
Integration of In Silico Strategies for Drug Repositioning towards P38α Mitogen-Activated Protein Kinase (MAPK) at the Allosteric Site



Abstract
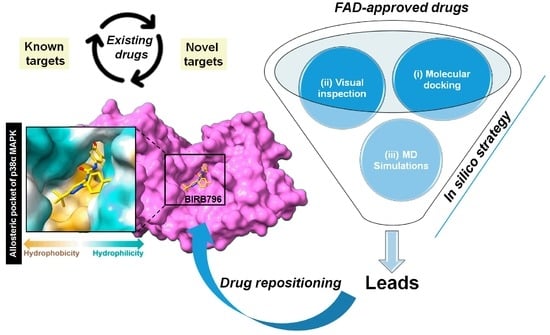
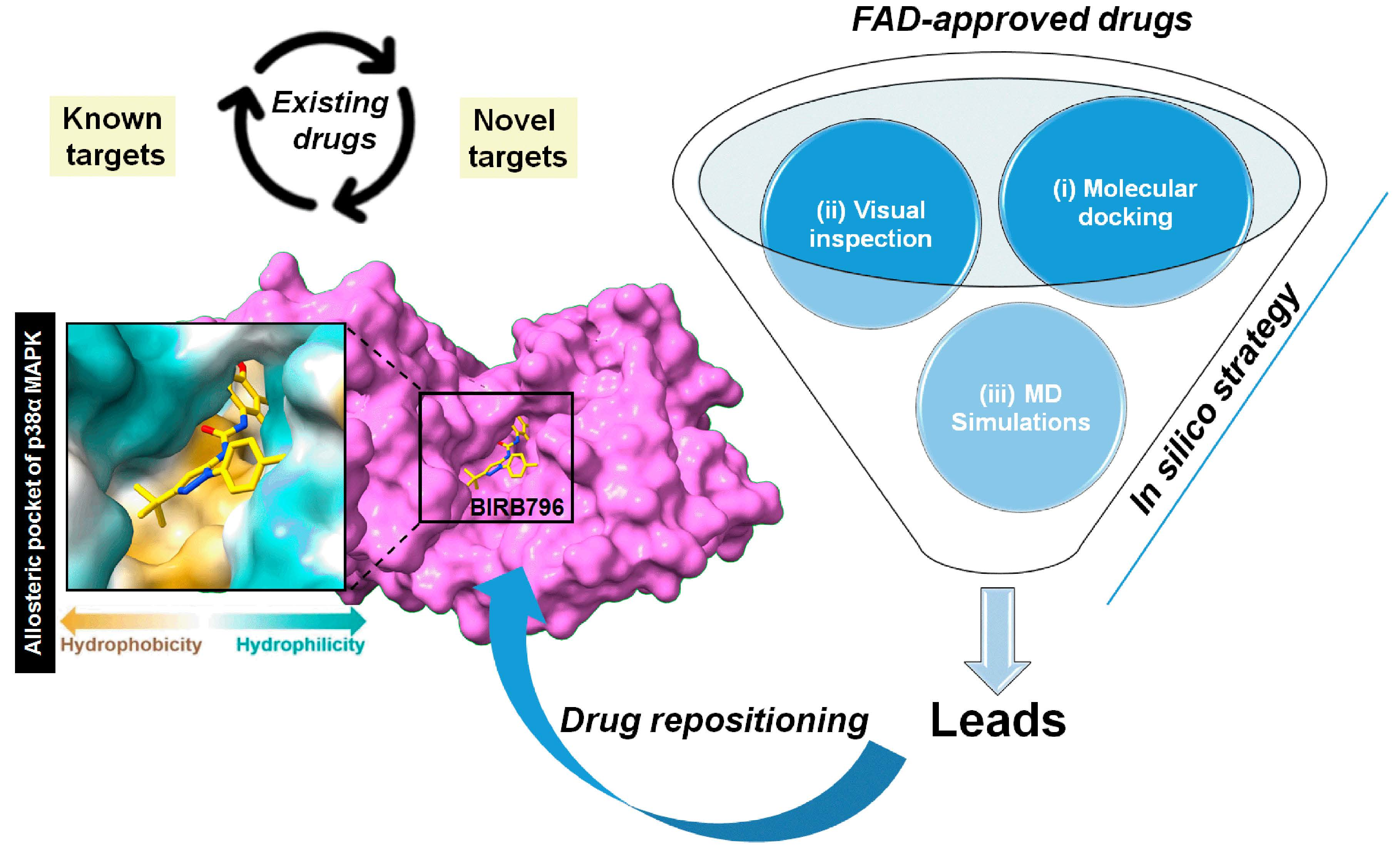
:
1. Introduction
2. Materials and Methods
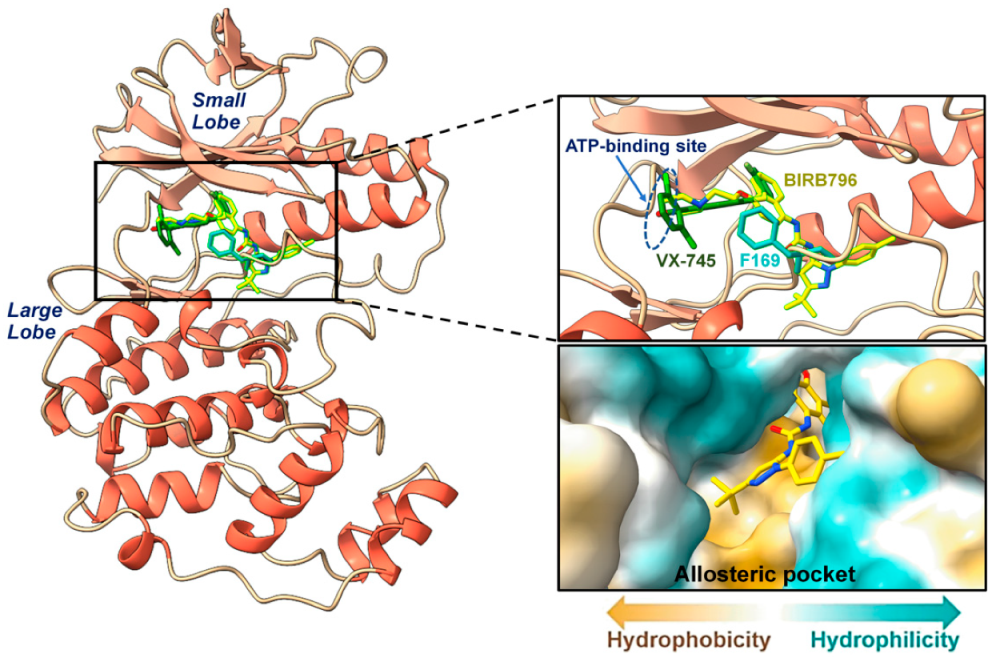
2.1. Preparation of the 3D Structure of P38α MAPK and Ligands
2.2. Molecular Docking and Visual Inspection
2.3. Molecular Dynamics (MD) Simulations
2.4. End-Point Binding Energy Calculations
2.5. QM-Based ONIOM Binding Energy Calculations
3. Results and Discussion
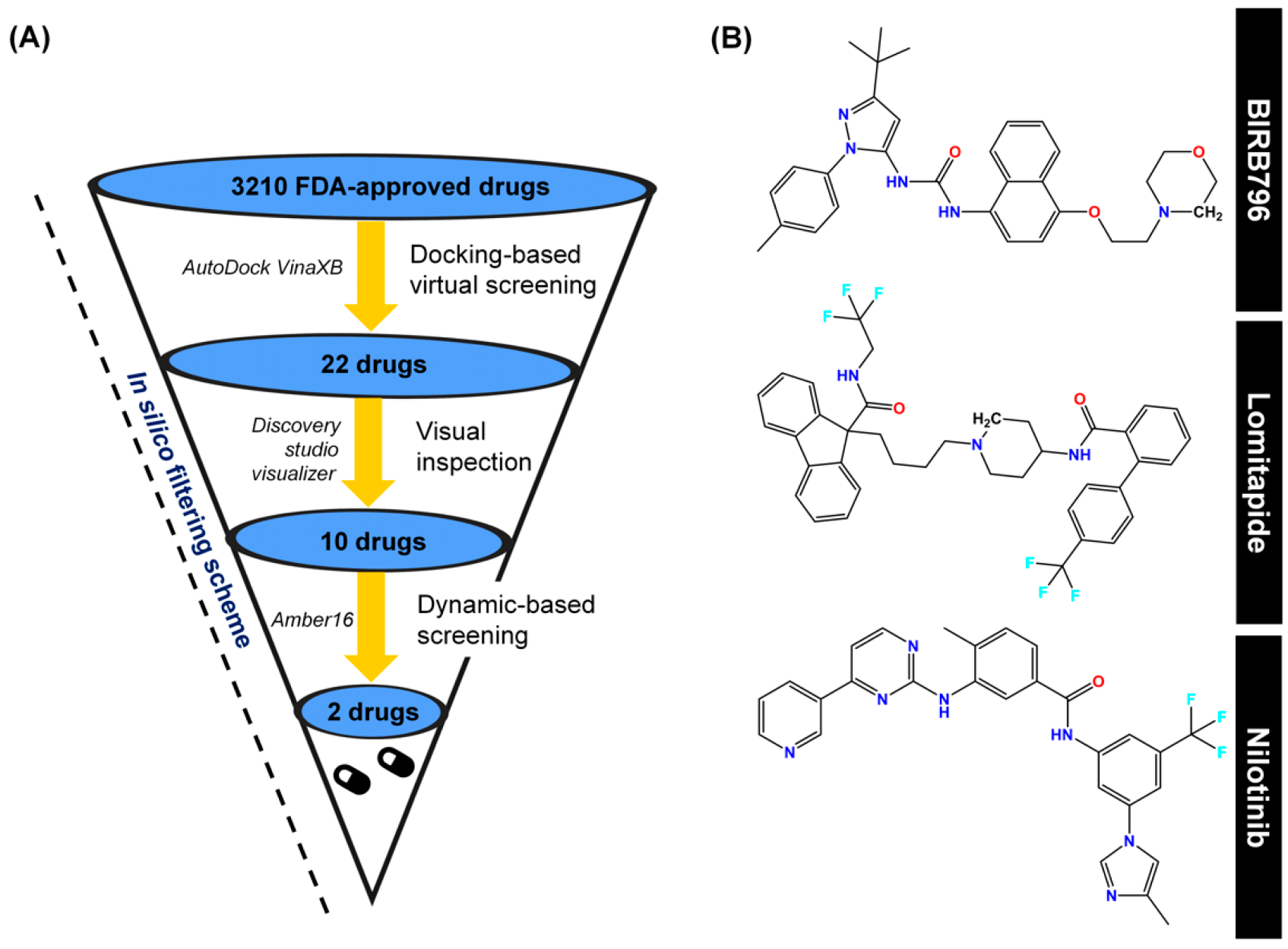
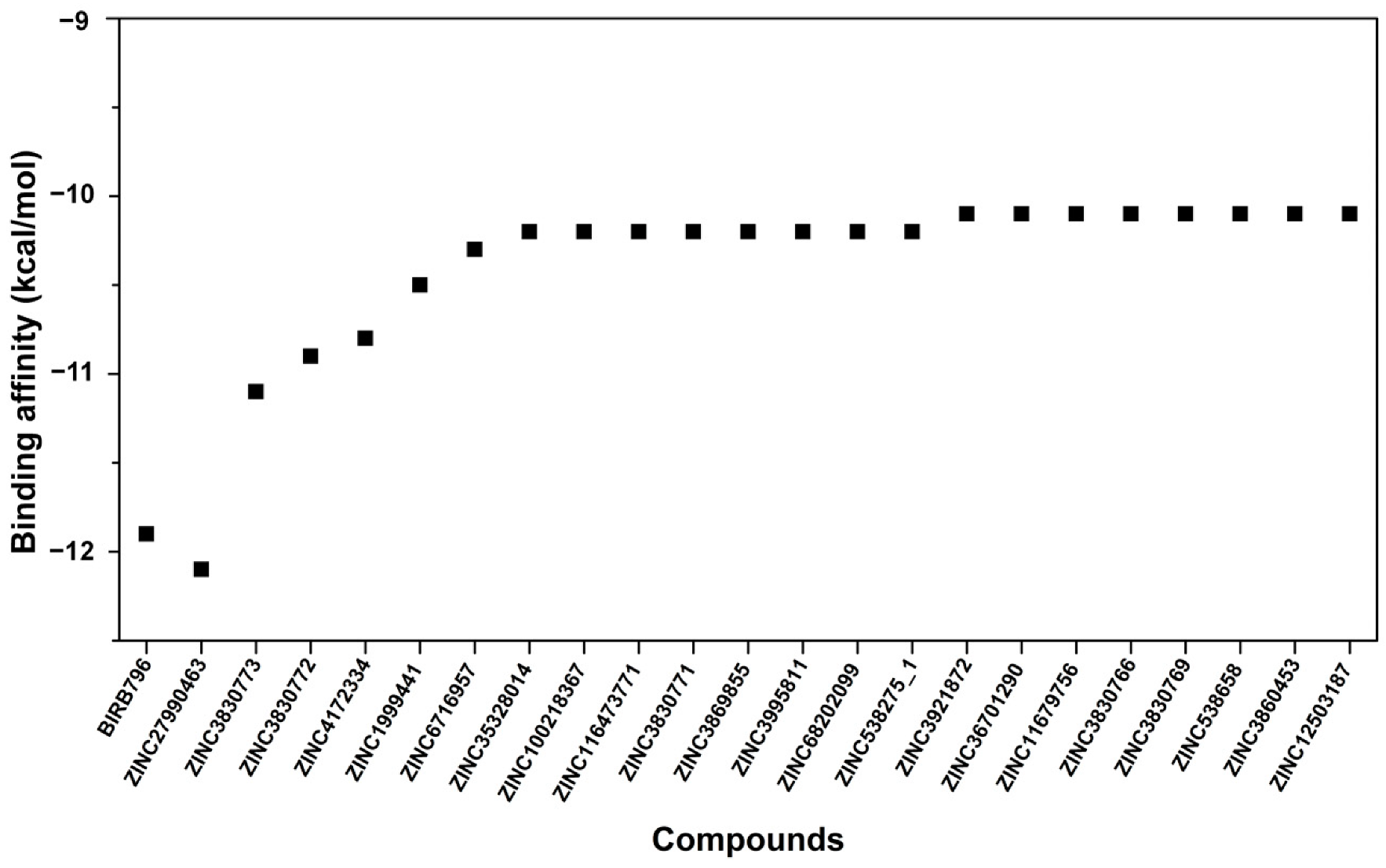
3.1. Docking-Based Screening and Visual Inspection
