of Microparticles and Nanoparticles
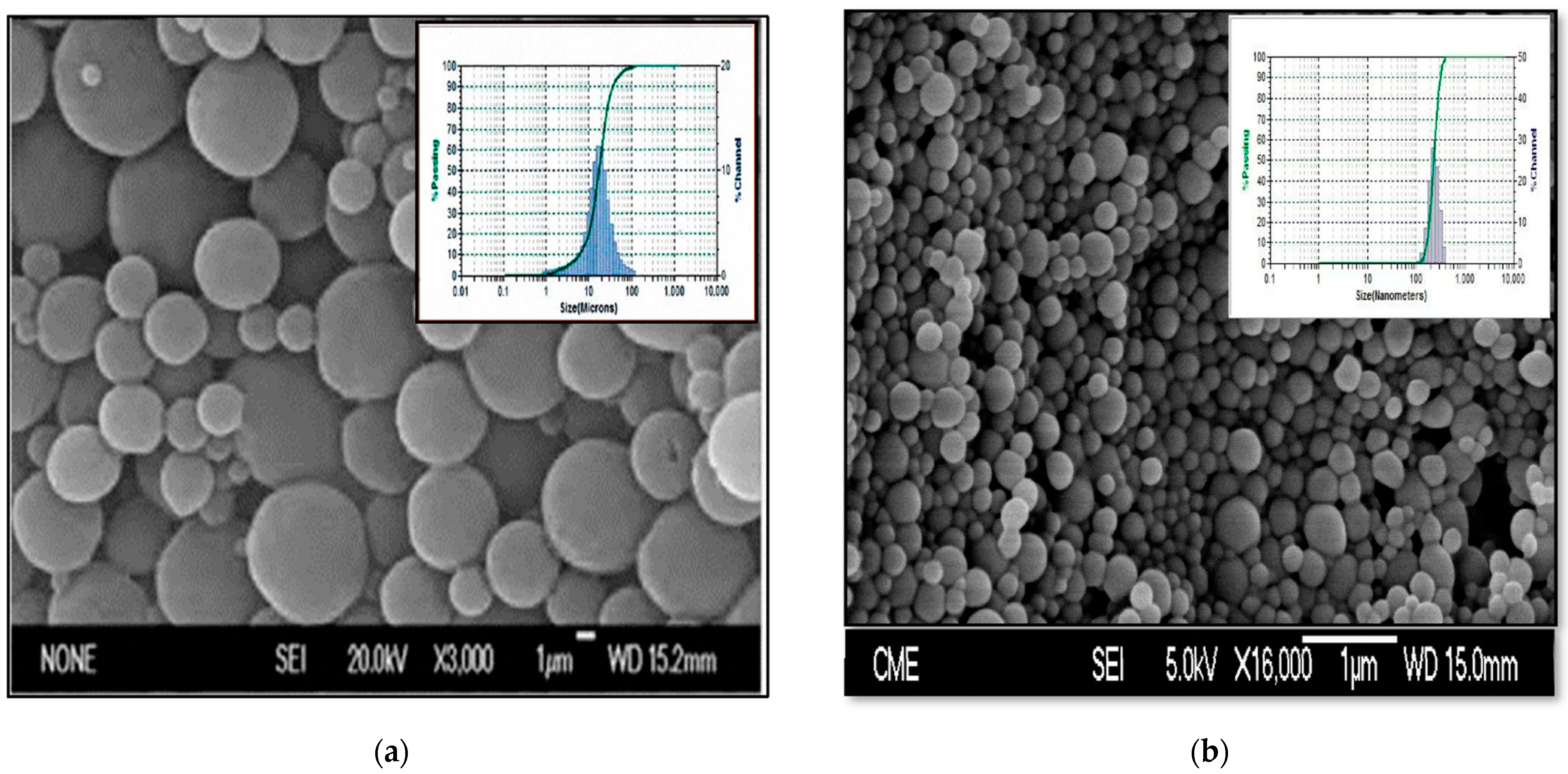
2.4.1. Morphological Characterization and Size Distribution
2.4.2. Calculation of Process Yield and Encapsulation Efficiency
amount of TC used in the preparation of MPs/NPs (mg)
2.4.3. Quantification of Tolcapone by HPLC
2.4.4. Zeta Potential
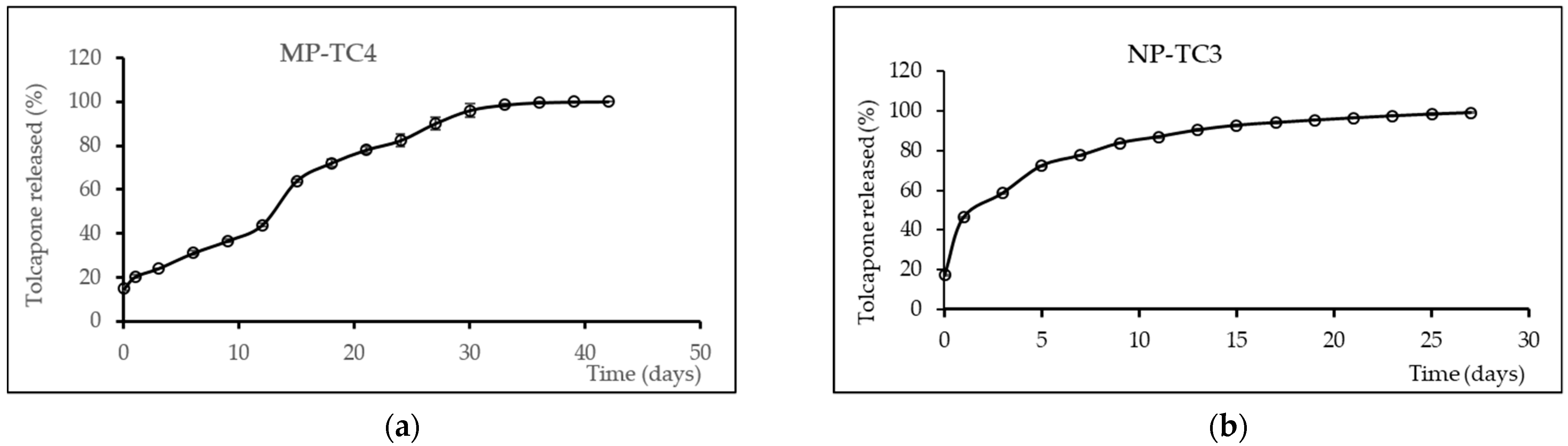
2.4.5. In Vitro Release Study
2.5. Animal Testing
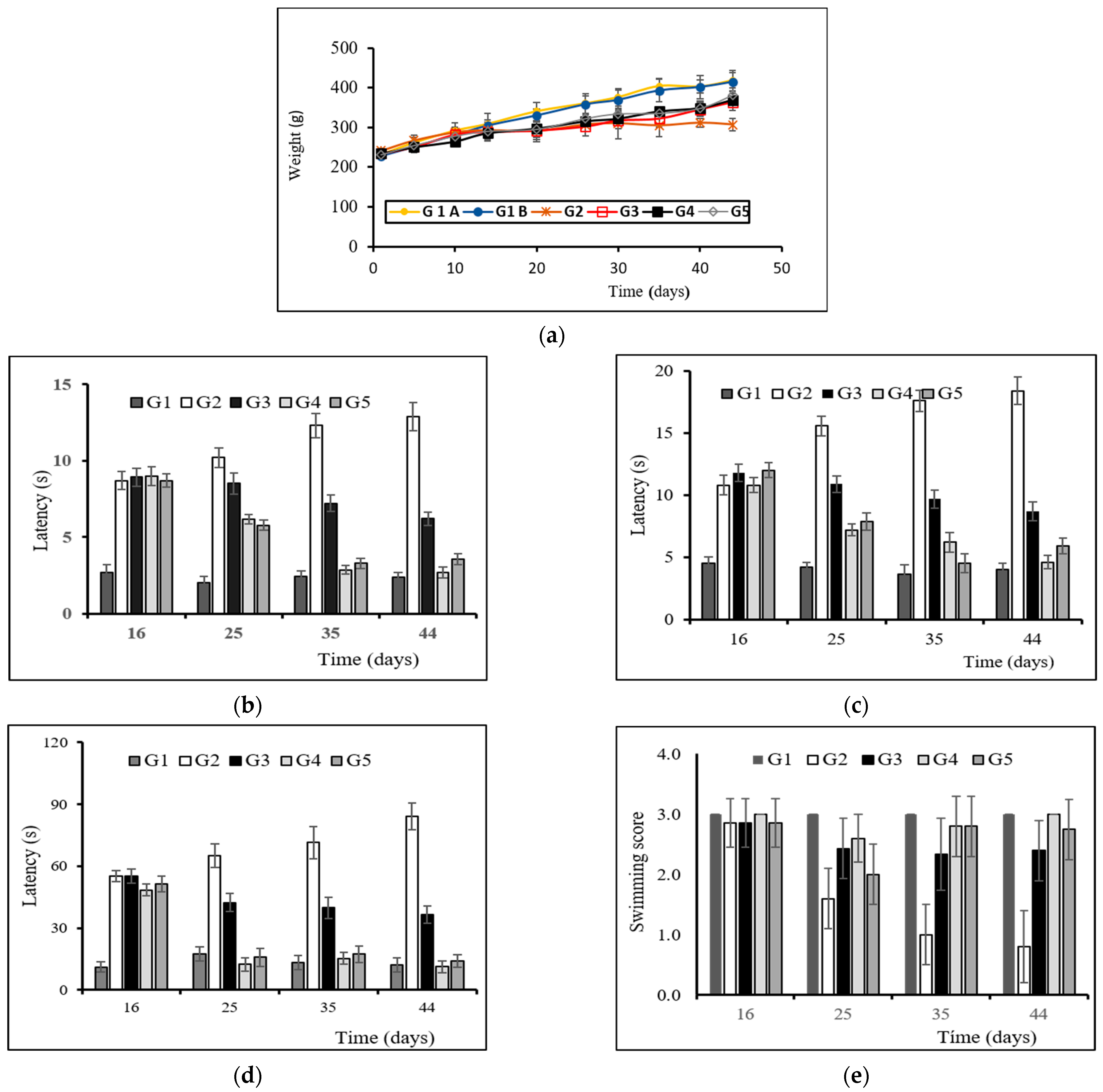
2.5.1. Treatments and Animal Groups
- -
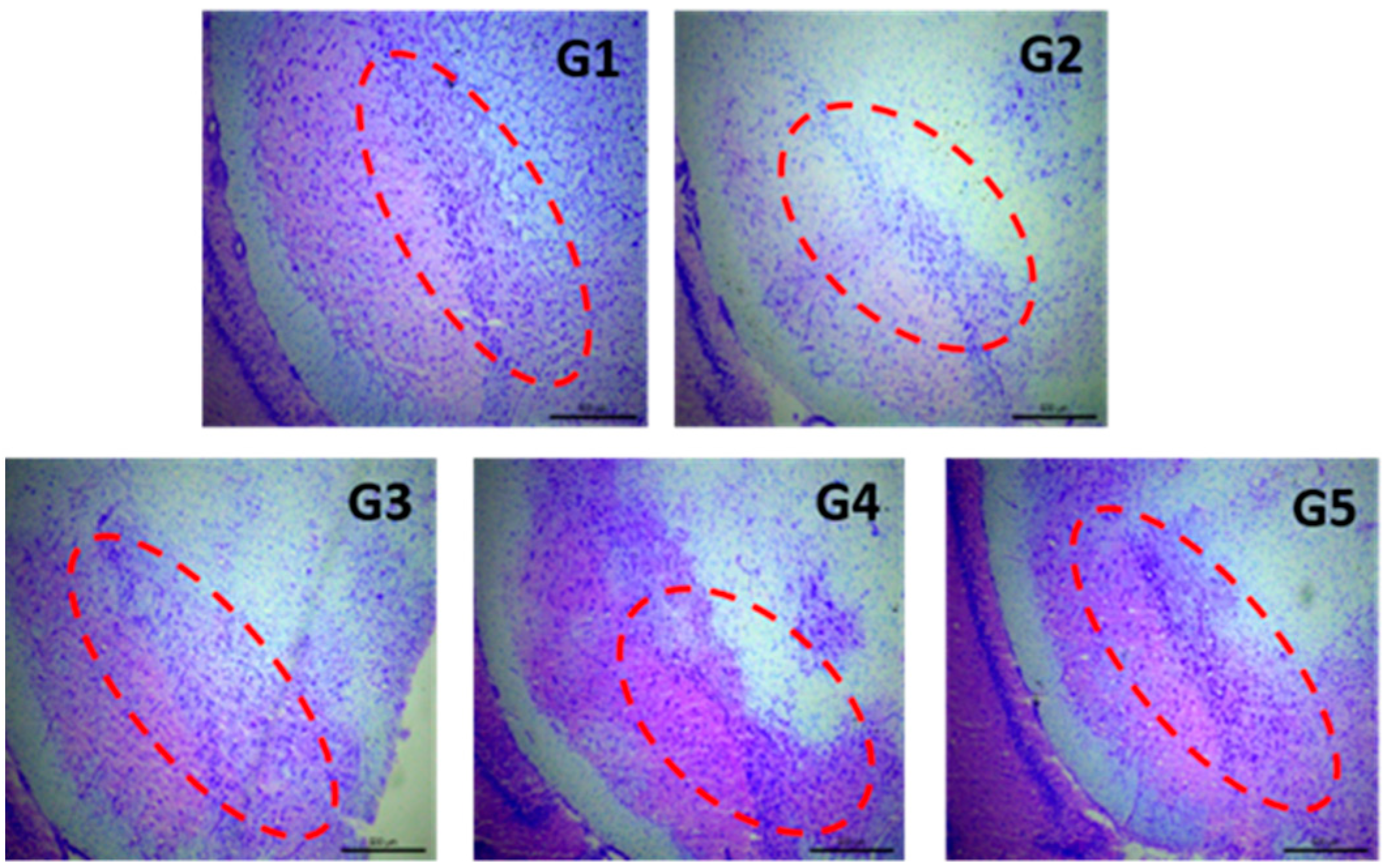
