
Transporter Regulation in Critical Protective Barriers: Focus on Brain and Placenta

 , , , and
, , , and
Abstract
:1. Introduction
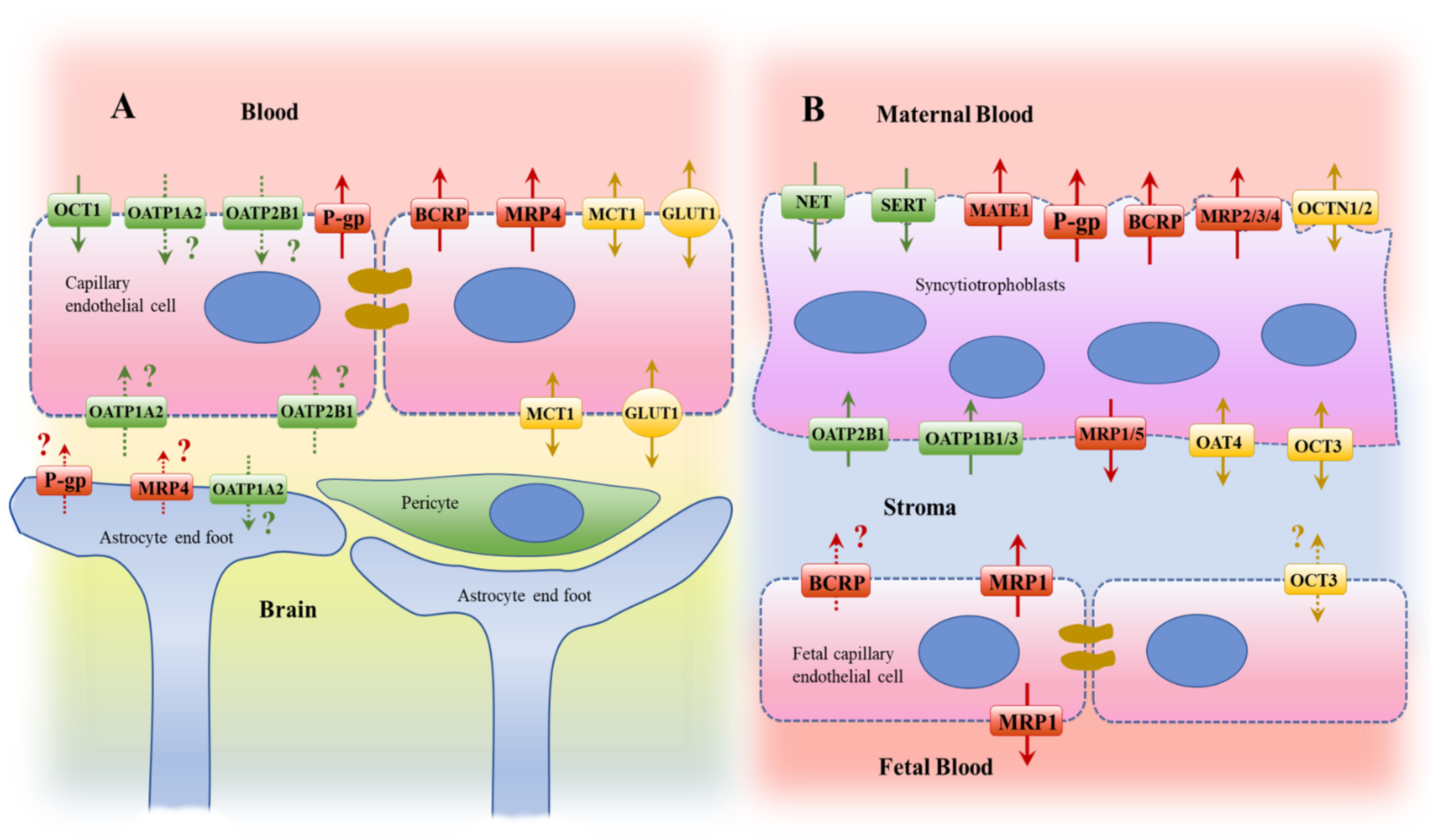
2. Histological and Transport Characteristics
2.1. Brain
2.2. Placenta
3. Transporter Regulation
3.1. Brain
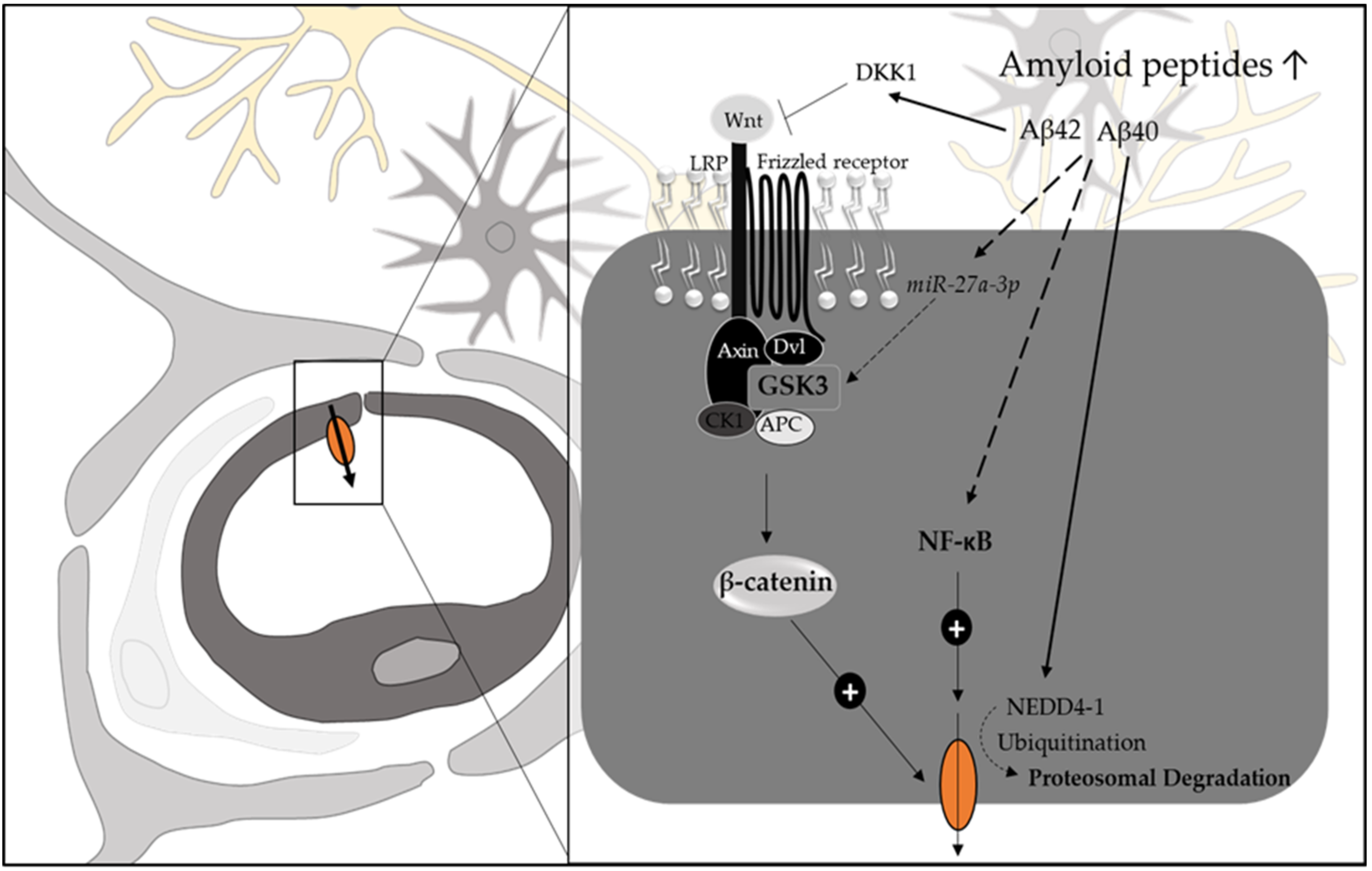
3.1.1. Alzheimer’s Disease
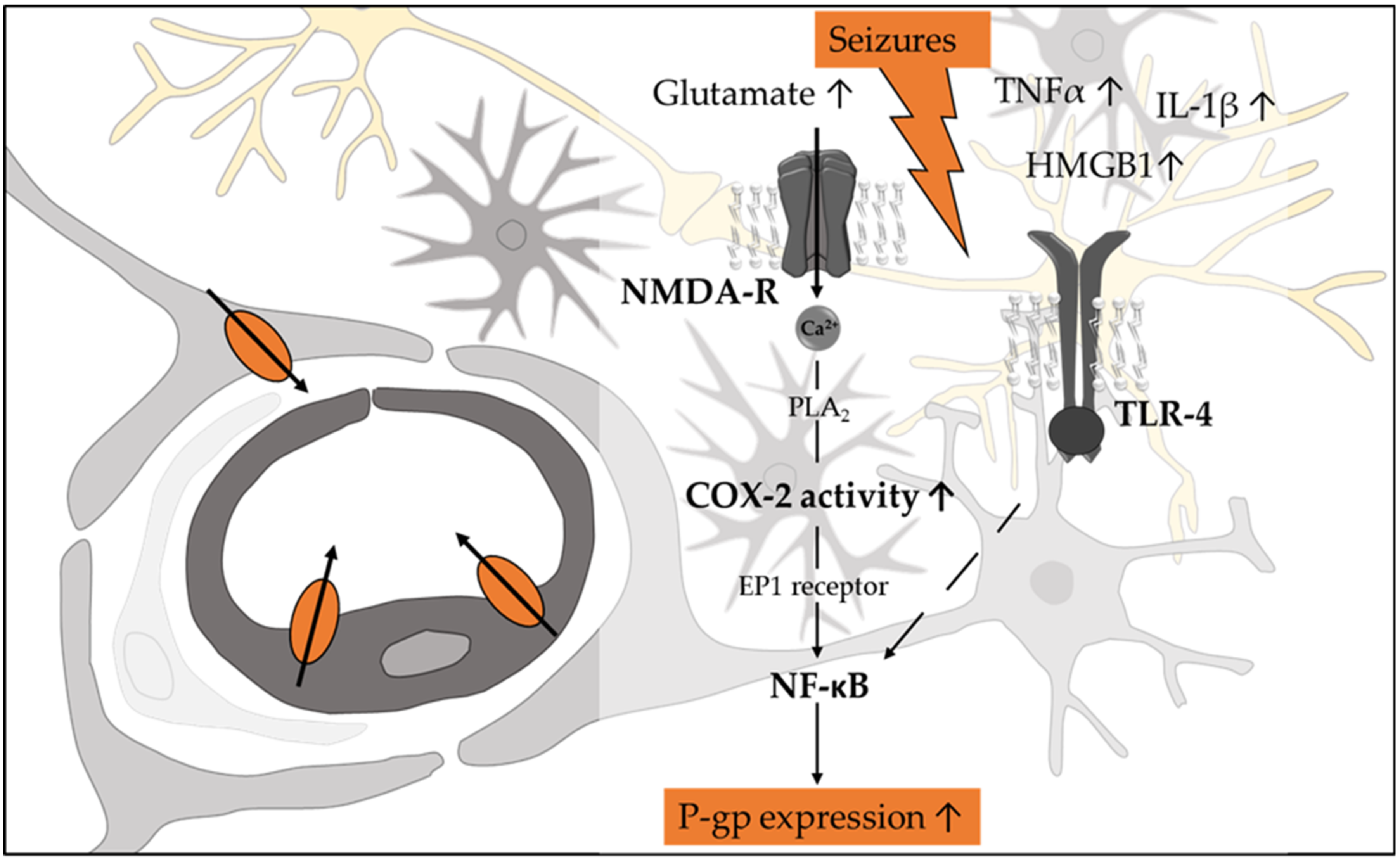
3.1.2. Epilepsy
3.1.3. Parkinson’s Disease
3.1.4. Human Immunodeficiency Virus Infection
3.2. Placenta

{kind=link}
{kind=link}
{kind=link}
| Condition/Factor | Effect | Reference | ||
|---|---|---|---|---|
| mRNA | Protein | Other | ||
| Based on Clinical Data | ||||
| Gestational Age | ↔ | ↓ ~2-fold from T1 to T2 (variable) | - | [27] |
| Gestational Age | - | ↓ 55% | - | [26] |
| Chorio (Preterm) | ↑ 1.72-fold | Ns (↑ trend) | mRNA + corr with Chorio degree | [171] |
| Chorio (Term) | ↓ 34% | ↔ | [185] | |
| Chorio (Preterm) | ↑ | Ns (↑ trend) | mRNA + corr to IL-8 | [170] |
| Preeclampsia | ↓ 40–60% | ↓ 45% | - | [174] |
| Preeclampsia—HELLP | ↓ | ↓ | No corr with maternal or umbilical cord TBA levels | [189] |
| IUFGR | ↓ | - | mRNA + corr with ABCB1 mRNA | [172] |
| Diabetes | ↔ | ↔ | ↑ + corr with ↑ HbA1c plasma) | [176] |
| HIV | ↓ 38% | ↔ | - | [177] |
| Hepatitis | ↑1.8 fold (trend, p = 011) | ↑ 2.3-fold | - | [178] |
| Estrogen | - | - | + corr with maternal estradiol at mid-gest not term | [185] |
| Nuclear factors | - | - | mRNA + corr with AHR and NRF2 mRNA | [190] |
| Prenatal Dexamethasone | - | ↓ 0.5-fold | GR activation | [191] |
| Based on Placental Explants | ||||
| LPS | ↓ (T1) | ↓ (T1) | - | [179] |
| Poly I:C | ↔ (Term) | ↔ (Term) | - | [179] |
| Hypoxia | ↔/↓ | ↔ (Term) | ↓ AHR, NRF2, RXRα, ↑PPAR-Υ mRNA | [180] |
| Th17 cytokines: IL-17/22/23 combo | ↓ | ↔ | - | [192] |
| Estradiol | ↓ (Term) | - | - | [193] |
| cART treatment | ↑ 1.6- to 1.9-fold | - | - | [177] |
| Based on Primary Trophoblasts | ||||
| TNF-a | ↓ >40% | ↓ 50% | ↓ function | [181] |
| IL-1b | ↓ >40% | ↓ 50% | - | [181] |
| Epidermal growth factor | ↑ >120% | ↑ ~90% | ↑ function | [181] |
| Insulin-like growth factor | ↑ ~70% | ↑ ~50% | [181] | |
| Estradiol | ↑ >50% | ↑ >50% | [181] | |
| Prostaglandin E2 | ↑ 1.5-fold | ↑ 1.6-fold | Via EP receptors in PHT | [183] |
| Condition/Factor | Effect | Reference | ||
|---|---|---|---|---|
| mRNA | Protein | Other | ||
| SLCO2B1/OATP2B1 | ||||
| Based on Clinical Data | ||||
| Gestational Age | - | ↓ 32% between T1 and T2 | [26] | |
| Chorio (Preterm) | ↓ 57% | ↓ 68% (trend, p = 0.05) | mRNA (-) corr with IL-1β and IL-6 | [169] |
| Chorio (Term) | - | ↓ 49% | - | [169] |
| HIV | ↓ 85–99% | - | - | [177] |
| Preeclampsia | ↑ 1.8-fold | ↑ | [174] | |
| Prenatal Dexamethasone | - | ↓ 0.75 to 0.5-fold | GR activation | [191] |
| Based on Placental Explants | ||||
| Th17 cytokines: | ||||
| IL-17, IL-22, IL-23 | ↓ | - | - | [192] |
| IL-23, and combo | - | ↓ | - | [192] |
| Hypoxia | ↓ 75% | - | - | [180] |
| SLC22A11/OAT4 | ||||
| Based on Clinical Data | ||||
| Gestational Age | - | ↑ 1.6-fold from T2 to Term | - | [26] |
| Preeclampsia | ↓ 50–70% | - | - | [174] |
| HIV | ↓ 85–99% | - | - | [177] |
| Based on Placental Explants | ||||
| Th17 cytokines: IL-23 | ↓ | - | - | [192] |
| Hypoxia | ↓ 25% | - | - | [180] |
| cART treatment | ↓ | - | - | [177] |
| SLC22A3/OCT3 | ||||
| Based on Clinical Data | ||||
| Gestational Age | - | ↑ 2-fold from T1 to Term | - | [26] |
| Preeclampsia | ↓ 50–70% | ↑ | - | [174] |
| HIV | ↓ 85–99% | ↓ 50% | - | [177] |
| Based on Placental Explants | ||||
| Th17 cytokines: IL-17/22/23 or combo | ↔ | ↔ | - | [192] |
3.2.1. Preeclampsia
3.2.2. Metabolic Disorders
3.2.3. Human Immunodeficiency Virus (HIV) and Hepatitis Infections
4. Summary Remarks and Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brouwer, K.L.; Evers, R.; Hayden, E.; Hu, S.; Li, C.Y.; zu Schwabedissen, H.E.M.; Neuhoff, S.; Oswald, S.; Piquette-Miller, M.; Saran, C.; et al. Regulation of Drug Transport Proteins-From Mechanisms to Clinical Impact: A White Paper on Behalf of the International Transporter Consortium. Clin. Pharmacol. Ther. 2022. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneman, R. The blood-brain barrier in health and disease. Ann. Neurol. 2012, 72, 648–672. [Google Scholar] [CrossRef] [PubMed]
- Shawahna, R.; Uchida, Y.; Declèves, X.; Ohtsuki, S.; Yousif, S.; Dauchy, S.; Jacob, A.; Chassoux, F.; Daumas-Duport, C.; Couraud, P.-O.; et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol. Pharm. 2011, 8, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef]
- Al-Majdoub, Z.M.; Al Feteisi, H.; Achour, B.; Warwood, S.; Neuhoff, S.; Rostami-Hodjegan, A.; Barber, J. Proteomic Quantification of Human Blood-Brain Barrier SLC and ABC Transporters in Healthy Individuals and Dementia Patients. Mol. Pharm. 2019, 16, 1220–1233. [Google Scholar] [CrossRef] [Green Version]
- Billington, S.; Salphati, L.; Hop, C.E.C.A.; Chu, X.; Evers, R.; Burdette, D.; Rowbottom, C.; Lai, Y.; Xiao, G.; Humphreys, W.G.; et al. Interindividual and Regional Variability in Drug Transporter Abundance at the Human Blood-Brain Barrier Measured by Quantitative Targeted Proteomics. Clin. Pharmacol. Ther. 2019, 106, 228–237. [Google Scholar] [CrossRef]
- Uchida, Y. Quantitative Proteomics-Based Blood-Brain Barrier Study. Biol. Pharm. Bull. 2021, 44, 465–473. [Google Scholar] [CrossRef]
- Harwood, M.D.; Russell, M.R.; Neuhoff, S.; Warhurst, G.; Rostami-Hodjegan, A. Lost in centrifugation: Accounting for transporter protein losses in quantitative targeted absolute proteomics. Drug Metab. Dispos. 2014, 42, 1766–1772. [Google Scholar] [CrossRef] [Green Version]
- Cooray, H.C.; Blackmore, C.G.; Maskell, L.; Barrand, M.A. Localisation of breast cancer resistance protein in microvessel endothelium of human brain. Neuroreport 2002, 13, 2059–2063. [Google Scholar] [CrossRef] [PubMed]
- Bendayan, R.; Ronaldson, P.T.; Gingras, D.; Bendayan, M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J. Histochem. Cytochem. 2006, 54, 1159–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritzen, F.; de Lanerolle, N.C.; Lee, T.-S.W.; Spencer, D.D.; Kim, J.H.; Bergersen, L.H.; Eid, T. Monocarboxylate transporter 1 is deficient on microvessels in the human epileptogenic hippocampus. Neurobiol. Dis. 2011, 41, 577–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornford, E.M.; Hyman, S.; Swartz, B.E. The human brain GLUT1 glucose transporter: Ultrastructural localization to the blood-brain barrier endothelia. J. Cereb. Blood Flow Metab. 1994, 14, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, A.M.; Meyer zu Schwabedissen, H.E.; Grube, M. Expression and Function of Organic Anion Transporting Polypeptides in the Human Brain: Physiological and Pharmacological Implications. Pharmaceutics 2021, 13, 834. [Google Scholar] [CrossRef]
- Schäfer, A.M.; Meyer zu Schwabedissen, H.E.; Bien-Möller, S.; Hubeny, A.; Vogelgesang, S.; Oswald, S.; Grube, M. OATP1A2 and OATP2B1 Are Interacting with Dopamine-Receptor Agonists and Antagonists. Mol. Pharm. 2020, 17, 1987–1995. [Google Scholar] [CrossRef]
- Storelli, F.; Billington, S.; Kumar, A.R.; Unadkat, J.D. Abundance of P-Glycoprotein and Other Drug Transporters at the Human Blood-Brain Barrier in Alzheimer’s Disease: A Quantitative Targeted Proteomic Study. Clin. Pharmacol. Ther. 2021, 109, 667–675. [Google Scholar] [CrossRef]
