
New Strategies for Volume Control in Patients with Diabetes Mellitus, a Narrative Review

 ,
,  ,
,  , , and
, , and
Abstract
:
1. Introduction
2. Rationale for Diuretics in Diabetes Mellitus
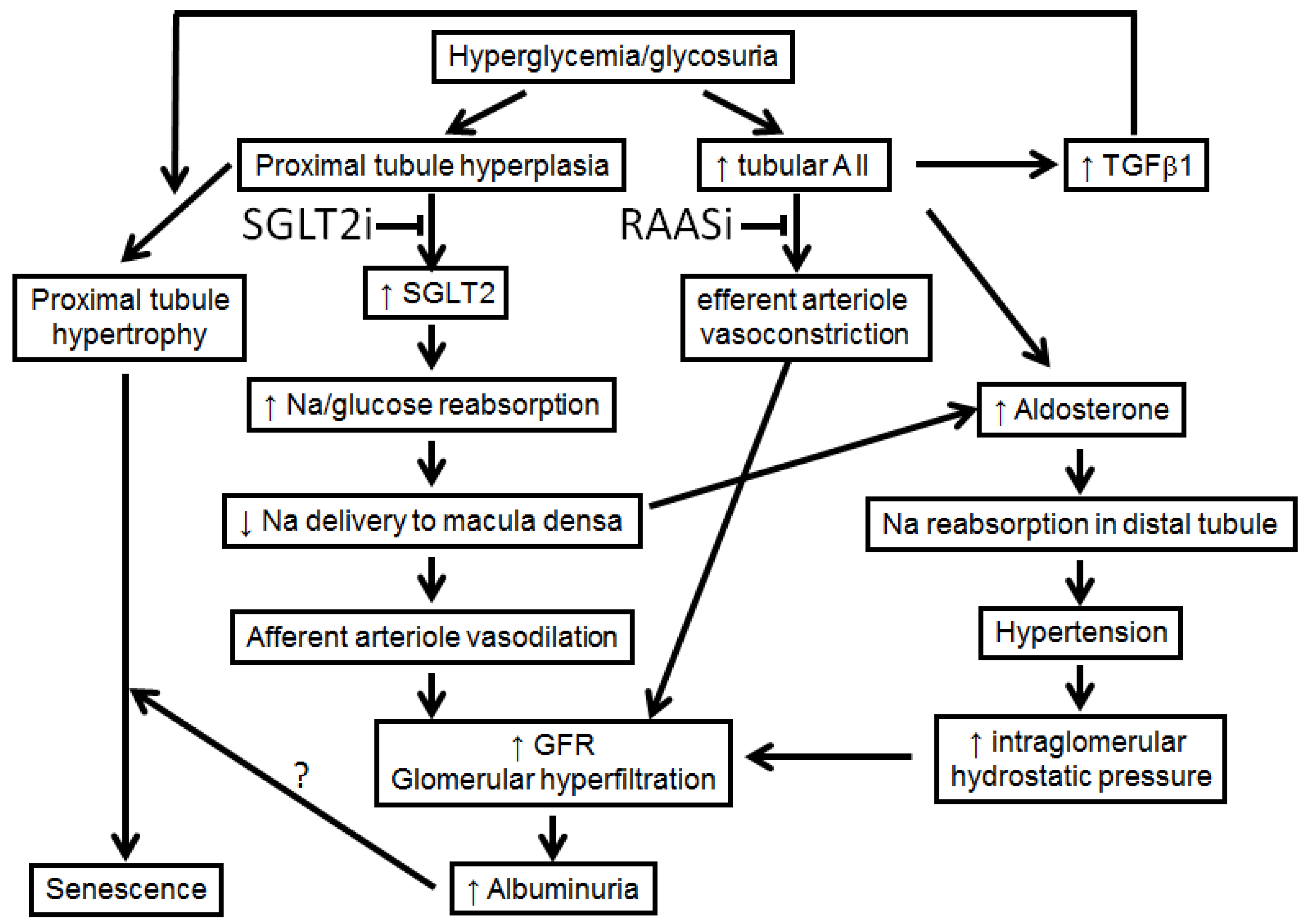
2.1. Sodium Retention in Diabetes Mellitus
2.2. Limitations of Monotherapies
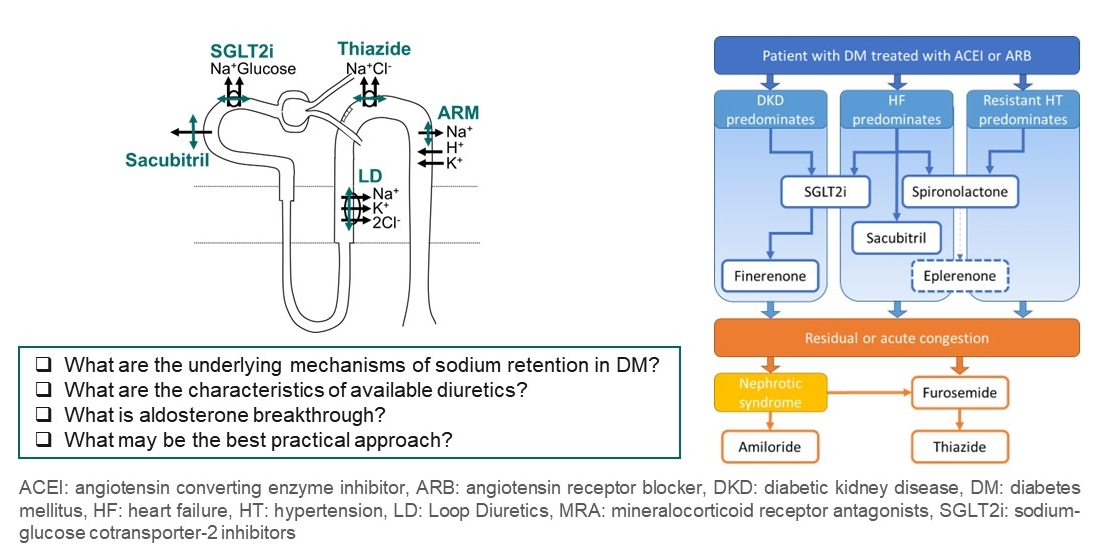
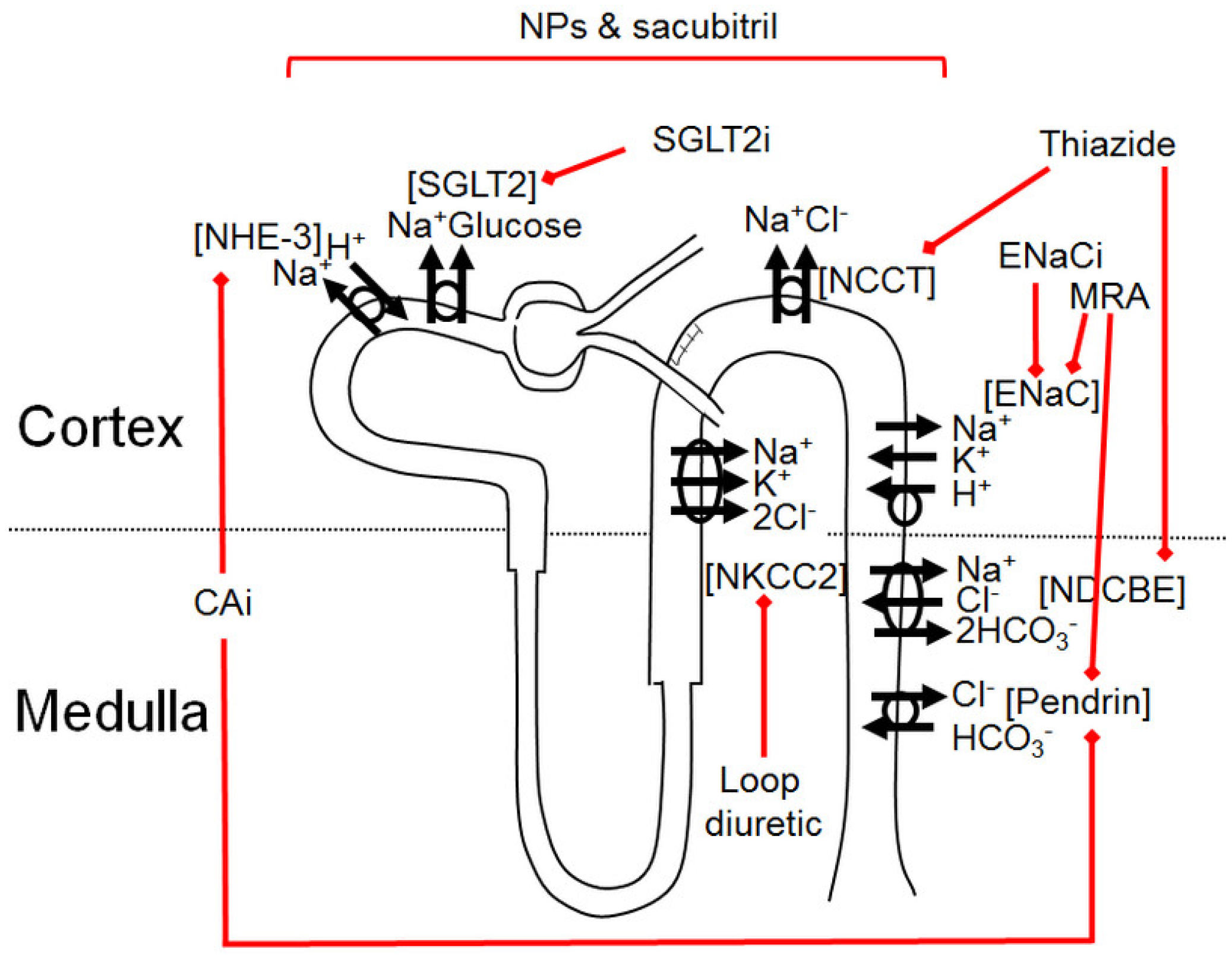
3. Available Natriuretic Drugs and Synergies
3.1. Carbonic Anhydrase Inhibitors
3.2. Sodium-Glucose Cotransporter-2 Inhibitors (SGLT2i)
3.3. Loop Diuretics
- (i)
- The short half-life of LD underlies the acute braking phenomenon, [41] with a decline in the filtered amount of sodium and an increase in proximal sodium reabsorption through glomerulotubular balance.