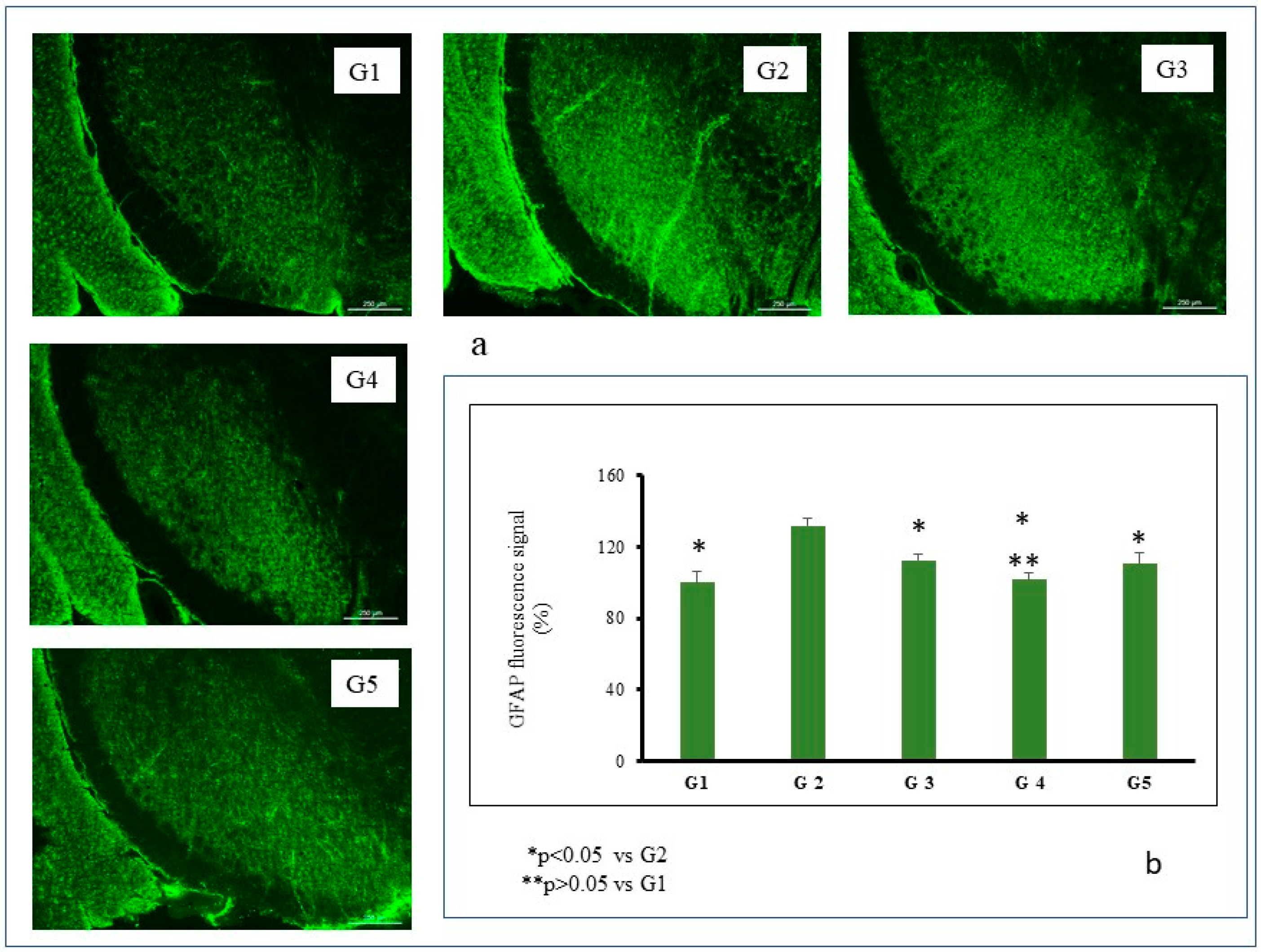
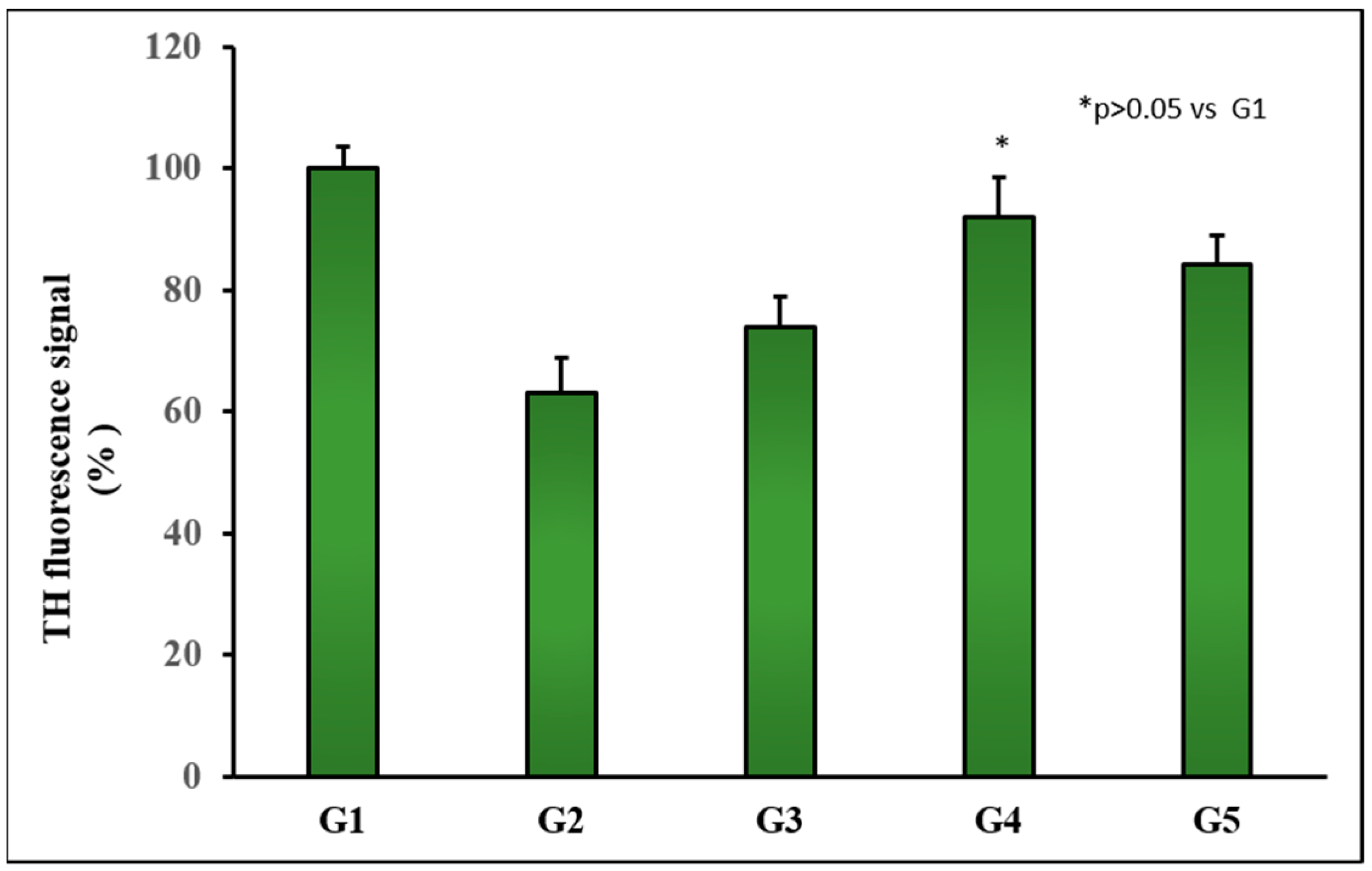
- Group 1 (G1): Control group. Animals (n = 8) receiving the vehicles; sunflower oil (subgroup G1A, n = 4) or saline (subgroup G1B, n = 4).
- -
- Group 2 (G2): Animals (n = 8) receiving only the neurotoxin RT (2 mg/kg/day) for 43 days.
- -
- Group 3 (G3): Animals (n = 8) receiving RT (2 mg/kg/day) for 43 days and the amount of MPs equivalent to 3 mg/kg/day of TC every 14 days from day 15.
- -