- Lee, W.; Glaeser, H.; Smith, L.H.; Roberts, R.L.; Moeckel, G.W.; Gervasini, G.; Leake, B.F.; Kim, R.B. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): Implications for altered drug disposition and central nervous system drug entry. J. Biol. Chem. 2005, 280, 9610–9617. [Google Scholar] [CrossRef] [Green Version]
- Nies, A.; Jedlitschky, G.; König, J.; Herold-Mende, C.; Steiner, H.; Schmitt, H.-P.; Keppler, D. Expression and immunolocalization of the multidrug resistance proteins, MRP1-MRP6 (ABCC1-ABCC6), in human brain. Neuroscience 2004, 129, 349–360. [Google Scholar] [CrossRef]
- Leggas, M.; Adachi, M.; Scheffer, G.L.; Sun, D.; Wielinga, P.; Du, G.; Mercer, K.E.; Zhuang, Y.; Panetta, J.C.; Johnston, B.; et al. Mrp4 confers resistance to topotecan and protects the brain from chemotherapy. Mol. Cell. Biol. 2004, 24, 7612–7621. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L.; Black, D.; Raman, C.; Woodford, K.; Zhou, M.; Haggerty, J.; Yan, A.; Cwirla, S.; Grindstaff, K. Subcellular localization of transporters along the rat blood-brain barrier and blood-cerebral-spinal fluid barrier by in vivo biotinylation. Neuroscience 2008, 155, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, D.; Kusuhara, H.; Taniguchi, H.; Ishikawa, S.; Nozaki, Y.; Aburatani, H.; Sugiyama, Y. Functional characterization of rat brain-specific organic anion transporter (Oatp14) at the blood-brain barrier: High affinity transporter for thyroxine. J. Biol. Chem. 2003, 278, 43489–43495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-J.; Tai, Y.; Huang, M.-T.; Tsai, Y.-F.; Hsu, H.-J.; Tzen, K.-Y.; Liou, H.-H. Cellular localization of the organic cation transporters, OCT1 and OCT2, in brain microvessel endothelial cells and its implication for MPTP transport across the blood-brain barrier and MPTP-induced dopaminergic toxicity in rodents. J. Neurochem. 2010, 114, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Dockery, P.; Bermingham, J.; Jenkins, D. Structure-function relations in the human placenta. Biochem. Soc. Trans. 2000, 28, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.; Markert, U.R. Overview of Drug Transporters in Human Placenta. Int. J. Mol. Sci. 2021, 22, 13149. [Google Scholar] [CrossRef]
- Anoshchenko, O.; Prasad, B.; Neradugomma, N.K.; Wang, J.; Mao, Q.; Unadkat, J.D. Gestational Age-Dependent Abundance of Human Placental Transporters as Determined by Quantitative Targeted Proteomics. Drug Metab. Dispos. 2020, 48, 735–741. [Google Scholar] [CrossRef]
- Mathias, A.A.; Hitti, J.; Unadkat, J.D. P-glycoprotein and breast cancer resistance protein expression in human placentae of various gestational ages. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R963–R969. [Google Scholar] [CrossRef]
- Zu Schwabedissen, H.E.M.; Grube, M.; Dreisbach, A.; Jedlitschky, G.; Meissner, K.; Linnemann, K.; Fusch, C.; Ritter, C.A.; Völker, U.; Kroemer, H.K.; et al. Epidermal growth factor-mediated activation of the map kinase cascade results in altered expression and function of ABCG2 (BCRP). Drug Metab. Dispos. 2006, 34, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, D.; Sibley, C.; Fairbairn, L.; Greenwood, S. MDR1 P-gp expression and activity in intact human placental tissue; upregulation by retroviral transduction. Placenta 2006, 27, 707–714. [Google Scholar] [CrossRef]
- Yeboah, D.; Sun, M.; Kingdom, J.; Baczyk, D.; Lye, S.; Matthews, S.; Gibb, W. Expression of breast cancer resistance protein (BCRP/ABCG2) in human placenta throughout gestation and at term before and after labor. Can. J. Physiol. Pharmacol. 2006, 84, 1251–1258. [Google Scholar] [CrossRef]
- St-Pierre, M.V.; Serrano, M.A.; Macias, R.I.R.; Dubs, U.; Hoechli, M.; Lauper, U.; Meier, P.J.; Marin, J.J.G. Expression of members of the multidrug resistance protein family in human term placenta. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1495–R1503. [Google Scholar] [CrossRef] [PubMed]
- Pascolo, L.; Fernetti, C.; Pirulli, D.; Crovella, S.; Amoroso, A.; Tiribelli, C. Effects of maturation on RNA transcription and protein expression of four MRP genes in human placenta and in BeWo cells. Biochem. Biophys. Res. Commun. 2003, 303, 259–265. [Google Scholar] [CrossRef]
- Granitzer, S.; Ellinger, I.; Khan, R.; Gelles, K.; Widhalm, R.; Hengstschläger, M.; Zeisler, H.; Desoye, G.; Tupova, L.; Ceckova, M.; et al. In vitro function and in situ localization of Multidrug Resistance-associated Protein (MRP)1 (ABCC1) suggest a protective role against methyl mercury-induced oxidative stress in the human placenta. Arch. Toxicol. 2020, 94, 3799–3817. [Google Scholar] [CrossRef] [PubMed]
- Ugele, B.; St-Pierre, M.V.; Pihusch, M.; Bahn, A.; Hantschmann, P. Characterization and identification of steroid sulfate transporters of human placenta. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E390–E398. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Reuther, S.; zu Schwabedissen, H.M.; Köck, K.; Draber, K.; Ritter, C.A.; Fusch, C.; Jedlitschky, G.; Kroemer, H.K. Organic anion transporting polypeptide 2B1 and breast cancer resistance protein interact in the transepithelial transport of steroid sulfates in human placenta. Drug Metab. Dispos. 2007, 35, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, E.; Kammala, A.K.; Benson, M.; Richardson, L.S.; Han, A.; Menon, R. Organic Anion Transporting Polypeptide 2B1 in Human Fetal Membranes: A Novel Gatekeeper for Drug Transport During Pregnancy? Front. Pharmacol. 2021, 12, 771818. [Google Scholar] [CrossRef]
- Lee, N.; Hebert, M.F.; Wagner, D.J.; Easterling, T.R.; Liang, C.J.; Rice, K.; Wang, J. Organic Cation Transporter 3 Facilitates Fetal Exposure to Metformin during Pregnancy. Mol. Pharmacol. 2018, 94, 1125–1131. [Google Scholar] [CrossRef]
- Grube, M.; zu Schwabedissen, H.M.; Draber, K.; Präger, D.; Möritz, K.-U.; Linnemann, K.; Fusch, C.; Jedlitschky, G.; Kroemer, H.K. Expression, localization, and function of the carnitine transporter octn2 (slc22a5) in human placenta. Drug Metab. Dispos. 2005, 33, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Anglada, M.; Perez-Torras, S. Emerging Roles of Nucleoside Transporters. Front. Pharmacol. 2018, 9, 606. [Google Scholar] [CrossRef]
- Govindarajan, R.; Bakken, A.H.; Hudkins, K.L.; Lai, Y.; Casado, F.J.; Anglada, M.P.; Tse, C.-M.; Hayashi, J.; Unadkat, J.D. In situ hybridization and immunolocalization of concentrative and equilibrative nucleoside transporters in the human intestine, liver, kidneys, and placenta. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1809–R1822. [Google Scholar] [CrossRef] [Green Version]
- Villegas, S.; Roda, A.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.F.; Elbert, D.L.; Kasten, T.P.; Patterson, B.W.; Sigurdson, W.C.; Connors, R.E.; Ovod, V.; Munsell, L.Y.; Mawuenyega, K.G.; Miller-Thomas, M.M.; et al. Amyloid-beta efflux from the central nervous system into the plasma. Ann. Neurol. 2014, 76, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.M.; Sharom, F.J.; Reiner, P.B. beta-Amyloid efflux mediated by p-glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [Google Scholar] [CrossRef]
