
Immune Checkpoint Inhibitor-Mediated Cancer Theranostics with Radiolabeled Anti-Granzyme B Peptide

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Syngeneic Colon Cancer Animal Model
2.2. Ga-68 and Y-90 Radiolabeling of GZP
2.3. PET/MR Imaging
2.4. Therapeutic Studies
2.5. Toxicity Evaluation
2.6. Histopathological Analysis of Tumor
2.6.1. Immunohistochemistry
2.6.2. Immunofluorescence
2.6.3. Western Blotting
2.7. Statistical Analysis
3. Results
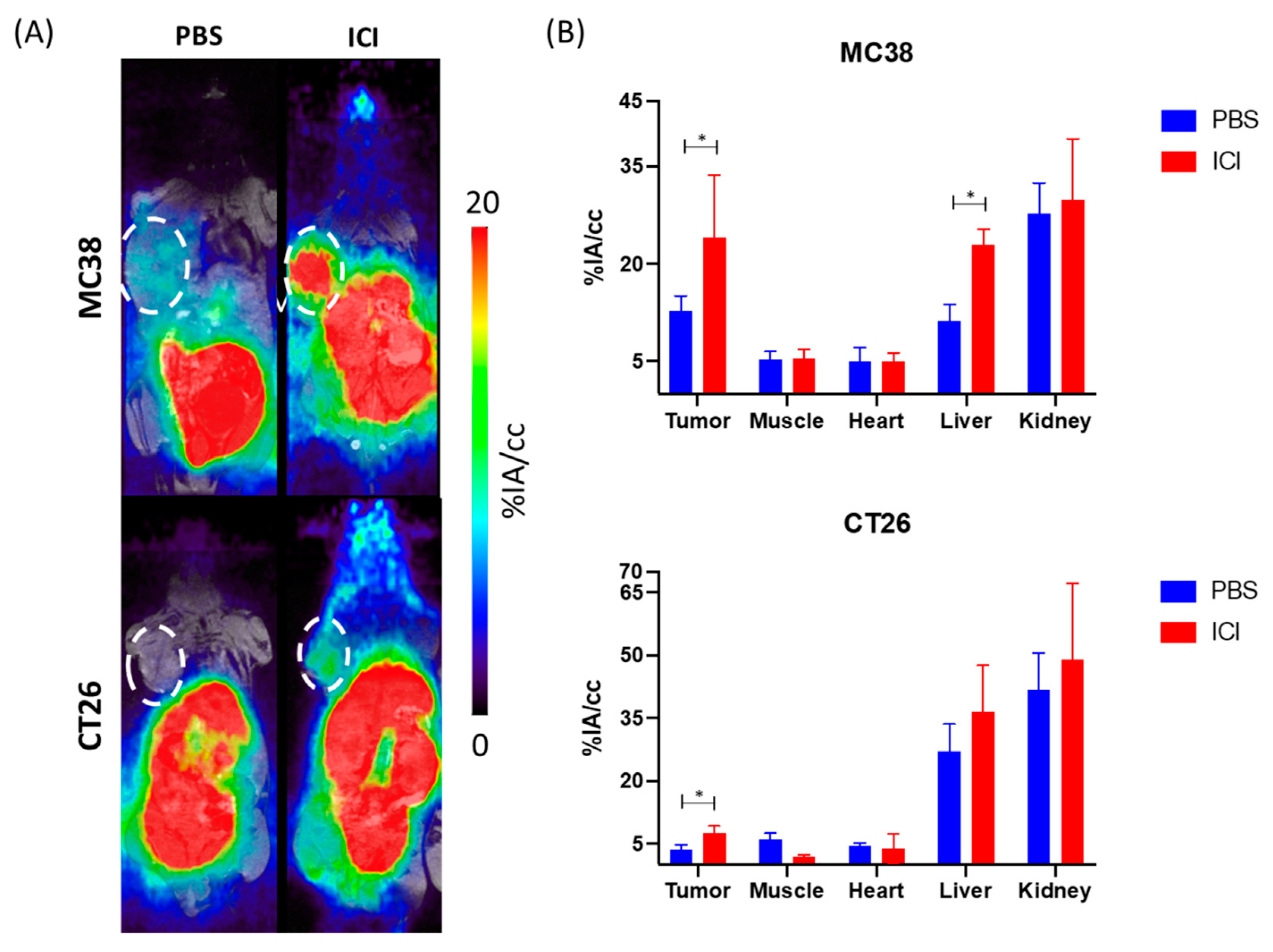
3.1. PET/MR Imaging
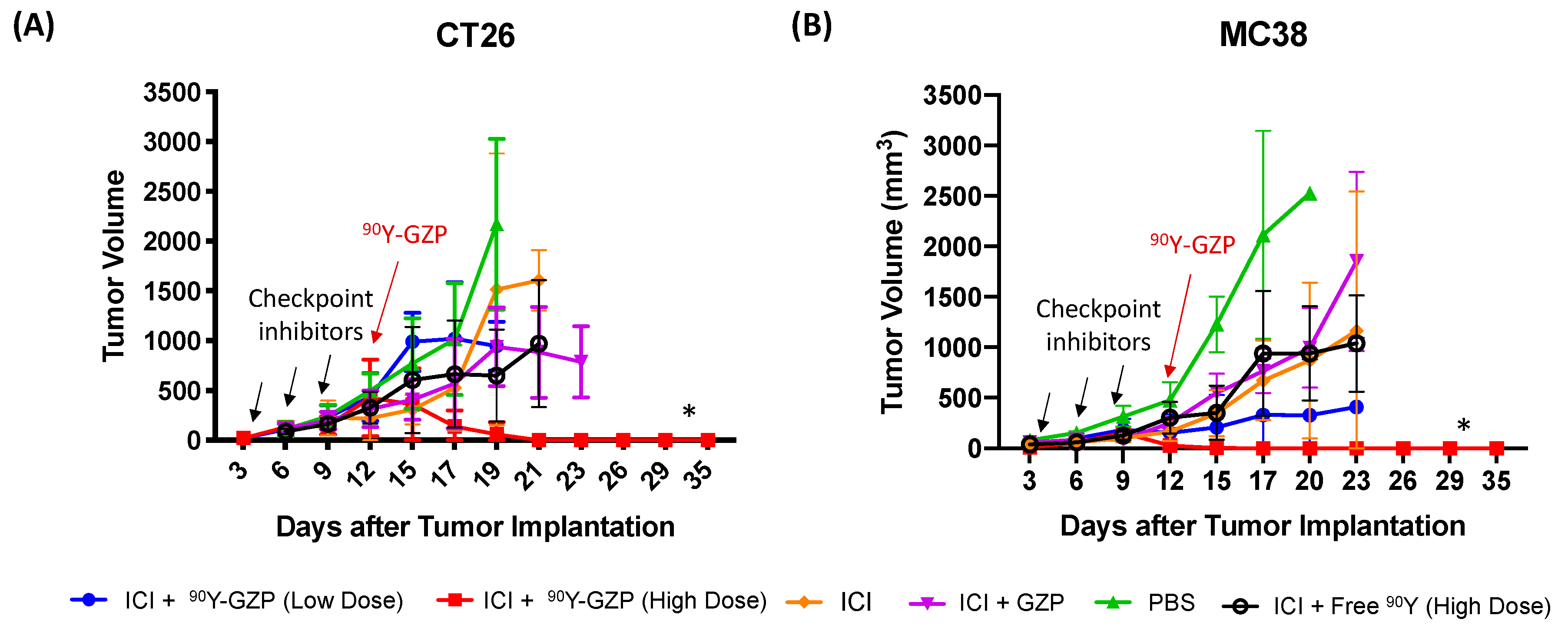
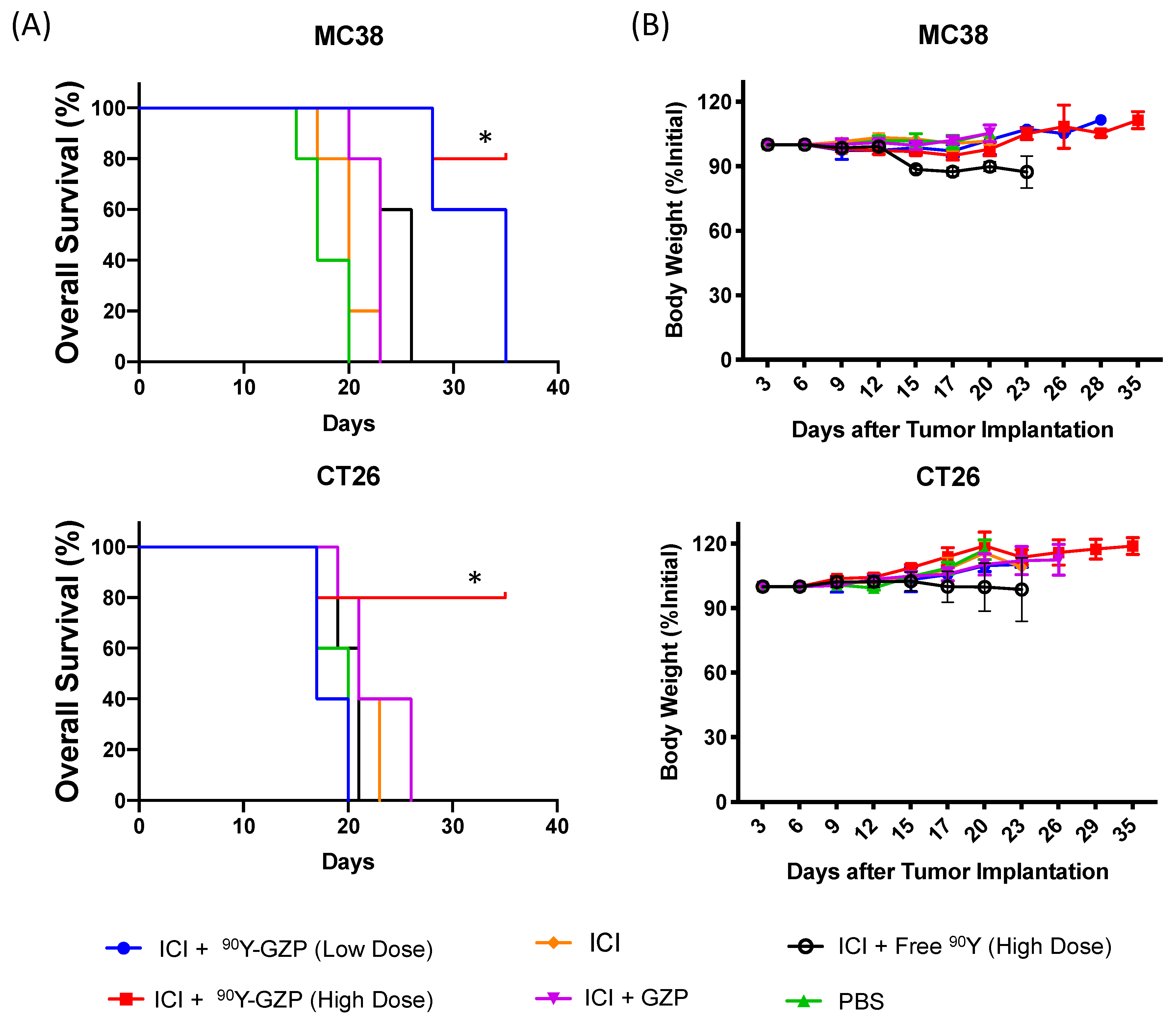
3.2. Therapeutic Studies
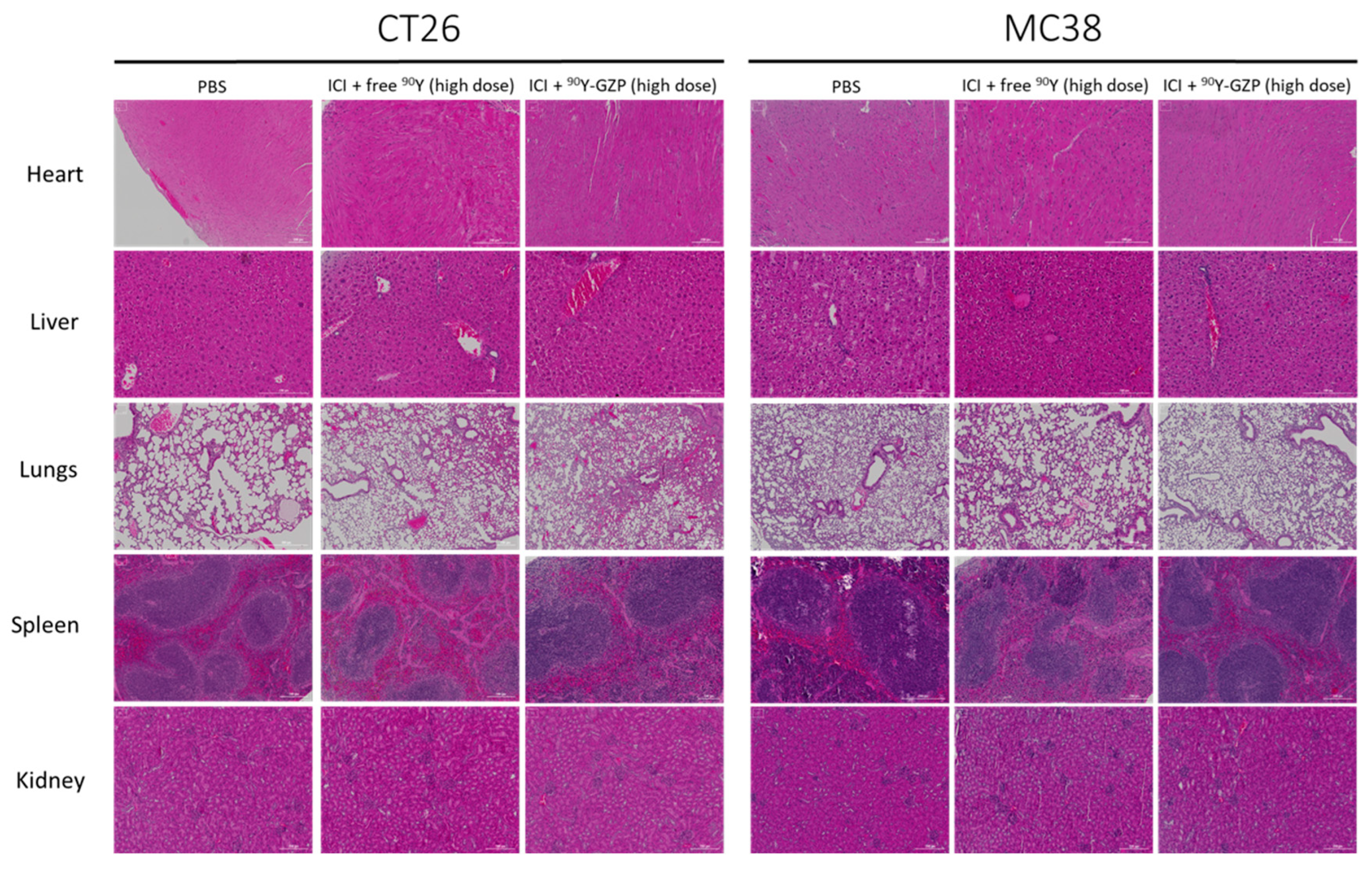
3.3. Toxicity Evaluation
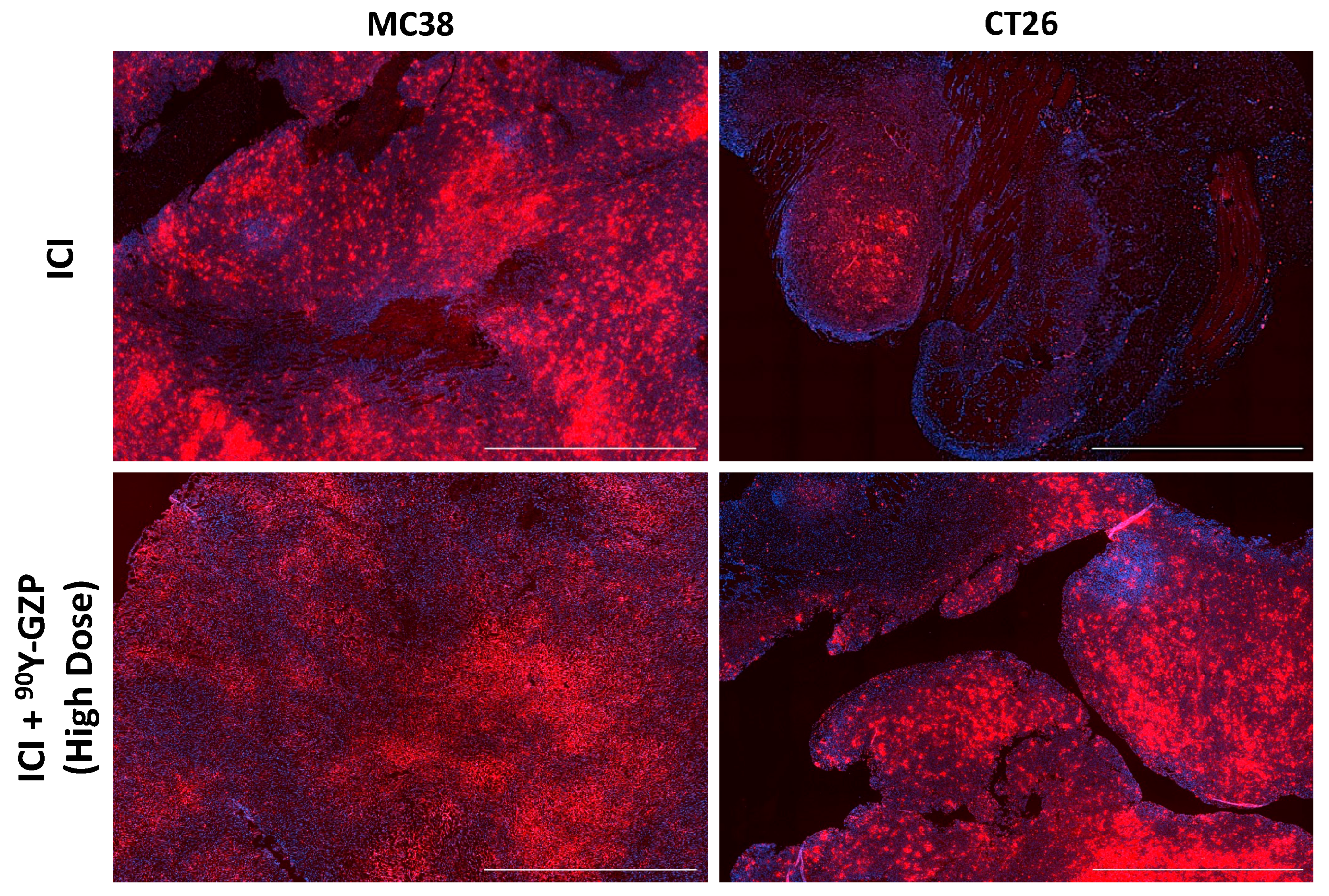
3.4. Histopathological Analysis of Tumor
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Cullen, S.P.; Brunet, M.; Martin, S.J. Granzymes in cancer and immunity. Cell Death Differ. 2010, 17, 616–623. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Willsmore, Z.N.; Coumbe, B.G.T.; Crescioli, S.; Reci, S.; Gupta, A.; Harris, R.J.; Chenoweth, A.; Chauhan, J.; Bax, H.J.; McCraw, A.; et al. Combined anti-PD-1 and anti-CTLA-4 checkpoint blockade: Treatment of melanoma and immune mechanisms of action. Eur. J. Immunol. 2021, 51, 544–556. [Google Scholar] [CrossRef]