- (ii)
- After several days, sodium depletion and rising prostacyclins activate RAAS. LD inhibit NKCC2 on macula densa, resulting in the inhibition of the tubuloglomerular feedback, [42,43] preventing afferent arteriole vasoconstriction. This may impair nephroprotection in patients with T2D when LD are used alone. RAAS inhibitors can prevent these important mechanisms of LD resistance and deleterious neuroendocrine activation [44]. Conversely, LD are synergistic with all drugs blocking RAAS, enhancing their antiproteinuric effect through sodium depletion [8,45].
- (iii)
- The long-term effect of LD is further chronically limited by the compensatory hypertrophy of early distal convoluted tubule. Thus, LD synergy with thiazides is expected. Thiazides limit the enhanced distal reabsorption of sodium and the prolong LD effect with their longer half-life, as a perfect example of sequential nephron blockade. However, the association of LD and thiazides exposes patients to a risk of hypokaiemia, which can be prevented by a triple combination with a potassium-sparing diuretic, especially since the activation of thiazide-sensitive NCC during LD treatment is partially aldosterone-induced [42,43].
3.4. Thiazide Diuretics
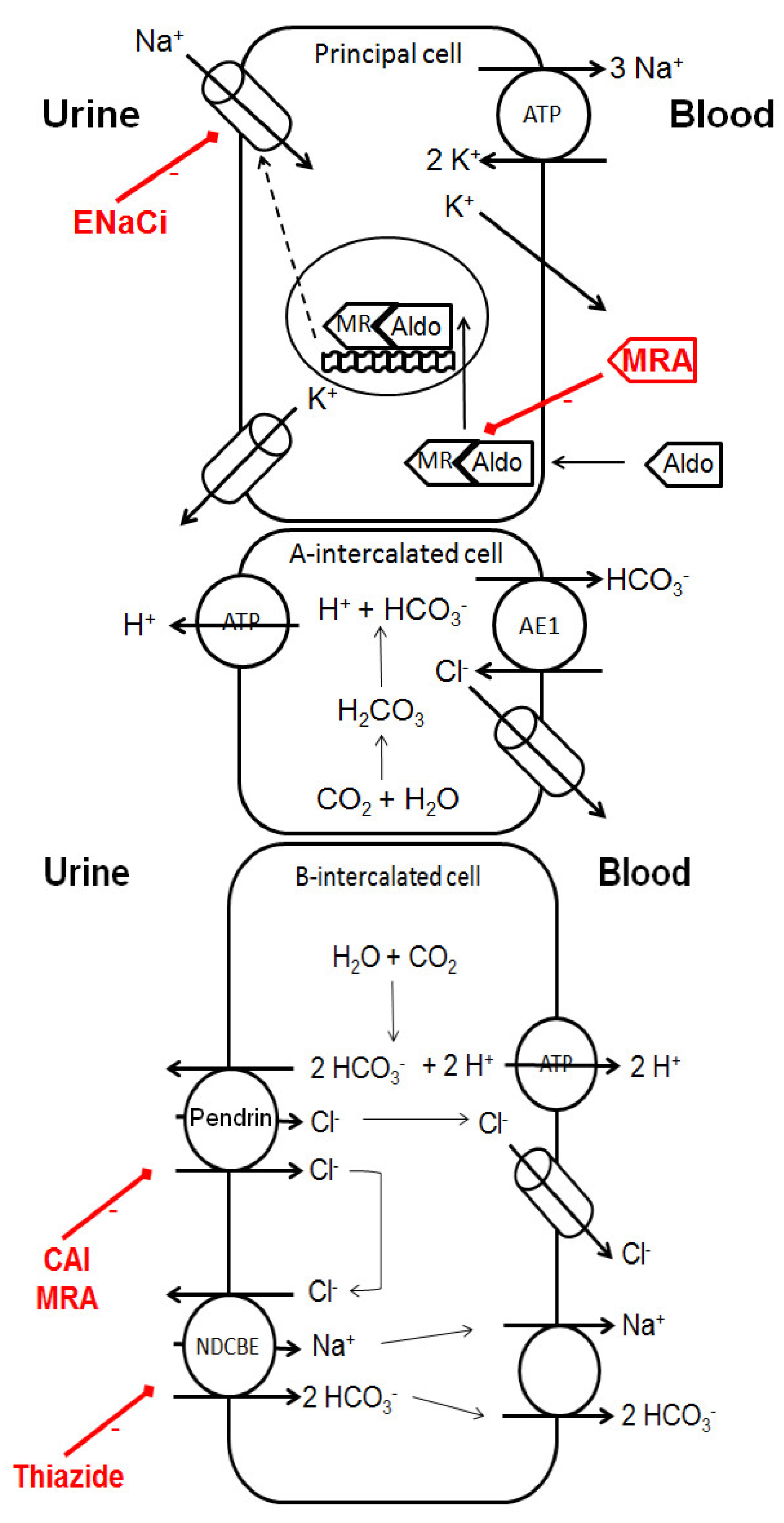
3.5. Mineralocorticoid Receptor Antagonists (MRA)
3.6. ENaC Inhibitors
3.7. Natriuretic Peptides and Angiotensin Receptor-Neprilysin Inhibitor (ARNI)
3.8. Vasopressin Receptor Antagonist
4. Aldosterone Breakthrough
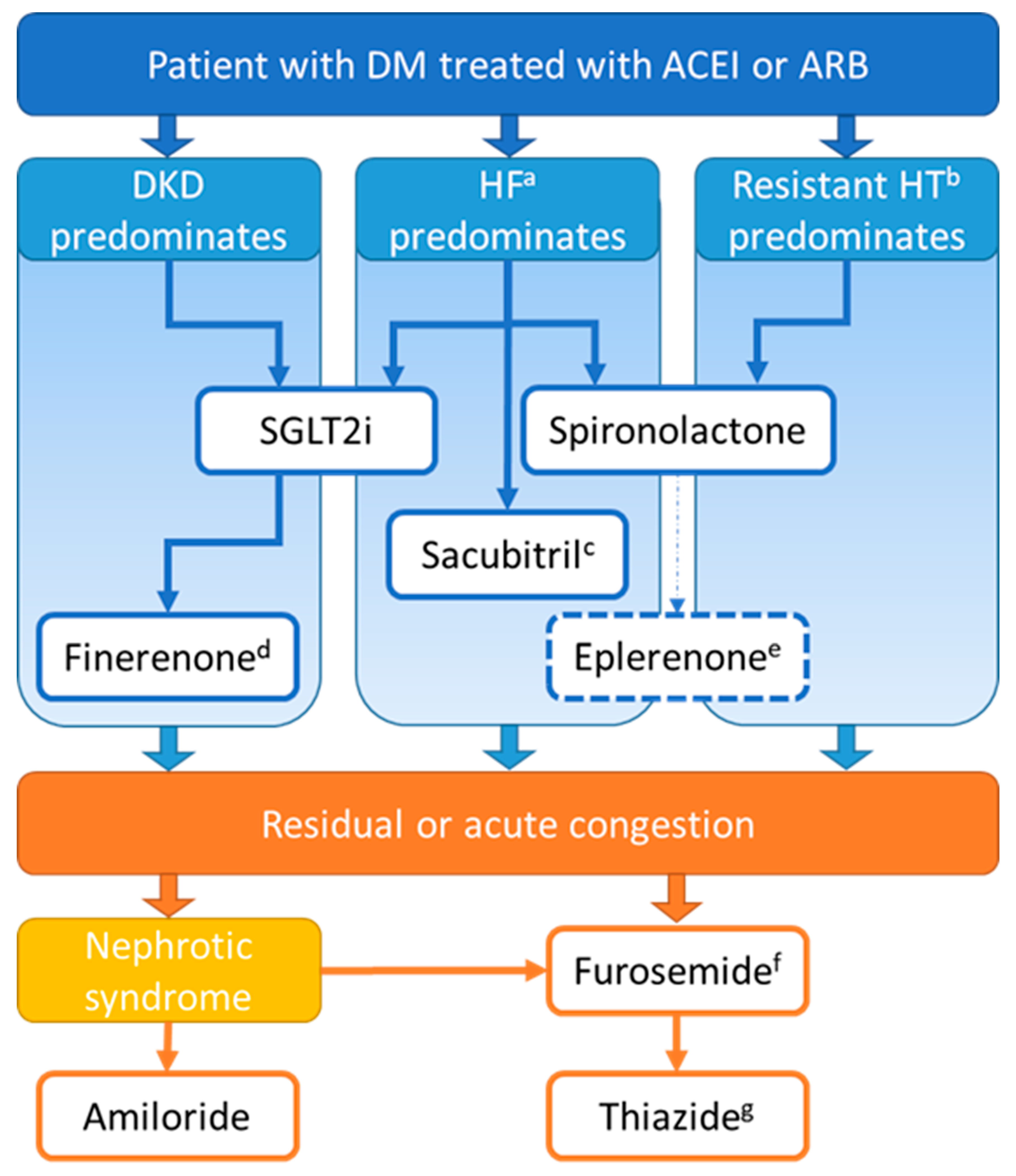
5. Practical Considerations
5.1. Preferential Indications of Each Diuretic
5.2. Safe Prescription
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Afkarian, M.; Zelnick, L.R.; Hall, Y.N.; Heagerty, P.J.; Tuttle, K.; Weiss, N.S.; de Boer, I.H. Clinical Manifestations of Kidney Disease Among US Adults With Diabetes, 1988-2014. JAMA 2016, 316, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Bommer, C.; Sagalova, V.; Heesemann, E.; Manne-Goehler, J.; Atun, R.; Bärnighausen, T.; Davies, J.; Vollmer, S. Global Economic Burden of Diabetes in Adults: Projections From 2015 to 2030. Diabetes Care 2018, 41, 963–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunwald, E. Diabetes, heart failure, and renal dysfunction: The vicious circles. Prog Cardiovasc. Dis. 2019, 62, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [Green Version]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, H.; Caravaca-Fontan, F.; Caro, J.; Morales, E.; Praga, M. The Forgotten Antiproteinuric Properties of Diuretics. Am. J. Nephrol. 2021, 52, 435–449. [Google Scholar] [CrossRef]
- Esnault, V.L.; Ekhlas, A.; Nguyen, J.M.; Moranne, O. Diuretic uptitration with half dose combined ACEI + ARB better decreases proteinuria than combined ACEI + ARB uptitration. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. -Eur. Ren. Assoc. 2010, 25, 2218–2224. [Google Scholar] [CrossRef] [Green Version]
- Asleh, R.; Sheikh-Ahmad, M.; Briasoulis, A.; Kushwaha, S.S. The influence of anti-hyperglycemic drug therapy on cardiovascular and heart failure outcomes in patients with type 2 diabetes mellitus. Heart Fail. Rev. 2018, 23, 445–459. [Google Scholar] [CrossRef]
- Clar, C.; Gill, J.A.; Court, R.; Waugh, N. Systematic review of SGLT2 receptor inhibitors in dual or triple therapy in type 2 diabetes. BMJ Open 2012, 2, e001007. [Google Scholar] [CrossRef]
