



Surface Modification of Biodegradable Microparticles with the Novel Host-Derived Immunostimulant CPDI-02 Significantly Increases Short-Term and Long-Term Mucosal and Systemic Antibodies against Encapsulated Protein Antigen in Young Naïve Mice after Respiratory Immunization

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. LPS Removal from Ovalbumin (OVA)
2.2. Synthesis of CGRR-CPDI-02 and CGRR-scCPDI-02 Peptides
2.3. Encapsulation of OVA in Biodegradable Microparticles Surface-Modified with CPDI-02
2.4. Quantitation of OVA Loading in MP by Ultra-Performance Liquid Chromatography (UPLC)
2.5. Quantitation of OVA Burst Release from MP
2.6. Quantitation of CPDI-02 and scCPDI-02 Surface Conjugation to MP by Kexin-Mediated Ultra-Performance Liquid Chromatography (UPLC)
2.7. Diameters and Zeta Potentials of Microparticles
2.8. Animals
2.9. Intranasal Administration
2.10. Isolation of Murine Lung Lymphocytes and Splenocytes
2.11. IgA, IgG, and IgM ELISpot Assays
2.12. Collection of Serum, BALF, and NLF from Mice
2.13. OVA-Specific Antibody Titers of Serum, BALF, and NLF
2.14. Lung Histology