- Group 4 (G4): Animals (n = 8) receiving RT (2 mg/kg/day) for 43 days and the amount of NPs equivalent to 3 mg/kg/day of TC every 3 days from day 15.
- -
- Group 5 (G5): Animals (n = 8) receiving RT (2 mg/kg/day) for 43 days and TC in saline (3 mg/kg/day) from day 15.
2.5.2. Body Weight Evaluation
2.5.3. Behavioral Testing
2.5.4. Histochemical Assessments
2.6. Statistical Analysis
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; Chapter 1. [Google Scholar] [CrossRef]
- GBD 2015 Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.V. Treatment Options in the Modern Management of Parkinson Disease. Arch. Neurol. 2007, 64, 1083–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leegwater-Kim, J.; Waters, C. Role of tolcapone in the treatment of Parkinson’s disease. Expert Rev. Neurother. 2007, 7, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.-L.; Wang, Y.-X.; Chen, Y.; Zhang, C.-H.; Li, C.-H. The efficacy of homemade tolcapone in the treatment of patients with Parkinson’s disease. Exp. Ther. Med. 2018, 15, 127–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olanow, C.W. Tolcapone and Hepatotoxic Effects. Arch. Neurol. 2000, 57, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchowersky, O.; Bailey, P.; Pourcher, E.; Bulger, L.; Facciponte, G. Comparison of Two Dosages of Tolcapone Added to Levodopa in Nonfluctuating Patients with PD. Clin. Neuropharmacol. 2001, 24, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Tolcapone and Hepatotoxic Effects. Tasmar Advisory Panel. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/tasmar (accessed on 3 March 2022).