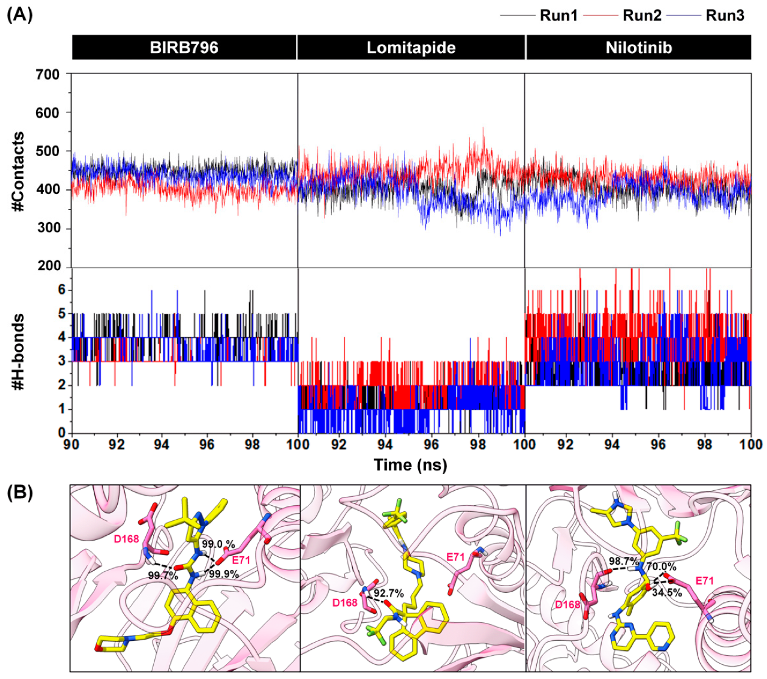
3.2. Dynamic-Based Screening and End-Point Binding Free Energy Calculations
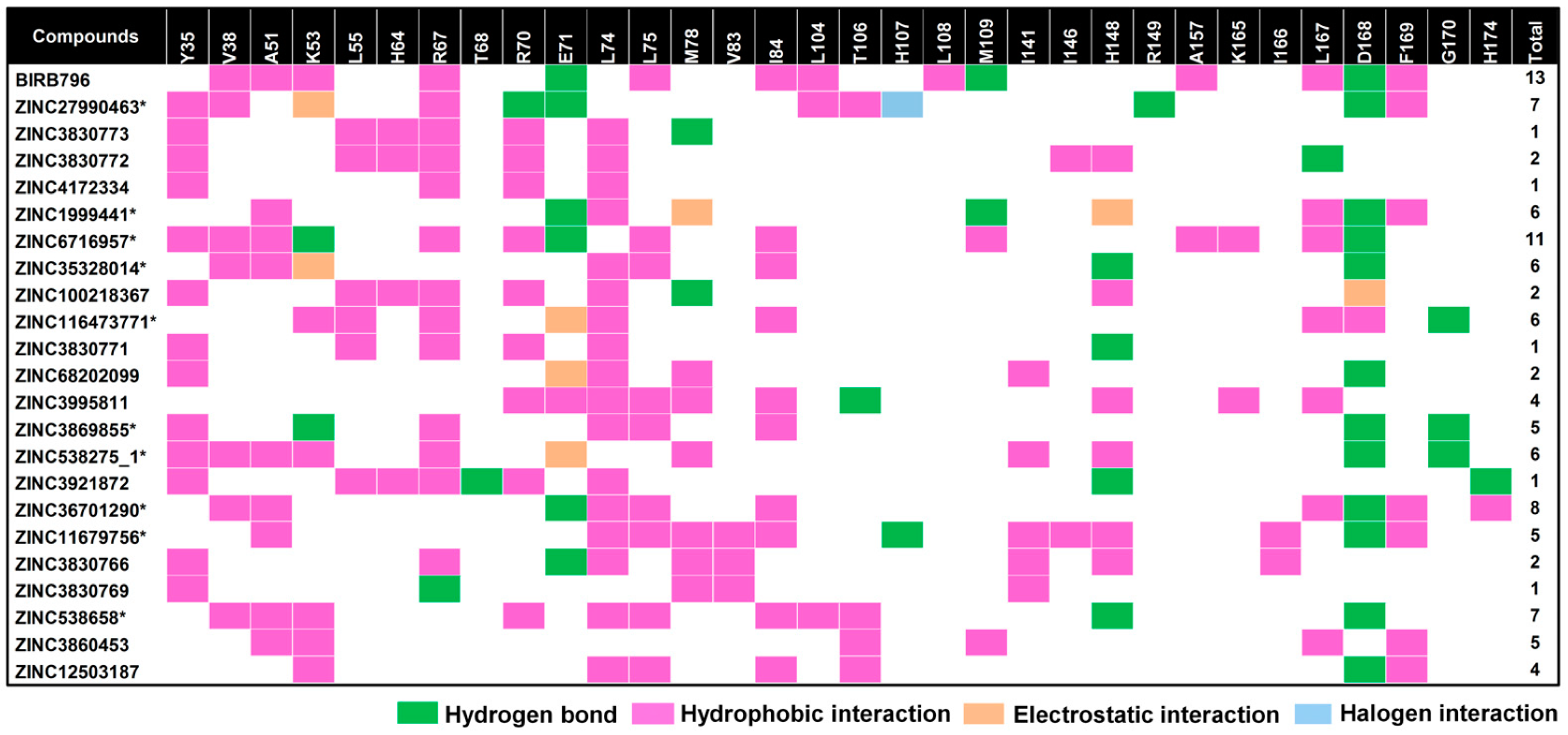
3.3. Contact Atoms and Numbers of Hydrogen Bond Formation
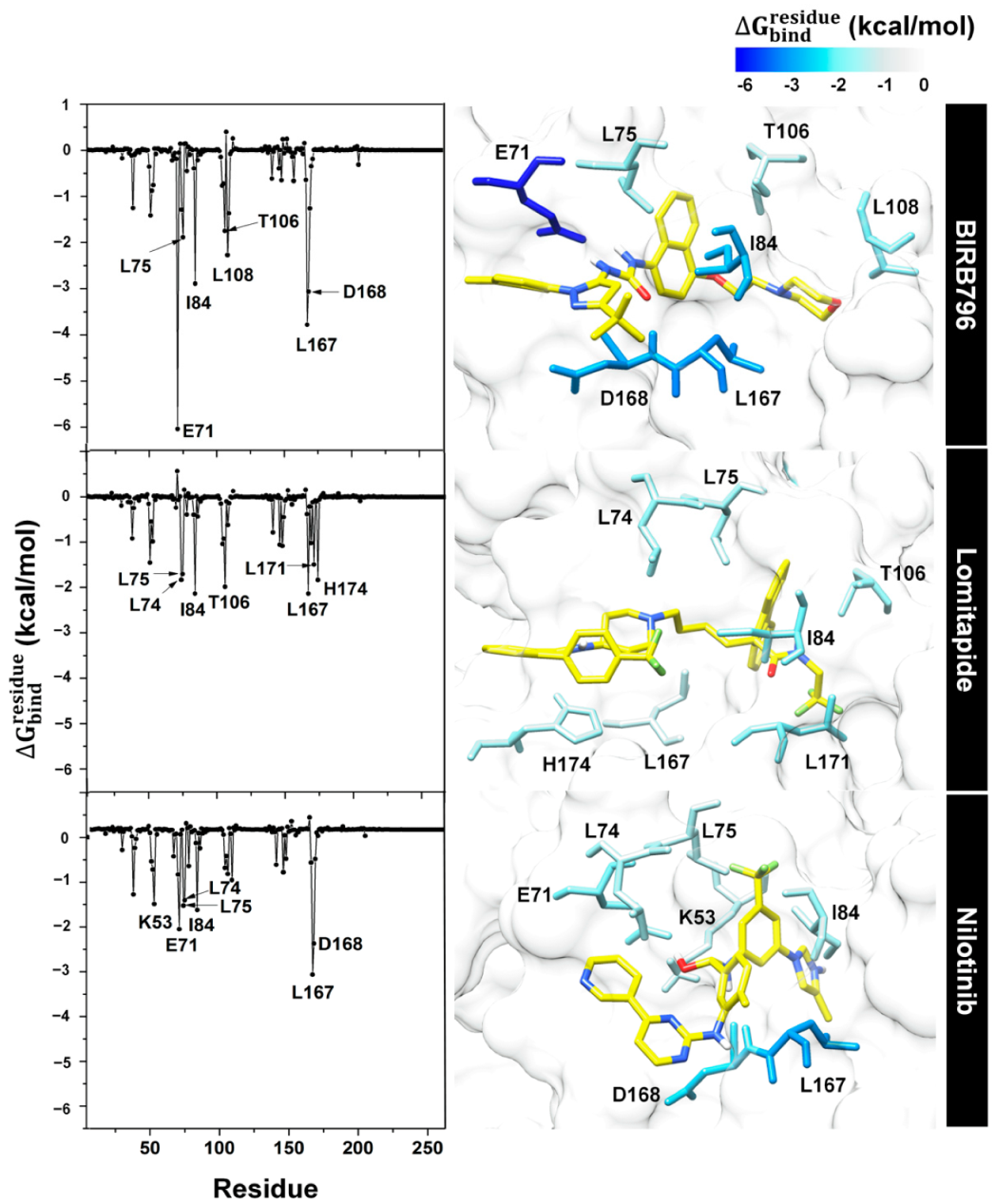
3.4. Key Binding Residues
3.5. QM-Based ONIOM Binding Energy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cargnello, M.; Roux, P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astolfi, A.; Manfroni, G.; Cecchetti, V.; Barreca, M. A comprehensive structural overview of p38α mitogen-activated protein kinase in complex with ATP-Site and non-ATP-site binders. ChemMedChem 2018, 13, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK signaling in cancer: Mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Boehm, J.; Lee, J. P38 MAP kinases: Key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2003, 2, 717–726. [Google Scholar] [CrossRef]
- Schett, G.; Zwerina, J.; Firestein, G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 909–916. [Google Scholar] [CrossRef] [Green Version]
- Dolado, I.; Swat, A.; Ajenjo, N.; De Vita, G.; Cuadrado, A.; Nebreda, A. P38α MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 2007, 11, 191–205. [Google Scholar] [CrossRef] [Green Version]
- Igea, A.; Nebreda, A. The stress kinase p38α as a target for cancer therapy. Cancer Res. 2015, 75, 3997. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Choi, E. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Grimes, J.; Grimes, K. P38 MAPK inhibition: A promising therapeutic approach for COVID-19. J. Mol. Cell. Cardiol. 2020, 144, 63–65. [Google Scholar] [CrossRef]
- Canovas, B.; Nebreda, A. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef]
- Haller, V.; Nahidino, P.; Forster, M.; Laufer, S. An updated patent review of p38 MAP kinase inhibitors (2014–2019). Expert Opin. Ther. Pat. 2020, 30, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Pargellis, C.; Tong, L.; Churchill, L.; Cirillo, P.; Gilmore, T.; Graham, A.; Grob, P.; Hickey, E.; Moss, N.; Pav, S.; et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 2002, 9, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Grant, S. Therapeutic protein kinase inhibitors. Cell. Mol. Life Sci. 2009, 66, 1163–1177. [Google Scholar] [CrossRef]
- Schindler, J.; Monahan, J.; Smith, W. p38 pathway kinases as anti-inflammatory drug targets. J. Dent. Res. 2007, 86, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Damjanov, N.; Kauffman, R.; Spencer-Green, G. Safety and Efficacy of VX-702, a p38 MAP Kinase Inhibitor, in Rheumatoid arthritis. OP-0246 European League Against Rheumatism. In Proceedings of the Annual Congress, Paris, France, 21 September 2008; pp. 11–14. [Google Scholar]
- Matthew, R.; Celia, D. MAP kinase p38 inhibitors: Clinical results and an intimate look at their interactions with p38alpha protein. Curr. Med. Chem. 2005, 12, 2979–2994. [Google Scholar]
- Sweeney, S.; Firestein, G. Mitogen activated protein kinase inhibitors: Where are we now and where are we going? Ann. Rheum. Dis. 2006, 65, 83. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.; Beer, T.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.; MacArthur, M.; Moss, D.; Thornton, J. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Olsson, M.; Søndergaard, C.; Rostkowski, M.; Jensen, J. Computation, PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 Revision D. 01, Gaussian Inc. 2009. Volume 112. Available online: http://www.gaussian.com (accessed on 31 January 2020).