- Twomey, J.D.; Zhang, B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. AAPS J. 2021, 23, 39. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Malouf, G.G.; Jacobs, I.; Chou, J.; Liu, L.; Johnson, M.L. Addressing resistance to immune checkpoint inhibitor therapy: An urgent unmet need. Future Oncol. 2021, 17, 1401–1439. [Google Scholar] [CrossRef]
- Gudkov, S.V.; Shilyagina, N.Y.; Vodeneev, V.A.; Zvyagin, A.V. Targeted Radionuclide Therapy of Human Tumors. Int. J. Mol. Sci. 2015, 17, 33. [Google Scholar] [CrossRef]
- Larimer, B.M.; Bloch, E.; Nesti, S.; Austin, E.E.; Wehrenberg-Klee, E.; Boland, G.; Mahmood, U. The Effectiveness of Checkpoint Inhibitor Combinations and Administration Timing Can Be Measured by Granzyme B PET Imaging. Clin. Cancer Res. 2019, 25, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Larimer, B.M.; Wehrenberg-Klee, E.; Dubois, F.; Mehta, A.; Kalomeris, T.; Flaherty, K.; Boland, G.; Mahmood, U. Granzyme B PET Imaging as a Predictive Biomarker of Immunotherapy Response. Cancer Res. 2017, 77, 2318–2327. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, C.A.; Heidari, P.; Ataeinia, B.; Sinevici, N.; Sise, M.E.; Colvin, R.B.; Wehrenberg-Klee, E.; Mahmood, U. Non-invasive Detection of Immunotherapy-Induced Adverse Events. Clin. Cancer Res. 2021, 27, 5353–5364. [Google Scholar] [CrossRef]