- Kuhnke, D.; Jedlitschky, G.; Grube, M.; Krohn, M.; Jucker, M.; Mosyagin, I.; Cascorbi, I.; Walker, L.C.; Kroemer, H.K.; Warzok, R.W.; et al. MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer’s amyloid-beta peptides--implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 2007, 17, 347–353. [Google Scholar] [CrossRef]
- Chai, A.B.; Hartz, A.M.; Gao, X.; Yang, A.; Callaghan, R.; Gelissen, I.C. New Evidence for P-gp-Mediated Export of Amyloid-beta PEPTIDES in Molecular, Blood-Brain Barrier and Neuronal Models. Int. J. Mol. Sci. 2020, 22, 246. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [Green Version]
- Chai, A.B.; Leung, G.K.; Callaghan, R.; Gelissen, I.C. P-glycoprotein: A role in the export of amyloid-beta in Alzheimer’s disease? FEBS J. 2020, 287, 612–625. [Google Scholar] [CrossRef] [Green Version]
- Vogelgesang, S.; Cascorbi, I.; Schroeder, E.; Pahnke, J.; Kroemer, H.K.; Siegmund, W.; Kunert-Keil, C.; Walker, L.C.; Warzok, R.W. Deposition of Alzheimer’s beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics 2002, 12, 535–541. [Google Scholar] [CrossRef]
- Vogelgesang, S.; Warzok, R.W.; Cascorbi, I.; Kunert-Keil, C.; Schroeder, E.; Kroemer, H.K.; Siegmund, W.; Walker, L.C.; Pahnke, J. The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2004, 1, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Wijesuriya, H.C.; Bullock, J.Y.; Faull, R.; Hladky, S.B.; Barrand, M.A. ABC efflux transporters in brain vasculature of Alzheimer’s subjects. Brain Res. 2010, 1358, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Jeynes, B.; Provias, J. P-Glycoprotein Altered Expression in Alzheimer’s Disease: Regional Anatomic Variability. J. Neurodegener. Dis. 2013, 2013, 257953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrano, A.; Snkhchyan, H.; Kooij, G.; van der Pol, S.; van Horssen, J.; Veerhuis, R.; Hoozemans, J.; Rozemuller, A.; de Vries, H.E. ATP-binding cassette transporters P-glycoprotein and breast cancer related protein are reduced in capillary cerebral amyloid angiopathy. Neurobiol. Aging 2014, 35, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Provias, J.; Jeynes, B. The role of the blood-brain barrier in the pathogenesis of senile plaques in Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2014, 2014, 191863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.; Miller, M.C.; Monahan, R.; Osgood, D.P.; Stopa, E.G.; Silverberg, G.D. P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer’s disease: Preliminary observations. Neurobiol. Aging 2015, 36, 2475–2482. [Google Scholar] [CrossRef]
- Kannan, P.; Schain, M.; Kretzschmar, W.W.; Weidner, L.; Mitsios, N.; Gulyás, B.; Blom, H.; Gottesman, M.M.; Innis, R.B.; Hall, M.D.; et al. An automated method measures variability in P-glycoprotein and ABCG2 densities across brain regions and brain matter. J. Cereb. Blood Flow Metab. 2017, 37, 2062–2075. [Google Scholar] [CrossRef] [Green Version]
- Van Assema, D.M.; Lubberink, M.; Bauer, M.; van der Flier, W.M.; Schuit, R.C.; Windhorst, A.D.; Comans, E.F.I.; Hoetjes, N.J.; Tolboom, N.; Langer, O.; et al. Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain 2012, 135, 181–189. [Google Scholar] [CrossRef]
- Van Assema, D.M.; Lubberink, M.; Rizzu, P.; van Swieten, J.C.; Schuit, R.C.; Eriksson, J.; Scheltens, P.; Koepp, M.; Lammertsma, A.A.; van Berckel, B.N.M. Blood-brain barrier P-glycoprotein function in healthy subjects and Alzheimer’s disease patients: Effect of polymorphisms in the ABCB1 gene. EJNMMI Res. 2012, 2, 57. [Google Scholar] [CrossRef]
- Deo, A.K.; Borson, S.; Link, J.M.; Domino, K.; Eary, J.F.; Ke, B.; Richards, T.L.; Mankoff, D.A.; Minoshima, S.; O’Sullivan, F.; et al. Activity of P-Glycoprotein, a beta-Amyloid Transporter at the Blood-Brain Barrier, Is Compromised in Patients with Mild Alzheimer Disease. J. Nucl. Med. 2014, 55, 1106–1111. [Google Scholar] [CrossRef] [Green Version]
- Frankfort, S.V.; Doodeman, V.D.; Bakker, R.; Tulner, L.R.; Van Campen, J.P.C.M.; Smits, P.H.M.; Beijnen, J.H. ABCB1 genotypes and haplotypes in patients with dementia and age-matched non-demented control patients. Mol. Neurodegener. 2006, 1, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohen, R.; Shofer, J.B.; Korvatska, O.; Petrie, E.C.; Wang, L.Y.; Schellenberg, G.D.; Peskind, E.R.; Wilkinson, C.W. ABCB1 genotype and CSF beta-amyloid in Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2011, 24, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Park, R.; Kook, S.Y.; Park, J.C.; Mook-Jung, I. Abeta1-42 reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-kappaB signaling. Cell Death Dis. 2014, 5, e1299. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, I.; Al-Shami, K.; Yang, E.; Kaddoumi, A. Blood-Brain Barrier Disruption Increases Amyloid-Related Pathology in TgSwDI Mice. Int. J. Mol. Sci. 2021, 22, 1231. [Google Scholar] [CrossRef] [PubMed]
- Akkaya, B.G.; Zolnerciks, J.K.; Ritchie, T.K.; Bauer, B.; Hartz, A.M.S.; Sullivan, J.A.; Linton, K.J. The multidrug resistance pump ABCB1 is a substrate for the ubiquitin ligase NEDD4-1. Mol. Membr. Biol. 2015, 32, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Chai, A.B.; Callaghan, R.; Gelissen, I.C. The Ubiquitin E3 Ligase Nedd4 Regulates the Expression and Amyloid-beta Peptide Export Activity of P-Glycoprotein. Int. J. Mol. Sci. 2022, 23, 1019. [Google Scholar] [CrossRef]
- Hartz, A.M.; Zhong, Y.; Wolf, A.; LeVine, H.; Miller, D.S.; Bauer, B. Abeta40 Reduces P-Glycoprotein at the Blood-Brain Barrier through the Ubiquitin-Proteasome Pathway. J. Neurosci. 2016, 36, 1930–1941. [Google Scholar] [CrossRef]
- Hartz, A.M.; Zhong, Y.; Shen, A.N.; Abner, E.L.; Bauer, B. Preventing P-gp Ubiquitination Lowers Abeta Brain Levels in an Alzheimer’s Disease Mouse Model. Front. Aging Neurosci. 2018, 10, 186. [Google Scholar] [CrossRef]
- Kania, K.D.; Wijesuriya, H.C.; Hladky, S.B.; Barrand, M.A. Beta amyloid effects on expression of multidrug efflux transporters in brain endothelial cells. Brain Res. 2011, 1418, 1–11. [Google Scholar] [CrossRef]
- Brenn, A.; Grube, M.; Peters, M.; Fischer, A.; Jedlitschky, G.; Kroemer, H.K.; Warzok, R.W.; Vogelgesang, S. Beta-Amyloid Downregulates MDR1-P-Glycoprotein (Abcb1) Expression at the Blood-Brain Barrier in Mice. Int. J. Alzheimer’s Dis. 2011, 2011, 690121. [Google Scholar] [CrossRef] [Green Version]
- Menet, R.; Bourassa, P.; Calon, F.; ElAli, A. Dickkopf-related protein-1 inhibition attenuates amyloid-beta pathology associated to Alzheimer’s disease. Neurochem. Int. 2020, 141, 104881. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wan, W.; Xia, S.; Kalionis, B.; Li, Y. Dysfunctional Wnt/beta-catenin signaling contributes to blood-brain barrier breakdown in Alzheimer’s disease. Neurochem. Int. 2014, 75, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Laksitorini, M.D.; Yathindranath, V.; Xiong, W.; Hombach-Klonisch, S.; Miller, D.W. Modulation of Wnt/beta-catenin signaling promotes blood-brain barrier phenotype in cultured brain endothelial cells. Sci. Rep. 2019, 9, 19718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, D.-D.; Zhang, H.; Zhang, P.; Zheng, Y.-S.; Zhang, X.-J.; Han, B.-W.; Luo, X.-Q.; Xu, L.; Zhou, H.; Qu, L.-H.; et al. Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukaemia. J. Cell. Mol. Med. 2011, 15, 2164–2175. [Google Scholar] [CrossRef] [Green Version]