- Keating, G.M.; Lyseng-Williamson, K.A. Tolcapone: A review of its use in the management of Parkinson’s disease. CNS Drugs 2005, 19, 165–184. [Google Scholar] [CrossRef]
- Truong, D.D. Tolcapone: Review of its pharmacology and use as adjunctive therapy in patients with Parkinson’s disease. Clin. Interv. Aging 2009, 4, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Dingemanse, J.; Jorga, K.M.; Schmitt, M.; Gieschke, R.; Fotteler, B.; Zürcher, G.; Prada, M.; Van Brummelen, P. Integrated pharmacokinetics and pharmacodynamics of the novel catechol-O-methyltransferase inhibitor tolcapone during first administration to humans. Clin. Pharmacol. Ther. 1995, 57, 508–517. [Google Scholar] [CrossRef]
- Dankyi, B.O.; Amponsah, S.K.; Allotey-Babington, G.L.; Adams, I.; Goode, N.A.; Nettey, H. Chitosan-Coated Hydroxypropylmethyl Cellulose Microparticles of Levodopa (and Carbidopa): In Vitro and Rat Model Kinetic Characteristics. Curr. Ther. Res. 2020, 93, 100612. [Google Scholar] [CrossRef]
- Arisoy, S.; Sayiner, O.; Comoglu, T.; Onal, D.; Atalay, O.; Pehlivanoglu, B. In vitro and in vivo evaluation of levodopa-loaded nanoparticles for nose to brain delivery. Pharm. Dev. Technol. 2020, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Bali, N.R.; Salve, P.S. Impact of rasagiline nanoparticles on brain targeting efficiency via gellan gum based transdermal patch: A nanotheranostic perspective for Parkinsonism. Int. J. Biol. Macromol. 2020, 164, 1006–1024. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, X.; Mu, H.; Hua, H.; Duan, D.; Yan, X.; Wang, Y.; Meng, Q.; Lu, X.; Wang, A.; et al. Preparation and evaluation of injectable Rasagiline mesylate dual-controlled drug delivery system for the treatment of Parkinson’s disease. Drug Deliv. 2018, 25, 143–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Liu, W.; Xiong, S.; Li, D.; Fang, S.; Wu, Z.; Wang, Q.; Chen, X. Nanoparticles Mediating the Sustained Puerarin Release Facilitate Improved Brain Delivery to Treat Parkinson’s Disease. ACS Appl. Mater. Interfaces 2019, 11, 45276–45289. [Google Scholar] [CrossRef] [PubMed]
- Sa, F.; Zhang, L.Q.; Chong, C.M.; Guo, B.J.; Li, S.; Zhang, Z.J.; Zheng, Y.; Hoi, P.M.; Lee, S.M.Y. Discovery of novel anti-parkinsonian effect of schisantherin A in in vitro and in vivo. Neurosci. Lett. 2015, 593, 7–12. [Google Scholar] [CrossRef]
- Lu, C.-T.; Jin, R.-R.; Jiang, Y.-N.; Lin, Q.; Yu, W.-Z.; Mao, K.-L.; Tian, F.-R.; Zhao, Y.-P.; Zhao, Y.-Z. Gelatin nanoparticle-mediated intranasal delivery of substance P protects against 6-hydroxydopamine-induced apoptosis: An in vitro and in vivo study. Drug Des. Dev. Ther. 2015, 9, 1955–1962. [Google Scholar] [CrossRef] [Green Version]
- Negro, S.; Boeva, L.; Slowing, K.; Fernandez-Carballido, A.; García-García, L.; Barcia, E. Efficacy of Ropinirole-Loaded PLGA Microspheres for the Reversion of Rotenone-Induced Parkinsonism. Curr. Pharm. Des. 2017, 23, 3423–3431. [Google Scholar] [CrossRef]
- Regnier-Delplace, C.; du Boullay, O.T.; Siepmann, F.; Martin-Vaca, B.; Degrave, N.; Demonchaux, P.; Jentzer, O.; Bourissou, D.; Siepmann, J. PLGA microparticles with zero-order release of the labile anti-Parkinson drug apomorphine. Int. J. Pharm. 2013, 443, 68–79. [Google Scholar] [CrossRef]
- Linazasoro, G. Potential applications of nanotechnologies to Parkinson’s disease therapy. Park. Relat. Disord. 2008, 14, 383–392. [Google Scholar] [CrossRef]
- Spuch, C.; Saida, O.; Navarro, C. Advances in the Treatment of Neurodegenerative Disorders Employing Nanoparticles. Recent Pat. Drug Deliv. Formul. 2012, 6, 2–18. [Google Scholar] [CrossRef]