- Aslani, A.; Snowdon, G.M.; Bailey, D.L.; Schembri, G.P.; Bailey, E.A.; Roach, P.J. Gallium-68 DOTATATE Production with Automated PET Radiopharmaceutical Synthesis System: A Three Year Experience. Asia Ocean. J. Nucl. Med. Biol. 2014, 2, 75–86. [Google Scholar]
- Mokaleng, B.B.; Ebenhan, T.; Ramesh, S.; Govender, T.; Kruger, H.G.; Parboosing, R.; Hazari, P.P.; Mishra, A.K.; Marjanovic-Painter, B.; Zeevaart, J.R.; et al. Synthesis, 68Ga-radiolabeling, and preliminary in vivo assessment of a depsipeptide-derived compound as a potential PET/CT infection imaging agent. Biomed Res. Int. 2015, 2015, 284354. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.R.; Shi, Y.; Taylor, C.R. Antigen retrieval immunohistochemistry: Review and future prospects in research and diagnosis over two decades. J. Histochem. Cytochem. 2011, 59, 13–32. [Google Scholar] [CrossRef]
- Manz, S.M.; Losa, M.; Fritsch, R.; Scharl, M. Efficacy and side effects of immune checkpoint inhibitors in the treatment of colorectal cancer. Therap. Adv. Gastroenterol. 2021, 14, 17562848211002018. [Google Scholar] [CrossRef]
- Almquist, D.R.; Ahn, D.H.; Bekaii-Saab, T.S. The Role of Immune Checkpoint Inhibitors in Colorectal Adenocarcinoma. BioDrugs 2020, 34, 349–362. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Ooki, A.; Shinozaki, E.; Yamaguchi, K. Immunotherapy in Colorectal Cancer: Current and Future Strategies. J. Anus Rectum Colon 2021, 5, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Ellsworth, S.G. Field size effects on the risk and severity of treatment-induced lymphopenia in patients undergoing radiation therapy for solid tumors. Adv. Radiat. Oncol. 2018, 3, 512–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug. Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef] [PubMed]
- Dzunic, M.; Petkovic, I.; Cvetanovic, A.; Vrbic, S.; Pejcic, I. Current and future targets and therapies in metastatic colorectal cancer. J. BUON 2019, 24, 1785–1792. [Google Scholar]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Goggi, J.L.; Tan, Y.X.; Hartimath, S.V.; Jieu, B.; Hwang, Y.Y.; Jiang, L.; Boominathan, R.; Cheng, P.; Yuen, T.Y.; Chin, H.X.; et al. Granzyme B PET Imaging of Immune Checkpoint Inhibitor Combinations in Colon Cancer Phenotypes. Mol. Imaging Biol. 2020, 22, 1392–1402. [Google Scholar] [CrossRef]
- Corro, C.; Dutoit, V.; Koessler, T. Emerging Trends for Radio-Immunotherapy in Rectal Cancer. Cancers 2021, 13, 1374. [Google Scholar] [CrossRef]
- Monjazeb, A.M.; Schalper, K.A.; Villarroel-Espindola, F.; Nguyen, A.; Shiao, S.L.; Young, K. Effects of Radiation on the Tumor Microenvironment. Semin. Radiat. Oncol. 