2.15. Statistical Analyses
3. Results
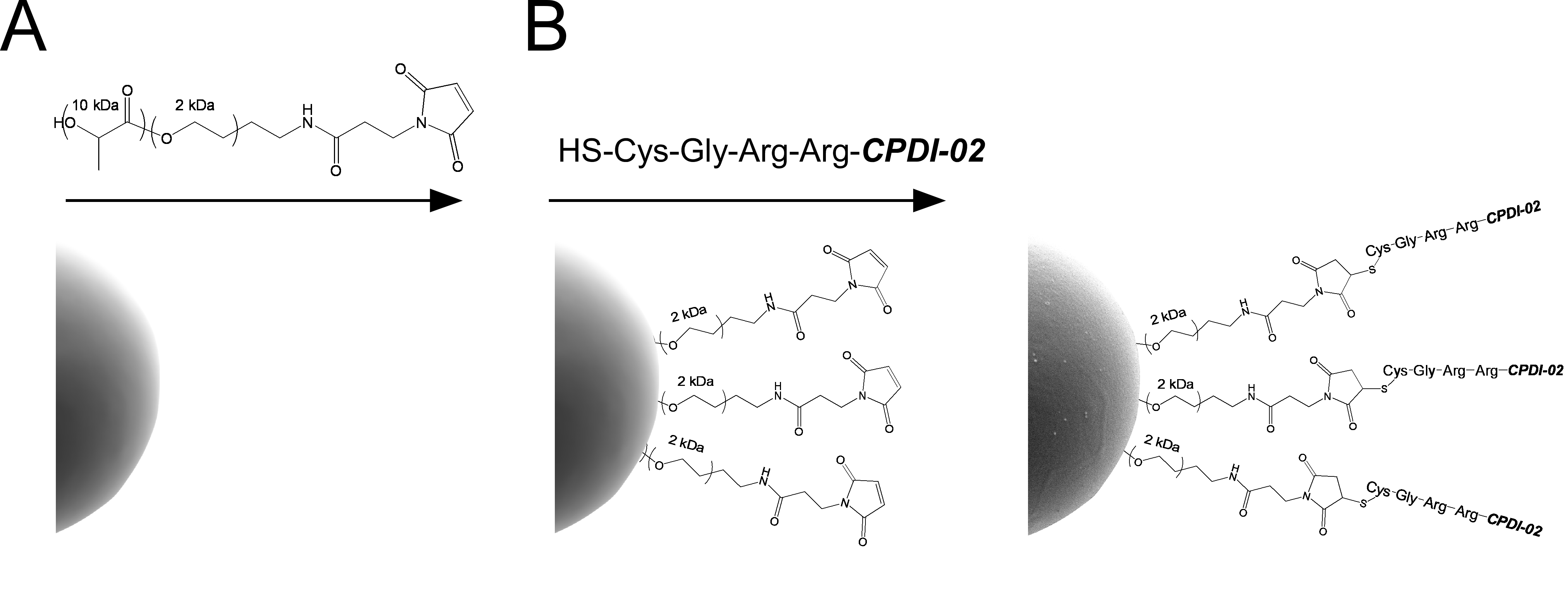
3.1. Encapsulation of LPS-Free OVA in ~1 μm Biodegradable Microparticles Surface-Modified with CPDI-02 or Inactive, Scrambled CPDI-02
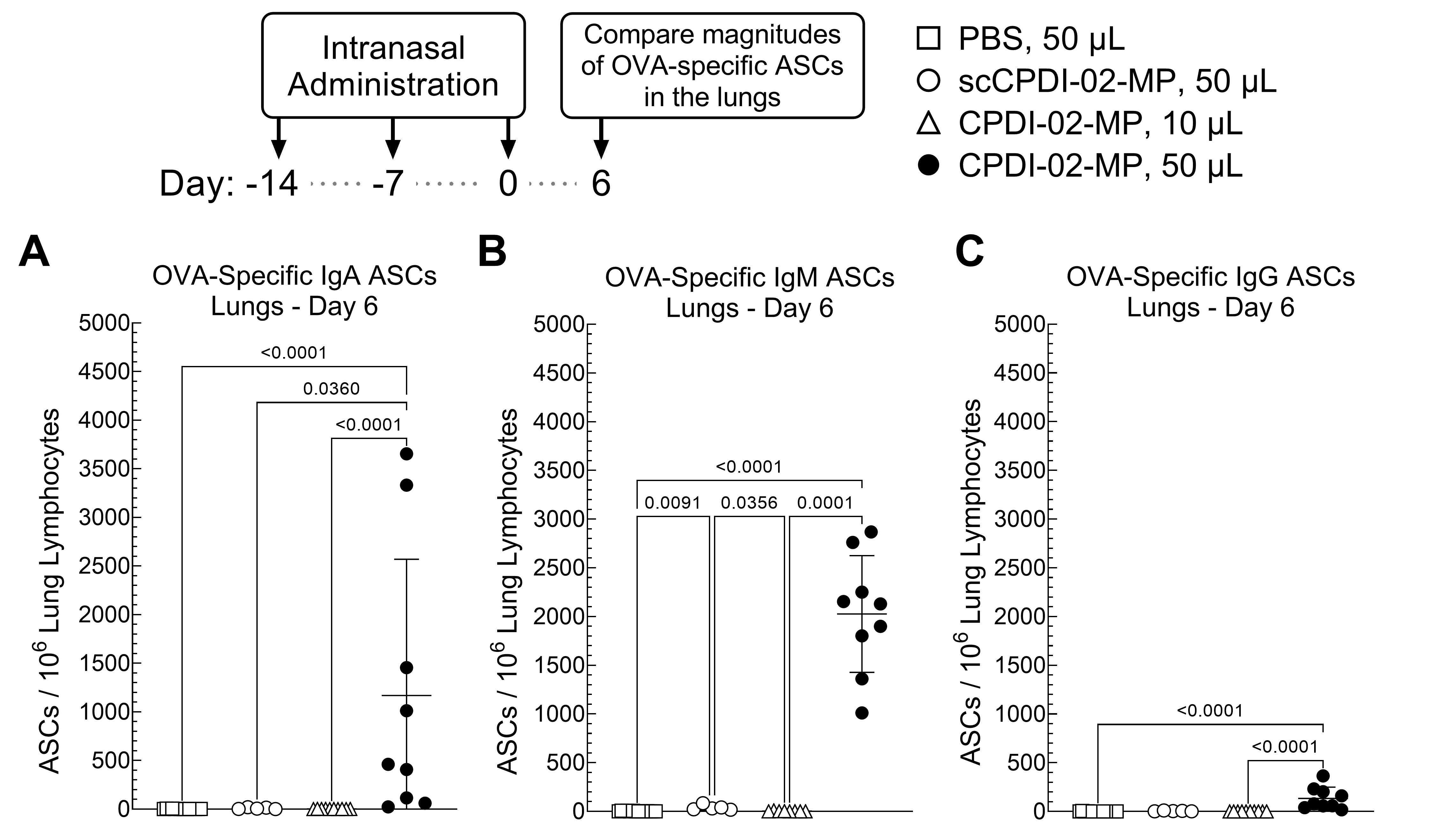
3.2. Surface Modification of ~1 μm Biodegradable Microparticles with CPDI-02 and Increased Pulmonary Delivery Increase the Magnitudes of Short-Term IgA and IgM Antibody-Secreting Cells (ASCs) against Encapsulated Protein Antigen in the Lungs of Young, Naïve Mice
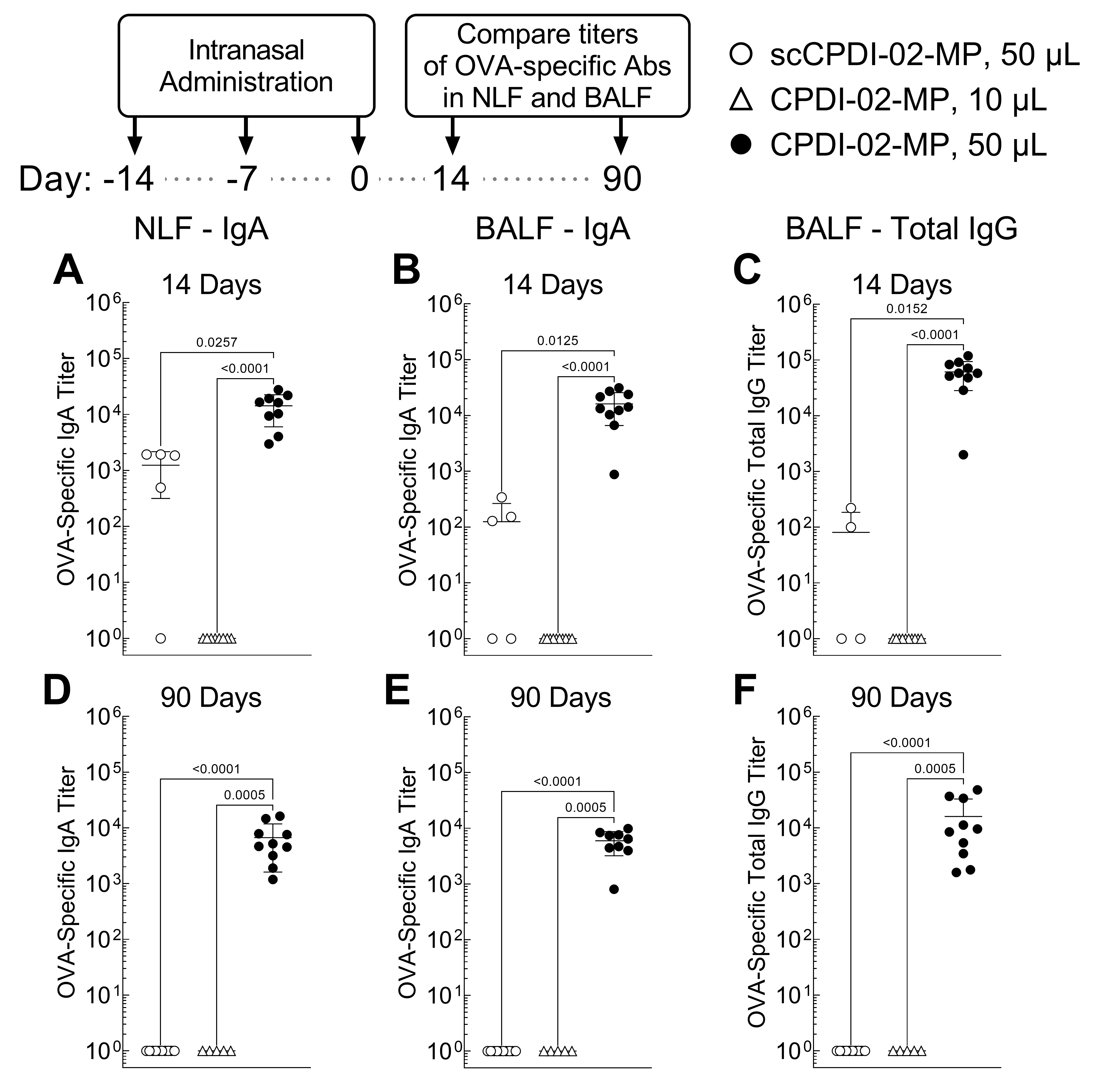