- Marcianes, P.; Negro, S.; Garcia-Garcia, L.; Montejo, C.; Barcia, E.; Fernandez-Carballido, A. Surface-modified gatifloxacin nanoparticles with potential for treating central nervous system tuberculosis. Int. J. Nanomed. 2017, 12, 1959–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcia, E.; Boeva, L.; García-García, L.; Slowing, K.; Fernández-Carballido, A.; Casanova, Y.; Negro, S. Nanotechnology-based drug delivery of ropinirole for Parkinson’s disease. Drug Deliv. 2017, 24, 1112–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katiyar, S.S.; Muntimadugu, E.; Rafeeqi, T.A.; Domb, A.J.; Khan, W. Co-delivery of rapamycin- and piperine-loaded polymeric nanoparticles for breast cancer treatment. Drug Deliv. 2016, 23, 2608–2616. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Barcia, E.; Fernández-Carballido, A.; Garcia, L.; Slowing, K.; Negro, S. Controlled release of rasagiline mesylate promotes neuroprotection in a rotenone-induced advanced model of Parkinson’s disease. Int. J. Pharm. 2012, 438, 266–278. [Google Scholar] [CrossRef]
- Haobam, R.; Sindhu, K.M.; Chandra, G.; Mohanakumar, K.P. Swim-test as a function of motor impairment in MPTP model of Parkinson’s disease: A comparative study in two mouse strains. Behav. Brain Res. 2005, 163, 159–167. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) As biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Freiberg, S.; Zhu, X.X. Polymer microspheres for controlled drug release. Int. J. Pharm. 2004, 282, 1–18. [Google Scholar] [CrossRef]
- Vert, M.; Schwach, G.; Engel, R.; Coudane, J. Something new in the field of PLA/GA bioresorbable polymers? J. Control. Release 1998, 53, 85–92. [Google Scholar] [CrossRef]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Hillaireau, H.; Couvreur, P. Nanocarriers’ entry into the cell: Relevance to drug delivery. Cell. Mol. Life Sci. 2009, 66, 2873–2896. [Google Scholar] [CrossRef]
- Wohlfart, S.; Gelperina, S.; Kreuter, J. Transport of drugs across the blood–brain barrier by nanoparticles. J. Control. Release 2012, 161, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, T.G.; Cohen, S.; Langer, R. Poly(L-lactic acid)/Pluronic blends: Characterization of phase separation behavior, degradation, and morphology and use as protein-releasing matrixes. Macromolecules 1992, 25, 116–122. [Google Scholar] [CrossRef]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Control. Release 2001, 73, 121–136. [Google Scholar] [CrossRef]
- Klose, D.; Siepmann, F.; Elkharraz, K.; Krenzlin, S.; Siepmann, J. How porosity and size affect the drug release mechanisms from PLGA-based microparticles. Int. J. Pharm. 2006, 314, 198–206. [Google Scholar] [CrossRef]
- Singh, R.; Lillard, J.W., Jr. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Lockman, P.; Koziara, J.M.; Mumper, R.J.; Allen, D.D. Nanoparticle Surface Charges Alter Blood–Brain Barrier Integrity and Permeability. J. Drug Target. 2004, 12, 635–641. [Google Scholar] [CrossRef]
- Fonseca, C.; Simões, S.; Gaspar, R. Paclitaxel-loaded PLGA nanoparticles: Preparation, physicochemical characterization and in vitro anti-tumoral activity. J. Control. Release 2002, 83, 273–286. [Google Scholar] [CrossRef] [Green Version]
- Kreuter, J. Mechanism of polymeric nanoparticle-based drug transport across the blood-brain barrier (BBB). J. Microencapsul. 2013, 30, 49–54. [Google Scholar] [CrossRef]
- Betarbet, R.; Canet-Aviles, R.M.; Sherer, T.; Mastroberardino, P.G.; McLendon, C.; Kim, J.-H.; Lund, S.; Na, H.-M.; Taylor, G.; Bence, N.F.; et al. Intersecting pathways to neurodegeneration in Parkinson’s disease: Effects of the pesticide rotenone on DJ-1, α-synuclein, and the ubiquitin–proteasome system. Neurobiol. Dis. 2006, 22, 404–420. [Google Scholar] [CrossRef]
- Sindhu, K.M.; Saravanan, K.S.; Mohanakumar, K.P. Behavioral differences in a rotenone-induced hemiparkinsonian rat model developed following intranigral or median forebrain bundle infusion. Brain Res. 2005, 1051, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilati, N.; Barker, M.; Panteleimonitis, S.; Donga, R.; Hamann, M. A Rapid Method Combining Golgi and Nissl Staining to Study Neuronal Morphology and Cytoarchitecture. J. Histochem. Cytochem. 2008, 56, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.-N.; Zhang, J.-S.; Xiang, J.; Yu, Z.-H.; Zhang, W.; Cai, M.; Li, X.-T.; Wu, T.; Li, W.-W.; Cai, D.-F. Subcutaneous rotenone rat model of Parkinson’s disease: Dose exploration study. Brain Res. 2017, 1655, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Tapias, V.; Greenamyre, J.T.; Watkins, S.C. Automated imaging system for fast quantitation of neurons, cell morphology and neurite morphometry in vivo and in vitro. Neurobiol. Dis. 2013, 54, 158–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Guo, H.; Guo, X.; Ge, D.; Shi, Y.; Lu, X.; Lu, J.; Chen, J.; Ding, F.; Zhang, Q. Involvement of Akt/mTOR in the Neurotoxicity of Rotenone-Induced Parkinson’s Disease Models. Int. J. Environ. Res. Public Health 2019, 16, 3811. [Google Scholar] [CrossRef] [Green Version]