- Wang, J.; Wang, W.; Kollman, P.; Case, D. ANTECHAMBER: An accessory software package for molecular mechanical calculations. J. Chem. Inf. Comput. Sci.—JCISD 2000, 222, U403. [Google Scholar]
- Koebel, M.R.; Schmadeke, G.; Posner, R.; Sirimulla, S. AutoDock VinaXB: Implementation of XBSF, new empirical halogen bond scoring function, into AutoDock Vina. J. Cheminform. 2016, 8, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanachai, K.; Mahalapbutr, P.; Sanghiran Lee, V.; Rungrotmongkol, T.; Hannongbua, S. In silico elucidation of potent inhibitors and rational drug design against SARS-CoV-2 Papain-like protease. J. Phys. Chem. B 2021, 125, 13644–13656. [Google Scholar] [CrossRef] [PubMed]
- Verma, K.; Mahalapbutr, P.; Suriya, U.; Somboon, T.; Aiebchun, T.; Shi, L.; Maitarad, P.; Rungrotmongkol, T. In silico screening of DNA gyrase B potent flavonoids for the treatment of clostridium difficile infection from phytoHub database. Braz. Arch. Biol. Technol. 2021, 64. [Google Scholar] [CrossRef]
- Sripattaraphan, A.; Sanachai, K.; Chavasiri, W.; Boonyasuppayakorn, S.; Maitarad, P.; Rungrotmongkol, T. Computational screening of newly designed compounds against coxsackievirus A16 and enterovirus A71. Molecules 2022, 27, 1908. [Google Scholar] [CrossRef]
- Thirunavukkarasu, M.; Suriya, U.; Rungrotmongkol, T.; Karuppasamy, R. In silico screening of available drugs targeting non-small cell lung cancer targets: A drug repurposing approach. Pharmaceutics 2022, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Mahalapbutr, P.; Wonganan, P.; Chavasiri, W.; Rungrotmongkol, T. Butoxy mansonone G inhibits STAT3 and akt signaling pathways in non-small cell lung cancers: Combined experimental and theoretical investigations. Cancers 2019, 11, 437. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.; Caldwell, J.; Kollman, P.; Case, D. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An n⋅ log (N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.; Ciccotti, G.; Berendsen, H. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Uberuaga, B.; Anghel, M.; Voter, A. Synchronization of trajectories in canonical molecular-dynamics simulations: Observation, explanation, and exploitation. J. Chem. Phys. 2004, 120, 6363–6374. [Google Scholar] [CrossRef]
- Berendsen, H.; Postma, J.; van Gunsteren, W.; DiNola, A.; Haak, J. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.; Klein, M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Naïm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated interaction energy (SIE) for scoring protein−ligand binding affinities. 1. exploring the parameter space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef]
- Dapprich, S.; Komáromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. THEOCHEM 1999, 461–462, 1–21. [Google Scholar] [CrossRef]
- Vreven, T.; Morokuma, K. Chapter 3 hybrid methods: ONIOM(QM:MM) and QM/MM. In Annual Reports in Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2006; pp. 35–51. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Li, Q.; Gusarov, S.; Evoy, S.; Kovalenko, A. Electronic structure, binding energy, and solvation structure of the streptavidin−biotin supramolecular complex: ONIOM and 3D-RISM study. J. Phys. Chem. B 2009, 113, 9958–9967. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M. Decision making in structure-based drug discovery: Visual inspection of docking results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef]
- Yang, Y.; Shen, Y.; Liu, H.; Yao, X. Molecular dynamics simulation and free energy calculation studies of the binding mechanism of allosteric inhibitors with p38α MAP kinase. J. Chem. Inf. Model. 2011, 51, 3235–3246. [Google Scholar] [CrossRef]