- Teo, Y.H.; Teo, Y.N.; Syn, N.L.; Kow, C.S.; Yoong, C.S.Y.; Tan, B.Y.Q.; Yeo, T.C.; Lee, C.H.; Lin, W.; Sia, C.H. Effects of Sodium/Glucose Cotransporter 2 (SGLT2) Inhibitors on Cardiovascular and Metabolic Outcomes in Patients Without Diabetes Mellitus: A Systematic Review and Meta-Analysis of Randomized-Controlled Trials. J. Am. Heart Assoc. 2021, 10, e019463. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Sattar, N.; McGuire, D.K. Pathways to Cardiorenal Complications in Type 2 Diabetes Mellitus: A Need to Rethink. Circulation 2018, 138, 7–9. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin-Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef] [Green Version]
- Moranne, O.; Bakris, G.; Fafin, C.; Favre, G.; Pradier, C.; Esnault, V.L. Determinants and changes associated with aldosterone breakthrough after angiotensin II receptor blockade in patients with type 2 diabetes with overt nephropathy. Clin. J. Am. Soc. Nephrol. 2013, 8, 1694–1701. [Google Scholar] [CrossRef] [Green Version]
- Effectiveness of spironolactone added to an angiotensin-converting enzyme inhibitor and a loop diuretic for severe chronic congestive heart failure (the Randomized Aldactone Evaluation Study [RALES]). Am. J. Cardiol. 1996, 78, 902–907. [CrossRef]
- Pitt, B.; Williams, G.; Remme, W.; Martinez, F.; Lopez-Sendon, J.; Zannad, F.; Neaton, J.; Roniker, B.; Hurley, S.; Burns, D.; et al. The EPHESUS trial: Eplerenone in patients with heart failure due to systolic dysfunction complicating acute myocardial infarction. Eplerenone Post-AMI Heart Failure Efficacy and Survival Study. Cardiovasc. Drugs Ther. 2001, 15, 79–87. [Google Scholar] [CrossRef]
- Zannad, F.; McMurray, J.J.; Krum, H.; van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B.; Group, E.-H.S. Eplerenone in patients with systolic heart failure and mild symptoms. N. Engl. J. Med. 2011, 364, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N.Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef]
- Weidmann, P.; Ferrari, P. Central role of sodium in hypertension in diabetic subjects. Diabetes Care 1991, 14, 220–232. [Google Scholar] [CrossRef]
- Saudek, C.D.; Boulter, P.R.; Knopp, R.H.; Arky, R.A. Sodium retention accompanying insulin treatment of diabetes mellitus. Diabetes 1974, 23, 240–246. [Google Scholar] [CrossRef]
- Reaven, G.M.; Lithell, H.; Landsberg, L. Hypertension and associated metabolic abnormalities--the role of insulin resistance and the sympathoadrenal system. N. Engl. J. Med. 1996, 334, 374–381. [Google Scholar] [CrossRef]
- Rocchini, A.P.; Moorehead, C.; DeRemer, S.; Goodfriend, T.L.; Ball, D.L. Hyperinsulinemia and the aldosterone and pressor responses to angiotensin II. Hypertension 1990, 15, 861–866. [Google Scholar] [CrossRef] [Green Version]
- Mandon, B.; Siga, E.; Chabardes, D.; Firsov, D.; Roinel, N.; De Rouffignac, C. Insulin stimulates Na+, Cl-, Ca2+, and Mg2+ transports in TAL of mouse nephron: Cross-potentiation with AVP. Am. J. Physiol. 1993, 265, F361–F369. [Google Scholar] [CrossRef]
- Nicco, C.; Wittner, M.; DiStefano, A.; Jounier, S.; Bankir, L.; Bouby, N. Chronic exposure to vasopressin upregulates ENaC and sodium transport in the rat renal collecting duct and lung. Hypertension 2001, 38, 1143–1149. [Google Scholar] [CrossRef] [Green Version]
- Kannenkeril, D.; Karg, M.V.; Bosch, A.; Ott, C.; Linz, P.; Nagel, A.M.; Uder, M.; Schmieder, R.E. Tissue sodium content in patients with type 2 diabetes mellitus. J. Diabetes Complicat. 2019, 33, 485–489. [Google Scholar] [CrossRef]