- Swarnkar, S.; Singh, S.; Goswami, P.; Mathur, R.; Patro, I.K.; Nath, C. Astrocyte Activation: A Key Step in Rotenone Induced Cytotoxicity and DNA Damage. Neurochem. Res. 2012, 37, 2178–2189. [Google Scholar] [CrossRef]
- Vila, M.; Jackson-Lewis, V.; Guégan, C.; Wu, D.C.; Teismann, P.; Choi, D.-K.; Tieu, K.; Przedborski, S. The role of glial cells in Parkinson’s disease. Curr. Opin. Neurol. 2001, 14, 483–489. [Google Scholar] [CrossRef]
- Gao, Z.; Zhu, Q.; Zhang, Y.; Zhao, Y.; Cai, L.; Shields, C.B.; Cai, J. Reciprocal Modulation Between Microglia and Astrocyte in Reactive Gliosis Following the CNS Injury. Mol. Neurobiol. 2013, 48, 690–701. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.M.; Walker, P.D.; Benjamins, J.A.; Geddes, T.J.; Kuhn, D.M. Methamphetamine Neurotoxicity in Dopamine Nerve Endings of the Striatum Is Associated with Microglial Activation. J. Pharmacol. Exp. Ther. 2004, 311, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.-S.; Kim, H.-W.; Xia, Z. JNK inhibition of VMAT2 contributes to rotenone-induced oxidative stress and dopamine neuron death. Toxicology 2015, 328, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, C.; Bandez, M.J.; Navarro, A. Pesticides and impairment of mitochondrial function in relation with the parkinsonian syndrome. Front. Biosci. 2007, 12, 1079–1093. [Google Scholar] [CrossRef] [Green Version]
- Bassani, T.B.; Gradowski, R.W.; Zaminelli, T.; Barbiero, J.K.; Santiago, R.M.; Boschen, S.L.; da Cunha, C.; Lima, M.M.S.; Andreatini, R.; Vital, M.A. Neuroprotective and antidepressant-like effects of melatonin in a rotenone-induced Parkinson’s disease model in rats. Brain Res. 2014, 1593, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Sherer, T.; Betarbet, R.; Kim, J.-H.; Greenamyre, J. Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci. Lett. 2003, 341, 87–90. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Amount of TC (mg) | Process Yield (%) ± SEM | EE (%) ± SEM | Mean Particle Size ± SEM |
|---|---|---|---|---|
| MP-TC1 | 70 | 62.33 ± 15.10 | 73.92 ± 10.17 | 27.73 ± 2.59 μm |
| MP-TC2 | 80 | 78.30 ± 11.96 | 79.63 ± 3.55 | 23.05 ± 3.53 μm |
| MP-TC3 | 100 | 84.58 ± 5.75 | 83.17 ± 4.82 | 16.35 ± 0.10 μm |
| MP-TC4 | 120 | 87.69 ± 7.04 | 85.13 ± 2.08 | 17.00 ± 0.04 μm |
| NP-TC1 | 6 | 55.78 ± 16.87 | 56.16 ± 4.65 | 197.39 ± 43.19 nm |
| NP-TC2 | 8 | 75.28 ± 5.73 | 55.99 ± 21.41 | 202.08 ± 48.70 nm |
| NP-TC3 | 10 | 70.35 ± 14.19 | 53.69 ± 9.09 | 182.59 ± 23.94 nm |
| NP-TC4 | 12 | 73.29 ± 4.50 | 46.16 ± 5.99 | 210.20 ± 7.92 nm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casanova, Y.; Negro, S.; Slowing, K.; García-García, L.; Fernández-Carballido, A.; Rahmani, M.; Barcia, E. Micro- and Nano-Systems Developed for Tolcapone in Parkinson’s Disease. Pharmaceutics 2022, 14, 1080. https://doi.org/10.3390/pharmaceutics14051080
Casanova Y, Negro S, Slowing K, García-García L, Fernández-Carballido A, Rahmani M, Barcia E. Micro- and Nano-Systems Developed for Tolcapone in Parkinson’s Disease. Pharmaceutics. 2022; 14(5):1080. https://doi.org/10.3390/pharmaceutics14051080
Chicago/Turabian StyleCasanova, Yaquelyn, Sofía Negro, Karla Slowing, Luis García-García, Ana Fernández-Carballido, Mahdieh Rahmani, and Emilia Barcia. 2022. "Micro- and Nano-Systems Developed for Tolcapone in Parkinson’s Disease" Pharmaceutics 14, no. 5: 1080. https://doi.org/10.3390/pharmaceutics14051080
APA StyleCasanova, Y., Negro, S., Slowing, K., García-García, L., Fernández-Carballido, A., Rahmani, M., & Barcia, E. (2022). Micro- and Nano-Systems Developed for Tolcapone in Parkinson’s Disease. Pharmaceutics, 14(5), 1080. https://doi.org/10.3390/pharmaceutics14051080