- Bruckmueller, H.; Martin, P.; Kähler, M.; Haenisch, S.; Ostrowski, M.; Drozdzik, M.; Siegmund, W.; Cascorbi, I.; Oswald, S. Clinically Relevant Multidrug Transporters Are Regulated by microRNAs along the Human Intestine. Mol. Pharm. 2017, 14, 2245–2253. [Google Scholar] [CrossRef] [Green Version]
- Hammad, S.; Mabondzo, A.; Hamoudi, R.; Harati, R. Regulation of P-glycoprotein by miR-27a-3p at the Brain Endothelial Barrier. J. Pharm. Sci. 2021, 111, 1470–1479. [Google Scholar] [CrossRef]
- Lim, J.C.; Kania, K.D.; Wijesuriya, H.; Chawla, S.; Sethi, J.K.; Pulaski, L.; Romero, I.A.; Couraud, P.O.; Weksler, B.B.; Hladky, S.B.; et al. Activation of beta-catenin signalling by GSK-3 inhibition increases p-glycoprotein expression in brain endothelial cells. J. Neurochem. 2008, 106, 1855–1865. [Google Scholar]
- Palomer, E.; Buechler, J.; Salinas, P.C. Wnt Signaling Deregulation in the Aging and Alzheimer’s Brain. Front. Cell. Neurosci. 2019, 13, 227. [Google Scholar] [CrossRef]
- Zetterberg, H.; Blennow, K. Moving fluid biomarkers for Alzheimer’s disease from research tools to routine clinical diagnostics. Mol. Neurodegener. 2021, 16, 10. [Google Scholar] [CrossRef]
- Margraf, N.G.; Jensen-Kondering, U.; Weiler, C.; Leypoldt, F.; Maetzler, W.; Philippen, S.; Bartsch, T.; Flüh, C.; Röcken, C.; Möller, B.; et al. Cerebrospinal Fluid Biomarkers in Cerebral Amyloid Angiopathy: New Data and Quantitative Meta-Analysis. Front. Aging Neurosci. 2022, 14, 783996. [Google Scholar] [CrossRef]
- Rao, V.V.; Dahlheimer, J.L.; Bardgett, M.E.; Snyder, A.Z.; Finch, R.A.; Sartorelli, A.C.; Piwnica-Worms, D. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fluid drug-permeability barrier. Proc. Natl. Acad. Sci. USA 1999, 96, 3900–3905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, K.; Cline, C.; Vogel, P.; Onciu, M.; Fatima, S.; Sorrentino, B.P.; Thirumaran, R.K.; Ekins, S.; Urade, Y.; Fujimori, K.; et al. Drug transporters on arachnoid barrier cells contribute to the blood-cerebrospinal fluid barrier. Drug Metab. Dispos. 2013, 41, 923–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, Y.; Zhang, Z.; Tachikawa, M.; Terasaki, T. Quantitative targeted absolute proteomics of rat blood-cerebrospinal fluid barrier transporters: Comparison with a human specimen. J. Neurochem. 2015, 134, 1104–1115. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Wang, N.; Zhang, Y.; Xue, D.; Lou, H.; Liu, X. Alteration in the Function and Expression of SLC and ABC Transporters in the Neurovascular Unit in Alzheimer’s Disease and the Clinical Significance. Aging Dis. 2020, 11, 390–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Rihani, S.B.; Darakjian, L.I.; Deodhar, M.; Dow, P.; Turgeon, J.; Michaud, V. Disease-Induced Modulation of Drug Transporters at the Blood-Brain Barrier Level. Int. J. Mol. Sci. 2021, 22, 3742. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, S.M.; Desai, S.Y.; Marroni, M.; Cucullo, L.; Goodrich, K.; Bingaman, W.; Mayberg, M.R.; Bengez, L.; Janigro, D. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia 2001, 42, 1501–1506. [Google Scholar] [CrossRef]
- Aronica, E.; Gorter, J.; Jansen, G.; van Veelen, C.; van Rijen, P.; Leenstra, S.; Ramkema, M.; Scheffer, G.; Scheper, R.; Troost, D. Expression and cellular distribution of multidrug transporter proteins in two major causes of medically intractable epilepsy: Focal cortical dysplasia and glioneuronal tumors. Neuroscience 2003, 118, 417–429. [Google Scholar] [CrossRef]
- Aronica, E.; Gorter, J.A.; Ramkema, M.; Redeker, S.; Gerceker, F.O.; van Vliet, E.; Scheffer, G.L.; Scheper, R.J.; Van Der Valk, P.; Baayen, J.C.; et al. Expression and cellular distribution of multidrug resistance-related proteins in the hippocampus of patients with mesial temporal lobe epilepsy. Epilepsia 2004, 45, 441–451. [Google Scholar] [CrossRef]
- Liu, J.; Thom, M.; Catarino, C.B.; Martinian, L.; Figarella-Branger, D.; Bartolomei, F.; Koepp, M.; Sisodiya, S.M. Neuropathology of the blood-brain barrier and pharmaco-resistance in human epilepsy. Brain 2012, 135, 3115–3133. [Google Scholar] [CrossRef] [Green Version]
- Sisodiya, S.M.; Lin, W.; Harding, B.N.; Squier, M.V.; Thom, M. Drug resistance in epilepsy: Expression of drug resistance proteins in common causes of refractory epilepsy. Brain 2002, 125, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Vogelgesang, S.; Kunert-Keil, C.; Cascorbi, I.; Mosyagin, I.; Schröder, E.; Runge, U.; Jedlitschky, G.; Kroemer, H.K.; Oertel, J.; Gaab, M.R.; et al. Expression of multidrug transporters in dysembryoplastic neuroepithelial tumors causing intractable epilepsy. Clin. Neuropathol. 2004, 23, 223–231. [Google Scholar] [PubMed]
- Ak, H.; Ay, B.; Tanriverdi, T.; Sanus, G.Z.; Is, M.; Sar, M.; Oz, B.; Ozkara, C.; Ozyurt, E.; Uzan, M. Expression and cellular distribution of multidrug resistance-related proteins in patients with focal cortical dysplasia. Seizure 2007, 16, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Brukner, A.M.; Billington, S.; Benifla, M.; Nguyen, T.B.; Han, H.; Bennett, O.; Gilboa, T.; Blatch, D.; Fellig, Y.; Volkov, O.; et al. Abundance of P-glycoprotein and Breast Cancer Resistance Protein Measured by Targeted Proteomics in Human Epileptogenic Brain Tissue. Mol. Pharm. 2021, 18, 2263–2273. [Google Scholar] [CrossRef]
- Loscher, W. Epilepsy and Alterations of the Blood-Brain Barrier: Cause or Consequence of Epileptic Seizures or Both? In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Loscher, W.; Friedman, A. Structural, Molecular, and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence, or Both? Int. J. Mol. Sci. 2020, 21, 591. [Google Scholar]
- Loscher, W.; Potschka, H. Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. J. Pharmacol. Exp. Ther. 2002, 301, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef]
- Gil-Martins, E.; Barbosa, D.J.; Silva, V.; Remião, F.; Silva, R. Dysfunction of ABC transporters at the blood-brain barrier: Role in neurological disorders. Pharmacol. Ther. 2020, 213, 107554. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Green, J.L.; Dos Santos, W.F.; Fontana, A.C.K. Role of glutamate excitotoxicity and glutamate transporter EAAT2 in epilepsy: Opportunities for novel therapeutics development. Biochem. Pharmacol. 2021, 193, 114786. [Google Scholar] [CrossRef]
- Zhu, H.J.; Liu, G.Q. Glutamate up-regulates P-glycoprotein expression in rat brain microvessel endothelial cells by an NMDA receptor-mediated mechanism. Life Sci. 2004, 75, 1313–1322. [Google Scholar] [CrossRef]
- Hartz, A.M.S.; Rempe, R.G.; Soldner, E.L.B.; Pekcec, A.; Schlichtiger, J.; Kryscio, R.; Bauer, B. Cytosolic phospholipase A2 is a key regulator of blood-brain barrier function in epilepsy. FASEB J. 2019, 33, 14281–14295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, B.; Hartz, A.M.S.; Pekcec, A.; Toellner, K.; Miller, D.S.; Potschka, H. Seizure-induced up-regulation of P-glycoprotein at the blood-brain barrier through glutamate and cyclooxygenase-2 signaling. Mol. Pharmacol. 2008, 73, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Zibell, G.; Unkrüer, B.; Pekcec, A.; Hartz, A.M.; Bauer, B.; Miller, D.S.; Potschka, H. Prevention of seizure-induced up-regulation of endothelial P-glycoprotein by COX-2 inhibition. Neuropharmacology 2009, 56, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Rawat, C.; Kutum, R.; Kukal, S.; Srivastava, A.; Dahiya, U.R.; Kushwaha, S.; Sharma, S.; Dash, D.; Saso, L.; Srivastava, A.K.; et al. Downregulation of peripheral PTGS2/COX-2 in response to valproate treatment in patients with epilepsy. Sci. Rep. 2020, 10, 2546. [Google Scholar] [CrossRef]