- Officers, A.; Coordinators for the, A.C.R.G.T.A.; Lipid-Lowering Treatment to Prevent Heart Attack, T. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Jama 2002, 288, 2981–2997. [Google Scholar] [CrossRef]
- Esnault, V.L. Angiotensin-converting-enzyme inhibitors and diuretics for hypertension. N. Engl. J. Med. 2003, 349, 90–93. [Google Scholar]
- group, P.c. Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack. Lancet 2001, 358, 1033–1041. [Google Scholar]
- Zahedi, K.; Barone, S.; Xu, J.; Soleimani, M. Potentiation of the effect of thiazide derivatives by carbonic anhydrase inhibitors: Molecular mechanisms and potential clinical implications. PLoS ONE 2013, 8, e79327. [Google Scholar] [CrossRef] [Green Version]
- Wall, S.M.; Lazo-Fernandez, Y. The role of pendrin in renal physiology. Annu. Rev. Physiol. 2015, 77, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Baker, W.L.; Smyth, L.R.; Riche, D.M.; Bourret, E.M.; Chamberlin, K.W.; White, W.B. Effects of sodium-glucose co-transporter 2 inhibitors on blood pressure: A systematic review and meta-analysis. J. Am. Soc. Hypertens. JASH 2014, 8, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Nespoux, J.; Vallon, V. SGLT2 inhibition and kidney protection. Clin. Sci. 2018, 132, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin. Pharmacokinet. 2014, 53, 213–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delanaye, P.; Scheen, A.J. The diuretic effects of SGLT2 inhibitors: A comprehensive review of their specificities and their role in renal protection. Diabetes Metab. 2021, 47, 101285. [Google Scholar] [CrossRef] [PubMed]
- Gerard, A.O.; Laurain, A.; Favre, G.; Drici, M.D.; Esnault, V.L. Activation of the tubulo-glomerular feedback by SGLT2 inhibitors in patients with type 2 diabetes and advanced chronic kidney disease: Towards the end of a myth? Diabetes Care 2022, in press. [Google Scholar]
- Vallon, V.; Thomson, S.C. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 317–336. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Kim, Y.C. Protecting the Kidney: The Unexpected Logic of Inhibiting a Glucose Transporter. Clin. Pharmacol. Ther. 2022. [Google Scholar] [CrossRef]
- De Bruyne, L.K. Mechanisms and management of diuretic resistance in congestive heart failure. Postgrad. Med. J. 2003, 79, 268–271. [Google Scholar] [CrossRef] [Green Version]
- Diuretic Treatment in Heart Failure. N. Engl. J. Med. 2018, 378, 492. [CrossRef] [Green Version]
- Ellison, D.H.; Felker, G.M. Diuretic Treatment in Heart Failure. N. Engl. J. Med. 2017, 377, 1964–1975. [Google Scholar] [CrossRef]
- Pellicori, P.; Fitchett, D.; Kosiborod, M.N.; Ofstad, A.P.; Seman, L.; Zinman, B.; Zwiener, I.; Wanner, C.; George, J.; Inzucchi, S.E.; et al. Use of diuretics and outcomes in patients with type 2 diabetes: Findings from the EMPA-REG OUTCOME trial. Eur. J. Heart Fail. 2021, 23, 1085–1093. [Google Scholar] [CrossRef]
- Esnault, V.L.; Ekhlas, A.; Delcroix, C.; Moutel, M.G.; Nguyen, J.M. Diuretic and enhanced sodium restriction results in improved antiproteinuric response to RAS blocking agents. J. Am. Soc. Nephrol. JASN 2005, 16, 474–481. [Google Scholar] [CrossRef]
- Ellison, D.H.; Loffing, J. Thiazide effects and adverse effects: Insights from molecular genetics. Hypertension 2009, 54, 196–202. [Google Scholar] [CrossRef]
- Wagner, C.A. Pendrin-A New Target for Diuretic Therapy? J. Am. Soc. Nephrol. JASN 2016, 27, 3499–3501. [Google Scholar] [CrossRef]