2020, 30, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, B.; Liu, S.; Zhang, A.; Wu, T.; Zhao, Y. 2-Gy whole-body irradiation significantly alters the balance of CD4+ CD25- T effector cells and CD4+ CD25+ Foxp3+ T regulatory cells in mice. Nature 2010, 7, 419–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Cao, Y.; Markelc, B.; Kaeppler, J.; Vermeer, J.A.; Muschel, R.J. Type I IFN protects cancer cells from CD8+ T cell-mediated cytotoxicity after radiation. J. Clin. Investig. 2019, 129, 4224–4238. [Google Scholar] [CrossRef] [PubMed]
- Benci, J.L.; Johnson, L.R.; Choa, R.; Xu, Y.; Qiu, J.; Zhou, Z.; Xu, B.; Ye, D.; Nathanson, K.L.; June, C.H.; et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 2019, 178, 933–948.e14. [Google Scholar] [CrossRef] [PubMed]
- Werner, R.A.; Bluemel, C.; Allen-Auerbach, M.S.; Higuchi, T.; Herrmann, K. 68Gallium- and 90Yttrium-/177Lutetium: “theranostic twins” for diagnosis and treatment of NETs. Ann. Nucl. Med. 2015, 29, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öksüz, M.Ö.; Winter, L.; Pfannenberg, C.; Reischl, G.; Müssig, K.; Bares, R.; Dittmann, H. Peptide receptor radionuclide therapy of neuroendocrine tumors with 90Y-DOTATOC: Is treatment response predictable by pre-therapeutic uptake of 68Ga-DOTATOC? Diagn. Interv. Imaging 2014, 95, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Bois, F.; Noirot, C.; Dietemann, S.; Mainta, I.C.; Zilli, T.; Garibotto, V.; Walter, M.A. [(68)Ga]Ga-PSMA-11 in prostate cancer: A comprehensive review. Am. J. Nucl. Med. Mol. Imaging 2020, 10, 349–374. [Google Scholar]