- Pekcec, A.; Unkrüer, B.; Schlichtiger, J.; Soerensen, J.; Hartz, A.M.S.; Bauer, B.; van Vliet, E.A.; Gorter, J.A.; Potschka, H. Targeting prostaglandin E2 EP1 receptors prevents seizure-associated P-glycoprotein up-regulation. J. Pharmacol. Exp. Ther. 2009, 330, 939–947. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Chen, Y. Inflammation: A Network in the Pathogenesis of Status Epilepticus. Front. Mol. Neurosci. 2018, 11, 341. [Google Scholar]
- Vishwakarma, S.; Singh, S.; Singh, T.G. Pharmacological modulation of cytokines correlating neuroinflammatory cascades in epileptogenesis. Mol. Biol. Rep. 2022, 49, 1437–1452. [Google Scholar] [CrossRef]
- Singh, S.; Singh, T.G. Role of Nuclear Factor Kappa B (NF-kappaB) Signalling in Neurodegenerative Diseases: An Mechanistic Approach. Curr. Neuropharmacol. 2020, 18, 918–935. [Google Scholar] [CrossRef]
- Bauer, B.; Hartz, A.M.; Miller, D.S. Tumor necrosis factor alpha and endothelin-1 increase P-glycoprotein expression and transport activity at the blood-brain barrier. Mol. Pharmacol. 2007, 71, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Poller, B.; Drewe, J.; Krähenbühl, S.; Huwyler, J.; Gutmann, H. Regulation of BCRP (ABCG2) and P-glycoprotein (ABCB1) by cytokines in a model of the human blood-brain barrier. Cell. Mol. Neurobiol. 2010, 30, 63–70. [Google Scholar] [CrossRef]
- Hoshi, Y.; Uchida, Y.; Tachikawa, M.; Ohtsuki, S.; Terasaki, T. Actin filament-associated protein 1 (AFAP-1) is a key mediator in inflammatory signaling-induced rapid attenuation of intrinsic P-gp function in human brain capillary endothelial cells. J. Neurochem. 2017, 141, 247–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imperio, G.E.D.; Lye, P.; Bloise, E.; Matthews, S.G. Function of Multidrug Resistance Transporters is Disrupted by Infection Mimics in Human Brain Endothelial Cells. Tissue Barriers 2021, 9, 1860616. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Chen, X.; Li, X.; Cheng, H.; Gan, J.; Liu, Z. The TLR4 mediated inflammatory signal pathway might be involved in drug resistance in drug-resistant epileptic rats. J. Neuroimmunol. 2022, 365, 577802. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xia, H.; Hu, J.; Zhang, B. MicroRNA-542-3p Regulates P-glycoprotein Expression in Rat Epilepsy via the Toll-like Receptor 4/Nuclear Factor-kappaB Signaling Pathway. Curr. Neurovasc. Res. 2019, 16, 433–440. [Google Scholar] [CrossRef]
- Beleznai, Z.; Szalay, L.; Jancsik, V. Ca2+ and Mg2+ as modulators of mitochondrial L-glycerol-3-phosphate dehydrogenase. Eur. J. Biochem. 1988, 170, 631–636. [Google Scholar] [CrossRef]
- Nishibori, M.; Wang, D.; Ousaka, D.; Wake, H. High Mobility Group Box-1 and Blood-Brain Barrier Disruption. Cells 2020, 9, 2650. [Google Scholar] [CrossRef]
- Xie, Y.; Yu, N.; Chen, Y.; Zhang, K.; Ma, H.Y.; Di, Q. HMGB1 regulates P-glycoprotein expression in status epilepticus rat brains via the RAGE/NF-kappaB signaling pathway. Mol. Med. Rep. 2017, 16, 1691–1700. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Huang, X.-J.; Yu, N.; Xie, Y.; Zhang, K.; Wen, F.; Liu, H.; Di, Q. HMGB1 Contributes to the Expression of P-Glycoprotein in Mouse Epileptic Brain through Toll-Like Receptor 4 and Receptor for Advanced Glycation End Products. PLoS ONE 2015, 10, e0140918. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, Y.; Xu, X. Upregulation of miR-142-3p Improves Drug Sensitivity of Acute Myelogenous Leukemia through Reducing P-Glycoprotein and Repressing Autophagy by Targeting HMGB1. Transl. Oncol. 2017, 10, 410–418. [Google Scholar] [CrossRef]
- Lai, W.; Li, X.; Kong, Q.; Chen, H.; Li, Y.; Xu, L.-H.; Fang, J. Extracellular HMGB1 interacts with RAGE and promotes chemoresistance in acute leukemia cells. Cancer Cell Int. 2021, 21, 700. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Hussen, B.M.; Abak, A.; Taheri, M.; Khoshnoud, R.J. Aberrant expression of miRNAs in epilepsy. Mol. Biol. Rep. 2022. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Shao, Y.; Deng, X.; Wang, M.; Chen, Y. MicroRNA-298 Reverses Multidrug Resistance to Antiepileptic Drugs by Suppressing MDR1/P-gp Expression in vitro. Front. Neurosci. 2018, 12, 602. [Google Scholar] [CrossRef] [PubMed]
- Leontariti, M.; Avgeris, M.; Katsarou, M.; Drakoulis, N.; Siatouni, A.; Verentzioti, A.; Alexoudi, A.; Fytraki, A.; Patrikelis, P.; Vassilacopoulou, D.; et al. Circulating miR-146a and miR-134 in predicting drug-resistant epilepsy in patients with focal impaired awareness seizures. Epilepsia 2020, 61, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qu, Y.; Wang, A. Antagonist targeting microRNA-146a protects against lithium-pilocarpine-induced status epilepticus in rats by nuclear factor-kappaB pathway. Mol. Med. Rep. 2018, 17, 5356–5361. [Google Scholar]
- Raoof, R.; Bauer, S.; El Naggar, H.; Connolly, N.M.; Brennan, G.P.; Brindley, E.; Hill, T.; McArdle, H.; Spain, E.; Forster, R.J.; et al. Dual-center, dual-platform microRNA profiling identifies potential plasma biomarkers of adult temporal lobe epilepsy. EBioMedicine 2018, 38, 127–141. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhou, N.; Yang, P.; Deng, L.; Liu, G. MicroRNA-27a-3p Downregulation Inhibits Inflammatory Response and Hippocampal Neuronal Cell Apoptosis by Upregulating Mitogen-Activated Protein Kinase 4 (MAP2K4) Expression in Epilepsy: In Vivo and In Vitro Studies. Med. Sci. Monit. 2019, 25, 8499–8508. [Google Scholar] [CrossRef]
- Wen, T.; Liu, Y.-C.; Yang, H.-W.; Liu, H.-Y.; Liu, X.-D.; Wang, G.-J.; Xie, L. Effect of 21-day exposure of phenobarbital, carbamazepine and phenytoin on P-glycoprotein expression and activity in the rat brain. J. Neurol. Sci. 2008, 270, 99–106. [Google Scholar] [CrossRef]
- Yang, H.-W.; Liu, H.-Y.; Liu, X.; Zhang, D.-M.; Liu, Y.-C.; Liu, X.-D.; Wang, G.-J.; Xie, L. Increased P-glycoprotein function and level after long-term exposure of four antiepileptic drugs to rat brain microvascular endothelial cells in vitro. Neurosci. Lett. 2008, 434, 299–303. [Google Scholar] [CrossRef]
- Rubinchik-Stern, M.; Shmuel, M.; Bar, J.; Kovo, M.; Eyal, S. Adverse placental effects of valproic acid: Studies in perfused human placentas. Epilepsia 2018, 59, 993–1003. [Google Scholar]
- Rubinchik-Stern, M.; Shmuel, M.; Eyal, S. Antiepileptic drugs alter the expression of placental carriers: An in vitro study in a human placental cell line. Epilepsia 2015, 56, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Tetro, N.; Imbar, T.; Wohl, D.; Eisenberg, I.; Yagel, S.; Shmuel, M.; Eyal, S. The effects of valproic acid on early pregnancy human placentas: Pilot ex vivo analysis in cultured placental villi. Epilepsia 2019, 60, e47–e51. [Google Scholar] [CrossRef] [PubMed]