- Fukuda, M.; Kimura, G. Pathophysiology of antihypertensive therapy with diuretics. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2006, 29, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Al-bataineh, M.M.; Pastor-Soler, N.M. Collecting duct intercalated cell function and regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 305–324. [Google Scholar] [CrossRef]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M.; et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Rossignol, P.; Menard, J.; Fay, R.; Gustafsson, F.; Pitt, B.; Zannad, F. Eplerenone survival benefits in heart failure patients post-myocardial infarction are independent from its diuretic and potassium-sparing effects. Insights from an EPHESUS (Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study) substudy. J. Am. Coll. Cardiol. 2011, 58, 1958–1966. [Google Scholar] [CrossRef] [Green Version]
- Gerard, A.O.; Laurain, A.; Esnault, V.L.M. Cardiovascular Events with Finerenone in CKD and Diabetes. N. Engl. J. Med. 2022, 386, e43. [Google Scholar] [CrossRef]
- Epstein, M.; Duprez, D.A. Resistant Hypertension and the Pivotal Role for Mineralocorticoid Receptor Antagonists: A Clinical Update 2016. Am. J. Med. 2016, 129, 661–666. [Google Scholar] [CrossRef] [Green Version]
- Pitt, B.; Kober, L.; Ponikowski, P.; Gheorghiade, M.; Filippatos, G.; Krum, H.; Nowack, C.; Kolkhof, P.; Kim, S.Y.; Zannad, F. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: A randomized, double-blind trial. Eur. Heart J. 2013, 34, 2453–2463. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Staehr, M.; Buhl, K.B.; Andersen, R.F.; Svenningsen, P.; Nielsen, F.; Hinrichs, G.R.; Bistrup, C.; Jensen, B.L. Aberrant glomerular filtration of urokinase-type plasminogen activator in nephrotic syndrome leads to amiloride-sensitive plasminogen activation in urine. Am. J. Physiol. Ren. Physiol. 2015, 309, F235–F241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doucet, A.; Favre, G.; Deschenes, G. Molecular mechanism of edema formation in nephrotic syndrome: Therapeutic implications. Pediatric Nephrol. 2007, 22, 1983–1990. [Google Scholar] [CrossRef] [Green Version]
- Volpe, M.; Carnovali, M.; Mastromarino, V. The natriuretic peptides system in the pathophysiology of heart failure: From molecular basis to treatment. Clin. Sci. 2016, 130, 57–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, F.; Mascolo, A.; Mollace, V. The pathophysiological role of natriuretic peptide-RAAS cross talk in heart failure. Int. J. Cardiol. 2017, 226, 121–125. [Google Scholar] [CrossRef]
- Packer, M.; Claggett, B.; Lefkowitz, M.P.; McMurray, J.J.V.; Rouleau, J.L.; Solomon, S.D.; Zile, M.R. Effect of neprilysin inhibition on renal function in patients with type 2 diabetes and chronic heart failure who are receiving target doses of inhibitors of the renin-angiotensin system: A secondary analysis of the PARADIGM-HF trial. Lancet Diabetes Endocrinol. 2018, 6, 547–554. [Google Scholar] [CrossRef]
- Damman, K.; Gori, M.; Claggett, B.; Jhund, P.S.; Senni, M.; Lefkowitz, M.P.; Prescott, M.F.; Shi, V.C.; Rouleau, J.L.; Swedberg, K.; et al. Renal Effects and Associated Outcomes During Angiotensin-Neprilysin Inhibition in Heart Failure. JACC Heart Fail. 2018, 6, 489–498. [Google Scholar] [CrossRef]
- Peikert, A.; Vaduganathan, M.; Mc Causland, F.; Claggett, B.L.; Chatur, S.; Packer, M.; Pfeffer, M.A.; Zannad, F.; Lefkowitz, M.P.; Pieske, B.; et al. Effects of sacubitril/valsartan versus valsartan on renal function in patients with and without diabetes and heart failure with preserved ejection fraction: Insights from PARAGON-HF. Eur. J. Heart Fail. 2022, 24, 794–803. [Google Scholar] [CrossRef]