- Heinzel, A.A.-O.; Boghos, D.; Mottaghy, F.M.; Gaertner, F.; Essler, M.; von Mallek, D.; Ahmadzadehfar, H. (68)Ga-PSMA PET/CT for monitoring response to (177)Lu-PSMA-617 radioligand therapy in patients with metastatic castration-resistant prostate cancer. Eur. J. Nucl. Med. Mol. Imaging. 2019, 46, 1054–1062. [Google Scholar] [CrossRef]
- Hennrich, U.A.-O.; Kopka, K.A.-O. Lutathera(®): The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- FDA Approves Pluvicto/Locametz for Metastatic Castration-Resistant Prostate Cancer. J. Nucl. Med. 2022, 63, 13N.
- Kleinendorst, S.A.-O.; Oosterwijk, E.; Bussink, J.A.-O.; Westdorp, H.A.-O.; Konijnenberg, M.A.-O.; Heskamp, S.A.-O. Combining targeted radionuclide therapy and immune checkpoint inhibition for cancer treatment. Clin. Cancer Res. 2022; ahead of print. [Google Scholar] [CrossRef]
- Boivin, W.A.; Cooper, D.M.; Hiebert, P.R.; Granville, D.J. Intracellular versus extracellular granzyme B in immunity and disease: Challenging the dogma. Lab. Investig. 2009, 89, 1195–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Couturier, J.; Yu, X.; Medina, M.A.; Kozinetz, C.A.; Lewis, D.E. Granzyme B secretion by human memory CD4 T cells is less strictly regulated compared to memory CD8 T cells. BMC Immunol. 