- Gibb, W.R.; Lees, A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, C.G. The history of Parkinson’s disease: Early clinical descriptions and neurological therapies. Cold Spring Harb. Perspect. Med. 2011, 1, a008862. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Guan, J.; Pavlovic, D.; Dalkie, N.; Waldvogel, H.J.; O’Carroll, S.J.; Green, C.R.; Nicholson, L.F. Vascular degeneration in Parkinson’s disease. Brain Pathol. 2013, 23, 154–164. [Google Scholar] [CrossRef]
- Pan, Y.; Nicolazzo, J.A. Impact of aging, Alzheimer’s disease and Parkinson’s disease on the blood-brain barrier transport of therapeutics. Adv. Drug Deliv. Rev. 2018, 135, 62–74. [Google Scholar] [CrossRef]
- Vautier, S.; Milane, A.; Fernandez, C.; Chacun, H.; Lacomblez, L.; Farinotti, R. Role of two efflux proteins, ABCB1 and ABCG2 in blood-brain barrier transport of bromocriptine in a murine model of MPTP-induced dopaminergic degeneration. J. Pharm. Pharm. Sci. 2009, 12, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Carvey, P.M.; Zhao, C.H.; Hendey, B.; Lum, H.; Trachtenberg, J.; Desai, B.S.; Snyder, J.; Zhu, Y.G.; Ling, Z.D. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur. J. Neurosci. 2005, 22, 1158–1168. [Google Scholar] [CrossRef]
- Kim, H.; Shin, J.-Y.; Lee, Y.-S.; Yun, S.P.; Maeng, H.-J.; Lee, Y. Brain Endothelial P-Glycoprotein Level Is Reduced in Parkinson’s Disease via a Vitamin D Receptor-Dependent Pathway. Int. J. Mol. Sci. 2020, 21, 8538. [Google Scholar] [CrossRef]
- Huang, L.; Deng, M.; He, Y.; Lu, S.; Ma, R.; Fang, Y. Beta-asarone and levodopa co-administration increase striatal dopamine level in 6-hydroxydopamine induced rats by modulating P-glycoprotein and tight junction proteins at the blood-brain barrier and promoting levodopa into the brain. Clin. Exp. Pharmacol. Physiol. 2016, 43, 634–643. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuan, W.-L.; Bennett, N.; He, X.; Skepper, J.N.; Martynyuk, N.; Wijeyekoon, R.; Moghe, P.V.; Williams-Gray, C.; Barker, R.A. Alpha-Synuclein pre-formed fibrils impair tight junction protein expression without affecting cerebral endothelial cell function. Exp. Neurol. 2016, 285, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabezas, R.; Ávila, M.; Gonzalez, J.; El-Bachá, R.S.; Báez, E.; García-Segura, L.M.; Jurado Coronel, J.C.; Capani, F.; Cardona-Gomez, G.P.; Barreto, G.E. Astrocytic modulation of blood brain barrier: Perspectives on Parkinson’s disease. Front. Cell. Neurosci. 2014, 8, 211. [Google Scholar] [CrossRef] [Green Version]
- Farkas, E.; De Jong, G.I.; De Vos, R.A.I.; Jansen Steur, E.N.H.; Luiten, P.G.M. Pathological features of cerebral cortical capillaries are doubled in Alzheimer’s disease and Parkinson’s disease. Acta Neuropathol. 2000, 100, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerlund, M.; Belin, A.C.; Olson, L.; Galter, D. Expression of multi-drug resistance 1 mRNA in human and rodent tissues: Reduced levels in Parkinson patients. Cell Tissue Res. 2008, 334, 179–185. [Google Scholar] [CrossRef]
- Kortekaas, R.; Leenders, K.L.; Van Oostrom, J.C.H.; Vaalburg, W.; Bart, J.; Willemsen, A.T.M.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef]
- Bartels, A.; van Berckel, B.; Lubberink, M.; Luurtsema, G.; Lammertsma, A.; Leenders, K. Blood-brain barrier P-glycoprotein function is not impaired in early Parkinson’s disease. Park. Relat. Disord. 2008, 14, 505–508. [Google Scholar] [CrossRef]
- Bartels, A.L.; Willemsen, A.T.M.; Kortekaas, R.; De Jong, B.M.; De Vries, R.; De Klerk, O.; Van Oostrom, J.C.H.; Portman, A.; Leenders, K.L. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J. Neural Transm. 2008, 115, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Dahl, V.; Josefsson, L.; Palmer, S. HIV reservoirs, latency, and reactivation: Prospects for eradication. Antivir. Res. 2010, 85, 286–294. [Google Scholar] [CrossRef]
- Wang, B. Drug Transporters: Molecular Characterization and Role in Drug Disposition, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Langford, D.; Grigorian, A.; Hurford, R.; Adame, A.; Ellis, R.J.; Hansen, L.; Masliah, E. Altered P-glycoprotein expression in AIDS patients with HIV encephalitis. J. Neuropathol. Exp. Neurol. 2004, 63, 1038–1047. [Google Scholar] [CrossRef] [Green Version]
- Robillard, K.R.; Hoque, M.T.; Bendayan, R. Expression of ATP-binding cassette membrane transporters in a HIV-1 transgenic rat model. Biochem. Biophys. Res. Commun. 2014, 444, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Pu, H.; Andras, E.I.; Eum, S.Y.; Yamauchi, A.; Hennig, B.; Toborek, M. HIV-TAT protein upregulates expression of multidrug resistance protein 1 in the blood-brain barrier. J. Cereb. Blood Flow Metab. 2006, 26, 1052–1065. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Pu, H.; Tian, J.; Andras, I.E.; Lee, Y.W.; Hennig, B.; Toborek, M. HIV-Tat protein induces P-glycoprotein expression in brain microvascular endothelial cells. J. Neurochem. 2005, 93, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Hennig, B.; Toborek, M. Intact lipid rafts regulate HIV-1 Tat protein-induced activation of the Rho signaling and upregulation of P-glycoprotein in brain endothelial cells. J. Cereb. Blood Flow Metab. 2010, 30, 522–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashraf, T.; Ronaldson, P.T.; Persidsky, Y.; Bendayan, R. Regulation of P-glycoprotein by human immunodeficiency virus-1 in primary cultures of human fetal astrocytes. J. Neurosci. Res. 2011, 89, 1773–1782. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Leibrand, C.R.; Palasuberniam, P.; Couraud, P.-O.; Weksler, B.; Jahr, F.M.; McClay, J.L.; Hauser, K.F.; McRae, M. Effects of HIV-1 Tat and Methamphetamine on Blood-Brain Barrier Integrity and Function In Vitro. Antimicrob. Agents Chemother. 2017, 61, e01307–e01317. [Google Scholar] [CrossRef] [Green Version]
- Whyte-Allman, S.K.; Kaul, R.; Bendayan, R. Regulation of ABC Drug Efflux Transporters in Human T-Cells Exposed to an HIV Pseudotype. Front. Pharmacol. 2021, 12, 711999. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Bendayan, R. HIV-1 viral envelope glycoprotein gp120 triggers an inflammatory response in cultured rat astrocytes and regulates the functional expression of P-glycoprotein. Mol. Pharmacol. 2006, 70, 1087–1098. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Verma, A.S.; Patel, K.H.; Noel, R.; Rivera-Amill, V.; Silverstein, P.S.; Chaudhary, S.; Bhat, H.K.; Stamatatos, L.; Singh, D.P.; et al. HIV-1 gp120 induces expression of IL-6 through a nuclear factor-kappa B-dependent mechanism: Suppression by gp120 specific small interfering RNA. PLoS ONE 2011, 6, e21261. [Google Scholar] [CrossRef] [Green Version]
- Ilyin, S.E.; Plata-Salaman, C.R. HIV-1 envelope glycoprotein 120 regulates brain IL-1beta system and TNF-alpha mRNAs in vivo. Brain Res. Bull. 1997, 44, 67–73. [Google Scholar] [CrossRef]