- Seferovic, J.P.; Claggett, B.; Seidelmann, S.B.; Seely, E.W.; Packer, M.; Zile, M.R.; Rouleau, J.L.; Swedberg, K.; Lefkowitz, M.; Shi, V.C.; et al. Effect of sacubitril/valsartan versus enalapril on glycaemic control in patients with heart failure and diabetes: A post-hoc analysis from the PARADIGM-HF trial. Lancet Diabetes Endocrinol. 2017, 5, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Seferovic, J.P.; Solomon, S.D.; Seely, E.W. Potential mechanisms of beneficial effect of sacubitril/valsartan on glycemic control. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820970444. [Google Scholar] [CrossRef]
- Knepper, M.A. Molecular physiology of urinary concentrating mechanism: Regulation of aquaporin water channels by vasopressin. Am. J. Physiol. 1997, 272, F3–F12. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S.; et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2012, 367, 2407–2418. [Google Scholar] [CrossRef] [Green Version]
- Kleindienst, A.; Georgiev, S.; Schlaffer, S.M.; Buchfelder, M. Tolvaptan Versus Fluid Restriction in the Treatment of Hyponatremia Resulting from SIADH Following Pituitary Surgery. J. Endocr. Soc. 2020, 4, bvaa068. [Google Scholar] [CrossRef]
- Masuda, T.; Ohara, K.; Nagayama, I.; Matsuoka, R.; Murakami, T.; Nakagawa, S.; Oka, K.; Asakura, M.; Igarashi, Y.; Fukaya, Y.; et al. Impact of serum albumin levels on the body fluid response to tolvaptan in chronic kidney disease patients. Int. Urol. Nephrol. 2019, 51, 1623–1629. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Meijer, E.; Chapman, A.B.; Czerwiec, F.S.; Devuyst, O.; Grantham, J.J.; Higashihara, E.; Krasa, H.B.; Ouyang, J.; Perrone, R.D.; et al. Albuminuria and tolvaptan in autosomal-dominant polycystic kidney disease: Results of the TEMPO 3:4 Trial. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transplant. Assoc.-Eur. Ren. Assoc. 2016, 31, 1887–1894. [Google Scholar] [CrossRef] [Green Version]
- Boertien, W.E.; Meijer, E.; de Jong, P.E.; Bakker, S.J.; Czerwiec, F.S.; Struck, J.; Oberdhan, D.; Shoaf, S.E.; Krasa, H.B.; Gansevoort, R.T. Short-term renal hemodynamic effects of tolvaptan in subjects with autosomal dominant polycystic kidney disease at various stages of chronic kidney disease. Kidney Int. 2013, 84, 1278–1286. [Google Scholar] [CrossRef]
- Bardoux, P.; Bichet, D.G.; Martin, H.; Gallois, Y.; Marre, M.; Arthus, M.F.; Lonergan, M.; Ruel, N.; Bouby, N.; Bankir, L. Vasopressin increases urinary albumin excretion in rats and humans: Involvement of V2 receptors and the renin-angiotensin system. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2003, 18, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Takada, T.; Masaki, T.; Hoshiyama, A.; Toki, T.; Kamata, Y.; Shichiri, M. Tolvaptan alleviates excessive fluid retention of nephrotic diabetic renal failure unresponsive to furosemide. Nephrology 2018, 23, 883–886. [Google Scholar] [CrossRef]
- Konstam, M.A.; Gheorghiade, M.; Burnett, J.C., Jr.; Grinfeld, L.; Maggioni, A.P.; Swedberg, K.; Udelson, J.E.; Zannad, F.; Cook, T.; Ouyang, J.; et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: The EVEREST Outcome Trial. Jama 2007, 297, 1319–1331. [Google Scholar] [CrossRef]
- Bomback, A.S.; Klemmer, P.J. The incidence and implications of aldosterone breakthrough. Nat. Clin. Pract. Nephrol. 2007, 3, 486–492. [Google Scholar] [CrossRef]