2014, 15, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Aguiar Ferreira, C.; Heidari, P.; Ataeinia, B.; Sinevici, N.; Granito, A.; Kumar, H.M.; Wehrenberg-Klee, E.; Mahmood, U. Immune Checkpoint Inhibitor-Mediated Cancer Theranostics with Radiolabeled Anti-Granzyme B Peptide. Pharmaceutics 2022, 14, 1460. https://doi.org/10.3390/pharmaceutics14071460
de Aguiar Ferreira C, Heidari P, Ataeinia B, Sinevici N, Granito A, Kumar HM, Wehrenberg-Klee E, Mahmood U. Immune Checkpoint Inhibitor-Mediated Cancer Theranostics with Radiolabeled Anti-Granzyme B Peptide. Pharmaceutics. 2022; 14(7):1460. https://doi.org/10.3390/pharmaceutics14071460
Chicago/Turabian Stylede Aguiar Ferreira, Carolina, Pedram Heidari, Bahar Ataeinia, Nicoleta Sinevici, Alyssa Granito, Hritik Mahajan Kumar, Eric Wehrenberg-Klee, and Umar Mahmood. 2022. "Immune Checkpoint Inhibitor-Mediated Cancer Theranostics with Radiolabeled Anti-Granzyme B Peptide" Pharmaceutics 14, no. 7: 1460. https://doi.org/10.3390/pharmaceutics14071460
APA Stylede Aguiar Ferreira, C., Heidari, P., Ataeinia, B., Sinevici, N., Granito, A., Kumar, H. M., Wehrenberg-Klee, E., & Mahmood, U. (2022). Immune Checkpoint Inhibitor-Mediated Cancer Theranostics with Radiolabeled Anti-Granzyme B Peptide. Pharmaceutics, 14(7), 1460. https://doi.org/10.3390/pharmaceutics14071460