- Garden, G.A.; Budd, S.L.; Tsai, E.; Hanson, L.; Kaul, M.; D’Emilia, D.M.; Friedlander, R.M.; Yuan, J.; Masliah, E.; Lipton, S.A. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J. Neurosci. 2002, 22, 4015–4024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashraf, T.; Jiang, W.; Hoque, T.; Henderson, J.; Wu, C.; Bendayan, R. Role of anti-inflammatory compounds in human immunodeficiency virus-1 glycoprotein120-mediated brain inflammation. J. Neuroinflamm. 2014, 11, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-J.; Yeo, I.J.; Choi, D.Y.; Yun, J.; Son, D.J.; Han, S.-B.; Hong, J.T. Amyloidogenic, neuroinflammatory and memory dysfunction effects of HIV-1 gp120. Arch. Pharm. Res. 2021, 44, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zeng, Y.; Zhou, Y.; Yu, J.; Liang, M.; Qin, L.; Zhou, Y. Win55,212-2 improves neural injury induced by HIV-1 glycoprotein 120 in rats by exciting CB2R. Brain Res. Bull. 2022, 182, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Persidsky, Y.; Gendelman, H.E. Mononuclear phagocyte immunity and the neuropathogenesis of HIV-1 infection. J. Leukoc. Biol. 2003, 74, 691–701. [Google Scholar] [CrossRef]
- Hodyl, N.; Stark, M.; Butler, M.; Clifton, V. Placental P-glycoprotein is unaffected by timing of antenatal glucocorticoid therapy but reduced in SGA preterm infants. Placenta 2013, 34, 325–330. [Google Scholar] [CrossRef]
- Petrovic, V.; Kojovic, D.; Cressman, A.; Piquette-Miller, M. Maternal bacterial infections impact expression of drug transporters in human placenta. Int. Immunopharmacol. 2015, 26, 349–356. [Google Scholar] [CrossRef]
- Mason, C.W.; Buhimschi, I.; Buhimschi, C.S.; Dong, Y.; Weiner, C.P.; Swaan, P.W. ATP-binding cassette transporter expression in human placenta as a function of pregnancy condition. Drug Metab. Dispos. 2011, 39, 1000–1007. [Google Scholar] [CrossRef] [Green Version]
- Império, G.E.D.; Bloise, E.; Javam, M.; Lye, P.; Constantinof, A.; Dunk, C.; Dos Reisf, F.M.; Lye, S.J.; Gibb, W.; Ortiga-Carvalho, T.M.; et al. Chorioamnionitis Induces a Specific Signature of Placental ABC Transporters Associated with an Increase of miR-331-5p in the Human Preterm Placenta. Cell. Physiol. Biochem. 2018, 45, 591–604. [Google Scholar] [CrossRef]
- Evseenko, D.A.; Murthi, P.; Paxton, J.W.; Reid, G.; Emerald, B.S.; Mohankumar, K.M.; Lobie, P.E.; Brennecke, S.P.; Kalionis, B.; Keelan, J.A. The ABC transporter BCRP/ABCG2 is a placental survival factor, and its expression is reduced in idiopathic human fetal growth restriction. FASEB J. 2007, 21, 3592–3605. [Google Scholar] [CrossRef]
- Wang, C.; Li, H.; Luo, C.; Li, Y.; Zhang, Y.; Yun, D.; Mu, D.; Zhou, K.; Hua, Y. The effect of maternal obesity on the expression and functionality of placental P-glycoprotein: Implications in the individualized transplacental digoxin treatment for fetal heart failure. Placenta 2015, 36, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Kojovic, D.; Workewych, N.V.; Piquette-Miller, M. Role of Elevated SFLT-1 on the Regulation of Placental Transporters in Women with Pre-Eclampsia. Clin. Transl. Sci. 2020, 13, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Dunk, C.E.; Pappas, J.J.; Lye, P.; Kibschull, M.; Javam, M.; Bloise, E.; Lye, S.J.; Szyf, M.; Matthews, S.G. P-Glycoprotein (P-gp)/ABCB1 plays a functional role in extravillous trophoblast (EVT) invasion and is decreased in the pre-eclamptic placenta. J. Cell. Mol. Med. 2018, 22, 5378–5393. [Google Scholar] [CrossRef]
- Anger, G.J.; Cressman, A.M.; Piquette-Miller, M. Expression of ABC Efflux transporters in placenta from women with insulin-managed diabetes. PLoS ONE 2012, 7, e35027. [Google Scholar] [CrossRef] [Green Version]
- Kojovic, D.; Ghoneim, R.H.; Serghides, L.; Piquette-Miller, M. Role of HIV and Antiretroviral Therapy on the Expression of Placental Transporters in Women with HIV. AAPS J. 2020, 22, 138. [Google Scholar] [CrossRef]
- Pfeifer, E.; Parrott, J.; Lee, G.T.; Domalakes, E.; Zhou, H.; He, L.; Mason, C.W. Regulation of human placental drug transporters in HCV infection and their influence on direct acting antiviral medications. Placenta 2018, 69, 32–39. [Google Scholar] [CrossRef]
- Lye, P.; Bloise, E.; Javam, M.; Gibb, W.; Lye, S.J.; Matthews, S. Impact of bacterial and viral challenge on multidrug resistance in first- and third-trimester human placenta. Am. J. Pathol. 2015, 185, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Javam, M.; Audette, M.; Iqbal, M.; Bloise, E.; Gibb, W.; Matthews, S. Effect of oxygen on multidrug resistance in term human placenta. Placenta 2014, 35, 324–330. [Google Scholar] [CrossRef] [Green Version]
- Evseenko, D.A.; Paxton, J.W.; Keelan, J.A. Independent regulation of apical and basolateral drug transporter expression and function in placental trophoblasts by cytokines, steroids, and growth factors. Drug Metab. Dispos. 2007, 35, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Beghin, D.; Forestier, F.; Noël-Hudson, M.-S.; Gavard, L.; Guibourdenche, J.; Farinotti, R.; Gil, S. Modulation of endocrine and transport functions in human trophoblasts by saquinavir and nelfinavir. Eur. J. Obstet. Gynecol. Reprod. Biol. 2010, 152, 55–59. [Google Scholar] [CrossRef]
- Mason, C.W.; Lee, G.T.; Dong, Y.; Zhou, H.; He, L.; Weiner, C.P. Effect of prostaglandin E2 on multidrug resistance transporters in human placental cells. Drug Metab. Dispos. 2014, 42, 2077–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Hong, M.; You, G. Regulation of human organic anion transporter 4 by progesterone and protein kinase C in human placental BeWo cells. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E57–E61. [Google Scholar] [CrossRef]
- Jaacks, L.M.; Young, M.F.; Essley, B.V.; McNanley, T.J.; Cooper, E.M.; Pressman, E.K.; McIntyre, A.W.; Orlando, M.S.; Abkowitz, J.L.; Guillet, R.; et al. Placental expression of the heme transporter, feline leukemia virus subgroup C receptor, is related to maternal iron status in pregnant adolescents. J. Nutr. 2011, 141, 1267–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lye, P.; Bloise, E.; Nadeem, L.; Gibb, W.; Lye, S.J.; Matthews, S.G. Glucocorticoids modulate multidrug resistance transporters in the first trimester human placenta. J. Cell. Mol. Med. 2018, 22, 3652–3660. [Google Scholar] [CrossRef] [Green Version]
- Gorczyca, L.; Du, J.; Bircsak, K.M.; Wen, X.; Vetrano, A.M.; Aleksunes, L.M. Low oxygen tension differentially regulates the expression of placental solute carriers and ABC transporters. FEBS Lett. 2021, 595, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Cressman, A.M.; Petrovic, V.; Piquette-Miller, M. Inflammation-mediated changes in drug transporter expression/activity: Implications for therapeutic drug response. Expert Rev. Clin. Pharmacol. 2012, 5, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Jebbink, J.; Veenboer, G.; Boussata, S.; Keijser, R.; Kremer, A.E.; Elferink, R.O.; van der Post, J.; Afink, G.; Ris-Stalpers, C. Total bile acids in the maternal and fetal compartment in relation to placental ABCG2 expression in preeclamptic pregnancies complicated by HELLP syndrome. Biochim. Biophys. Acta 2015, 1852, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Bircsak, K.M.; Moscovitz, J.E.; Wen, X.; Archer, F.; Yuen, P.Y.S.; Mohammed, M.; Memon, N.; Weinberger, B.I.; Saba, L.M.; Vetrano, A.M.; et al. Interindividual Regulation of the Breast Cancer Resistance Protein/ABCG2 Transporter in Term Human Placentas. Drug Metab. Dispos. 2018, 46, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Zhou, J.; Guo, J.; Hu, W.; Chen, G.; Li, B.; Wen, Y.; Jiang, Y.; Fu, K.; Bi, H.; et al. Dexamethasone induces an imbalanced fetal-placental-maternal bile acid circulation: Involvement of placental transporters. BMC Med. 2021, 19, 87. [Google Scholar] [CrossRef]