- Schjoedt, K.J.; Andersen, S.; Rossing, P.; Tarnow, L.; Parving, H.H. Aldosterone escape during blockade of the renin-angiotensin-aldosterone system in diabetic nephropathy is associated with enhanced decline in glomerular filtration rate. Diabetologia 2004, 47, 1936–1939. [Google Scholar] [CrossRef] [PubMed]
- Fried, L.F.; Emanuele, N.; Zhang, J.H.; Brophy, M.; Conner, T.A.; Duckworth, W.; Leehey, D.J.; McCullough, P.A.; O’Connor, T.; Palevsky, P.M.; et al. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N. Engl. J. Med. 2013, 369, 1892–1903. [Google Scholar] [CrossRef] [Green Version]
- Buse, J.B.; Wexler, D.J.; Tsapas, A.; Rossing, P.; Mingrone, G.; Mathieu, C.; D’Alessio, D.A.; Davies, M.J. 2019 update to: Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2020, 63, 221–228. [Google Scholar] [CrossRef] [Green Version]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Liuzzo, G.; Volpe, M. FIGARO-DKD adds new evidence to the cardiovascular benefits of finerenone across the spectrum of patients with type 2 diabetes and chronic kidney disease. Eur. Heart J. 2021, 42, 4789–4790. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Oh, M.S.; Carroll, H.J. Hyperkalemia in diabetes mellitus. J. Diabet Complicat. 1990, 4, 3–7. [Google Scholar] [CrossRef]
- Hunter, R.W.; Bailey, M.A. Hyperkalemia: Pathophysiology, risk factors and consequences. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2019, 34, iii2–iii11. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subclass | Examples | Main Site of Action | Main Hydroelectrolytic Disorders | Main Other Adverse Drug Reactions |
|---|---|---|---|---|
| Carbonic anhydrase inhibitors | Acetazolamide | PCT | HypoK, hyperUrc | Urolithiasis |
| SGLT2 inhibitors | Dapagliflozin, canagliflozin, empagliflozin | PCT | / | Urogenital infections, euglycemic ketoacidosis |
| Loop diuretics | Furosemide, bumetanide | LH | HypoK, hypoCa, hypoMg, hyperUrc | Hypoacusis |
| Thiazide diuretics | Hydrochlorothiazide, indapamide | DCT | HypoNa, hypoK, hyperCa, hypoMg, hyperUrc | Skin tumors |
| Mineralocorticoid receptor antagonists | Spironolactone, eplerenone, finerenone | CD | HyperK | Gynecomastia |
| ENaC inhibitors | Amiloride | CD | HyperK | / |
| Neprilysin inhibitor | Sacubitril | PCT | (HyperK in association with valsartan) | Angioedema |
| Vasopressin receptor antagonist | Tolvaptan | CD | HyperNa, hyperUrc | Hepatotoxicity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gérard, A.O.; Laurain, A.; Sicard, A.; Merino, D.; Pathak, A.; Drici, M.-D.; Favre, G.; Esnault, V.L.M. New Strategies for Volume Control in Patients with Diabetes Mellitus, a Narrative Review. Pharmaceutics 2022, 14, 1569. https://doi.org/10.3390/pharmaceutics14081569
Gérard AO, Laurain A, Sicard A, Merino D, Pathak A, Drici M-D, Favre G, Esnault VLM. New Strategies for Volume Control in Patients with Diabetes Mellitus, a Narrative Review. Pharmaceutics. 2022; 14(8):1569. https://doi.org/10.3390/pharmaceutics14081569
Chicago/Turabian StyleGérard, Alexandre O., Audrey Laurain, Antoine Sicard, Diane Merino, Atul Pathak, Milou-Daniel Drici, Guillaume Favre, and Vincent L. M. Esnault. 2022. "New Strategies for Volume Control in Patients with Diabetes Mellitus, a Narrative Review" Pharmaceutics 14, no. 8: 1569. https://doi.org/10.3390/pharmaceutics14081569
APA StyleGérard, A. O., Laurain, A., Sicard, A., Merino, D., Pathak, A., Drici, M. -D., Favre, G., & Esnault, V. L. M. (2022). New Strategies for Volume Control in Patients with Diabetes Mellitus, a Narrative Review. Pharmaceutics, 14(8), 1569. https://doi.org/10.3390/pharmaceutics14081569