- Contreras, O.; Villarreal, M.; Brandan, E. Nilotinib impairs skeletal myogenesis by increasing myoblast proliferation. Skeletal Muscle 2018, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Chung, F.; Yang, C. Molecular modeling of p38α mitogen-activated protein kinase inhibitors through 3D-QSAR and molecular dynamics simulations. J. Chem. Inf. Model. 2013, 53, 1775–1786. [Google Scholar] [CrossRef]
- Schneidman, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H. Pharmagist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [Green Version]
- Maier, S.; Thapa, B.; Erickson, J.; Raghavachari, K. Comparative assessment of QM-based and MM-based models for prediction of protein–ligand binding affinity trends. Phys. Chem. Chem. Phys. 2022, 24, 14525–14537. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs (ZINC ID) | Energy Components (kcal/mol) | ||||
|---|---|---|---|---|---|
| EVdW | Ecoul | ΔGRF | ΔGcavity | ΔGbind | |
| BIRB796 | experiment | −10.98 * | |||
| −78.53 ± 0.29 | −9.93 ± 0.15 | 15.60 ± 0.20 | −13.63 ± 0.04 | −11.95 ± 0.04 | |
| Lomitapide (ZINC27990463) | −77.77 ± 0.39 | −5.95 ± 0.18 | 17.01 ± 0.24 | −14.43 ± 0.04 | −11.39 ± 0.05 |
| Nebivolol (ZINC1999441) | −49.57 ± 0.29 | −12.66 ± 0.26 | 12.78 ± 0.21 | −10.19 ± 0.03 | −9.14 ± 0.03 |
| Nilotinib (ZINC6716957) | −69.53 ± 0.35 | −13.04 ± 0.17 | 15.54 ± 0.19 | −12.35 ± 0.03 | −11.21 ± 0.04 |
| Ibrutinib (ZINC35328014) | −61.87 ± 0.29 | −4.07 ± 0.17 | 13.21 ± 0.23 | −10.81 ± 0.04 | −9.55 ± 0.04 |
| Atovaquone (ZINC116473771) | −42.15 ± 0.27 | −2.43 ± 0.39 | 10.94 ± 0.23 | −7.90 ± 0.05 | −7.24 ± 0.03 |
| Dicumarol (ZINC3869855) | −39.68 ± 0.26 | −13.87 ± 0.40 | 20.55 ± 0.32 | −7.13 ± 0.03 | −7.09 ± 0.03 |
| Raloxifene (ZINC538275) | −51.19 ± 0.44 | −12.45 ± 0.50 | 18.50 ± 0.25 | −9.90 ± 0.05 | −8.66 ± 0.07 |
| Ponatinib (ZINC36701290) | −61.66 ± 0.25 | −4.92 ± 0.14 | 16.99 ± 0.24 | −12.08 ± 0.05 | −9.35 ± 0.03 |
| Eltrombopag (ZINC11679756) | −59.44 ± 0.33 | −5.39 ± 0.17 | 10.52 ± 0.17 | −10.97 ± 0.03 | −9.73 ± 0.04 |
| Samsca (ZINC538658) | −51.69 ± 0.26 | −16.63 ± 0.21 | 19.86 ± 0.18 | −10.38 ± 0.04 | −9.05 ± 0.03 |
| Drugs | Energy Terms | |||
|---|---|---|---|---|
(a.u.) | (a.u.) | (a.u.) | (kcal/mol) | |
| BIRB796 | −1706.396 | −3.391 | −1702.927 | −48.536 |
| Lomitapide | −2425.571 | −3.303 | −2422.203 | −41.159 |
| Nilotinib | −1841.536 | −3.240 | −1838.230 | −41.048 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suriya, U.; Mahalapbutr, P.; Rungrotmongkol, T. Integration of In Silico Strategies for Drug Repositioning towards P38α Mitogen-Activated Protein Kinase (MAPK) at the Allosteric Site. Pharmaceutics 2022, 14, 1461. https://doi.org/10.3390/pharmaceutics14071461
Suriya U, Mahalapbutr P, Rungrotmongkol T. Integration of In Silico Strategies for Drug Repositioning towards P38α Mitogen-Activated Protein Kinase (MAPK) at the Allosteric Site. Pharmaceutics. 2022; 14(7):1461. https://doi.org/10.3390/pharmaceutics14071461
Chicago/Turabian StyleSuriya, Utid, Panupong Mahalapbutr, and Thanyada Rungrotmongkol. 2022. "Integration of In Silico Strategies for Drug Repositioning towards P38α Mitogen-Activated Protein Kinase (MAPK) at the Allosteric Site" Pharmaceutics 14, no. 7: 1461. https://doi.org/10.3390/pharmaceutics14071461
APA StyleSuriya, U., Mahalapbutr, P., & Rungrotmongkol, T. (2022). Integration of In Silico Strategies for Drug Repositioning towards P38α Mitogen-Activated Protein Kinase (MAPK) at the Allosteric Site. Pharmaceutics, 14(7), 1461. https://doi.org/10.3390/pharmaceutics14071461