- Mirdamadi, K.; Kwok, J.; Nevo, O.; Berger, H.; Piquette-Miller, M. Impact of Th-17 Cytokines on the Regulation of Transporters in Human Placental Explants. Pharmaceutics 2021, 13, 881. [Google Scholar] [CrossRef]
- Sieppi, E.; Vähäkangas, K.; Rautio, A.; Ietta, F.; Paulesu, L.; Myllynen, P. The xenoestrogens, bisphenol A and para-nonylphenol, decrease the expression of the ABCG2 transporter protein in human term placental explant cultures. Mol. Cell. Endocrinol. 2016, 429, 41–49. [Google Scholar] [CrossRef]
- Behravan, J.; Piquette-Miller, M. Drug transport across the placenta, role of the ABC drug efflux transporters. Expert Opin. Drug Metab. Toxicol. 2007, 3, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, X. Contributions of Drug Transporters to Blood-Placental Barrier. Adv. Exp. Med. Biol. 2019, 1141, 505–548. [Google Scholar] [PubMed]
- American College of Obstetricians and Gynecologists. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar]
- Gathiram, P.; Moodley, J. Preeclampsia: Its pathogenesis and pathophysiolgy. Cardiovasc. J. Afr. 2016, 27, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Li, J.; Baker, P.N.; Tong, C. Revisiting preeclampsia: A metabolic disorder of the placenta. FEBS J. 2022, 289, 336–354. [Google Scholar] [CrossRef]
- Afrouzian, M.; Al-Lahham, R.; Patrikeeva, S.; Xu, M.; Fokina, V.; Fischer, W.G.; Abdel-Rahman, S.Z.; Costantine, M.; Ahmed, M.S.; Nanovskaya, T. Role of the efflux transporters BCRP and MRP1 in human placental bio-disposition of pravastatin. Biochem. Pharmacol. 2018, 156, 467–478. [Google Scholar] [CrossRef]
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennström, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Predictive Value of the sFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N. Engl. J. Med. 2016, 374, 13–22. [Google Scholar] [CrossRef]
- McIntyre, H.D.; Catalano, P.; Zhang, C.; Desoye, G.; Mathiesen, E.R.; Damm, P. Gestational diabetes mellitus. Nat. Rev. Dis. Primers 2019, 5, 47. [Google Scholar] [CrossRef]
- Anger, G.J.; Piquette-Miller, M. Mechanisms of reduced maternal and fetal lopinavir exposure in a rat model of gestational diabetes. Drug Metab. Dispos. 2011, 39, 1850–1859. [Google Scholar] [CrossRef] [Green Version]
- Kozłowska-Rup, D.; Czekaj, P.; Plewka, D.; Sikora, J. Immunolocalization of ABC drug transporters in human placenta from normal and gestational diabetic pregnancies. Ginekol. Pol. 2014, 85, 410–419. [Google Scholar] [CrossRef]
- Muñoz, G.; Martín, R.S.; Farías, M.; Cea, L.; Vecchiola, A.; Casanello, P.; Sobrevia, L. Insulin restores glucose inhibition of adenosine transport by increasing the expression and activity of the equilibrative nucleoside transporter 2 in human umbilical vein endothelium. J. Cell. Physiol. 2006, 209, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Westermeier, F.; Salomón, C.; Farías, M.; Arroyo, P.; Fuenzalida, B.; Sáez, T.; Salsoso, R.; Sanhueza, C.; Guzmán-Gutiérrez, E.; Pardo, F.; et al. Insulin requires normal expression and signaling of insulin receptor A to reverse gestational diabetes-reduced adenosine transport in human umbilical vein endothelium. FASEB J. 2015, 29, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomon, C.; Westermeier, F.; Puebla, C.; Arroyo, P.; Guzman-Gutierrez, E.; Pardo, F.; Leiva, A.; Casanello, P.; Sobrevia, L. Gestational diabetes reduces adenosine transport in human placental microvascular endothelium, an effect reversed by insulin. PLoS ONE 2012, 7, e40578. [Google Scholar] [CrossRef] [PubMed]
- Fuenzalida, B.; Cantin, C.; Kallol, S.; Carvajal, L.; Pastén, V.; Contreras-Duarte, S.; Albrecht, C.; Gutierrez, J.; Leiva, A. Cholesterol uptake and efflux are impaired in human trophoblast cells from pregnancies with maternal supraphysiological hypercholesterolemia. Sci. Rep. 2020, 10, 5264. [Google Scholar] [CrossRef]
- Ghoneim, R.H.; Kojovic, D.; Piquette-Miller, M. Impact of endotoxin on the expression of drug transporters in the placenta of HIV-1 transgenic (HIV-Tg) rats. Eur. J. Pharm. Sci. 2017, 102, 94–102. [Google Scholar] [CrossRef]
- Gilmore, J.C.; Zhang, G.; Cameron, D.W.; Serghides, L.; Bendayan, R. Impact of in-utero antiretroviral drug exposure on expression of membrane-associated transporters in mouse placenta and fetal brain. AIDS 2021, 35, 2249–2258. [Google Scholar] [CrossRef]
- Camus, M.; Deloménie, C.; Didier, N.; Faye, A.; Gil, S.; Dauge, M.-C.; Mabondzo, A.; Farinotti, R. Increased expression of MDR1 mRNAs and P-glycoprotein in placentas from HIV-1 infected women. Placenta 2006, 27, 699–706. [Google Scholar] [CrossRef]



| Condition/Factor | Effect | Reference | ||
|---|---|---|---|---|
| mRNA | Protein | Other | ||
| Based on Clinical Data: | ||||
| Gestational Age | ↓ 6.5-fold | ↓ 5.1-fold | mRNA + corr with hCG-β | [27] |
| Gestational Age | - | ↓ 69% | - | [26] |
| SGA (Preterm) | ↓ | ↓ | - | [168] |
| Chorio (Preterm) | ↑ (trend) | ↔ | mRNA + corr with IL-6, IL-1b, TNF-a | [169] |
| Chorio (Preterm) | ↑ | - | mRNA + corr with IL-8 | [170] |
| Chorio (Preterm) | ↑ 1.61-fold | ↔ | mRNA + corr with Chorio degree | [171] |
| IUFGR | ↓ | - | mRNA + corr with ABCG2 mRNA | [172] |
| Obesity | ↓ | ↓ | - | [173] |
| Preeclampsia | ↔ | Ns (↓ trend) | - | [174] |
| Severe early onset preeclampsia | ↔ | ↓ | - | [175] |
| Diabetes | ↑ | ↔ | - | [176] |
| HIV | ↑ 5.5-fold | ↑ (trend, p = 0.11) | mRNA + corr with estradiol | [177] |
| Hepatitis | ↑ 2.5-fold | ↑ 3.1-fold | - | [178] |
| Maternal betamethasone therapy | ↔ | - | - | [168] |
| Based Placental Explants: | ||||
| LPS | ↓ (T1) | ↓ (T1) | - | [179] |
| Poly I:C | ↓ (Term) | ↔ (Term) | mRNA + corr with TLR3 and TLR4 | [179] |
| Hypoxia | ↑ (Term) | ↑ (Term) | ↓ mRNA of VEGF | [180] |
| cART treatment | ↑1.6- to 2.5-fold | - | - | [177] |
| Based Primary Trophoblasts: | ||||
| TNF-a | ↓ 45% | ↓ 50% | - | [181] |
| IL-1b | ↓ 45% | ↓ 50% | - | [181] |
| Estradiol | ↑ ~50% | ↑ ~60% | ↑ function | [181] |
| Progesterone | ↔ | ↑ ~40% | - | [181] |
| cART treatment | - | ↑ 2-fold | ↑ function | [182] |
| Prostaglandin E2 | ↔ | ↔ | ↔ | [183] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taggi, V.; Riera Romo, M.; Piquette-Miller, M.; Meyer zu Schwabedissen, H.E.; Neuhoff, S. Transporter Regulation in Critical Protective Barriers: Focus on Brain and Placenta. Pharmaceutics 2022, 14, 1376. https://doi.org/10.3390/pharmaceutics14071376
Taggi V, Riera Romo M, Piquette-Miller M, Meyer zu Schwabedissen HE, Neuhoff S. Transporter Regulation in Critical Protective Barriers: Focus on Brain and Placenta. Pharmaceutics. 2022; 14(7):1376. https://doi.org/10.3390/pharmaceutics14071376
Chicago/Turabian StyleTaggi, Valerio, Mario Riera Romo, Micheline Piquette-Miller, Henriette E. Meyer zu Schwabedissen, and Sibylle Neuhoff. 2022. "Transporter Regulation in Critical Protective Barriers: Focus on Brain and Placenta" Pharmaceutics 14, no. 7: 1376. https://doi.org/10.3390/pharmaceutics14071376
APA StyleTaggi, V., Riera Romo, M., Piquette-Miller, M., Meyer zu Schwabedissen, H. E., & Neuhoff, S. (2022). Transporter Regulation in Critical Protective Barriers: Focus on Brain and Placenta. Pharmaceutics, 14(7), 1376. https://doi.org/10.3390/pharmaceutics14071376