3.3. Surface Modification of ~1 μm Biodegradable Microparticles with CPDI-02 and Increased Pulmonary Delivery Greatly Increase Titers of Short-Term and Long-Term Mucosal Antibodies against Encapsulated Protein Antigen in the Nasal Cavity and Lungs of Young, Naïve Mice
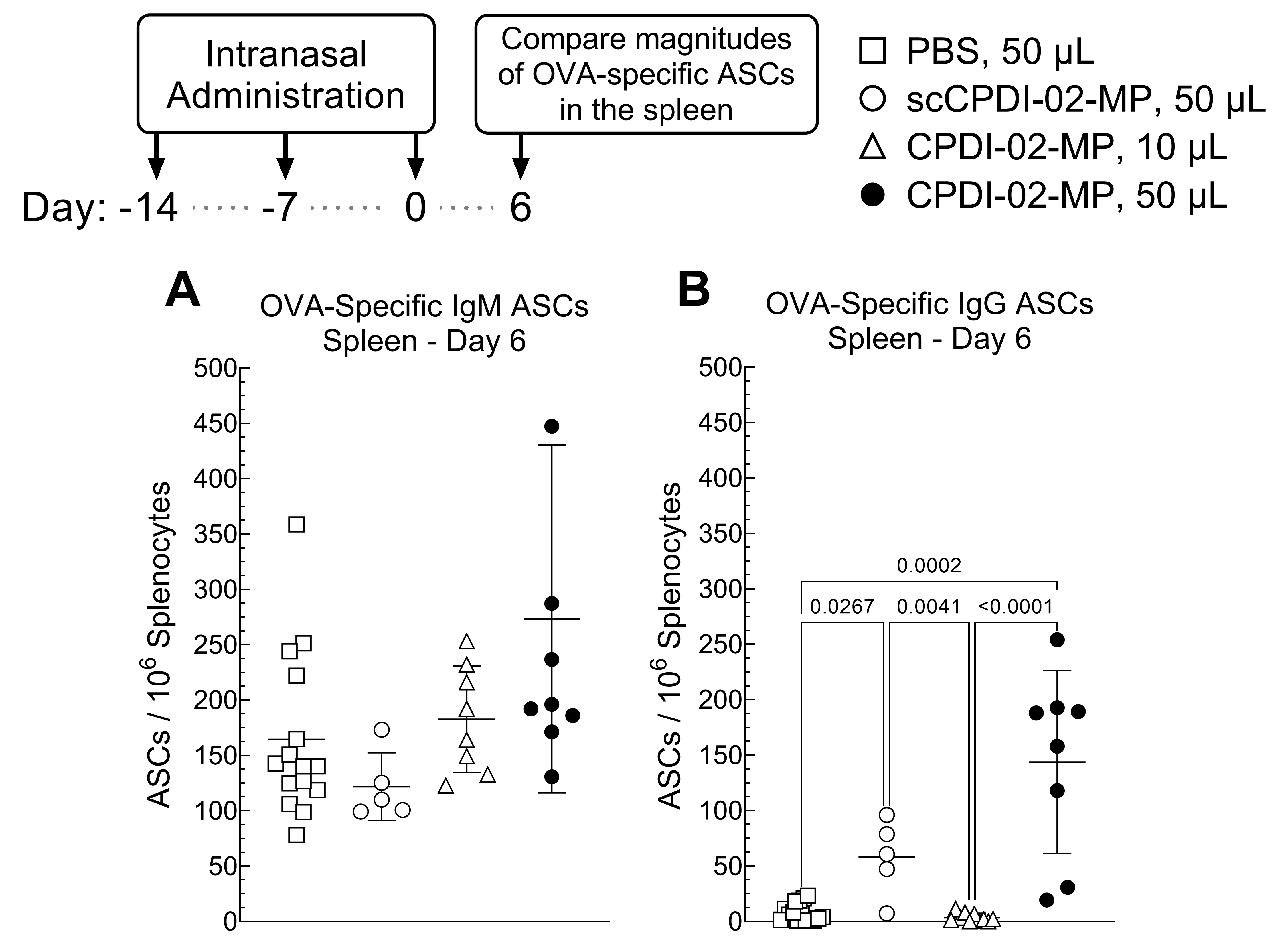
3.4. Effect of Surface Modification of ~1 μm Biodegradable Microparticles with CPDI-02 and Increased Pulmonary Delivery on Magnitudes of Short-Term Systemic Antibody-Secreting Cells (ASCs) against Encapsulated Protein Antigen in Young, Naïve Mice
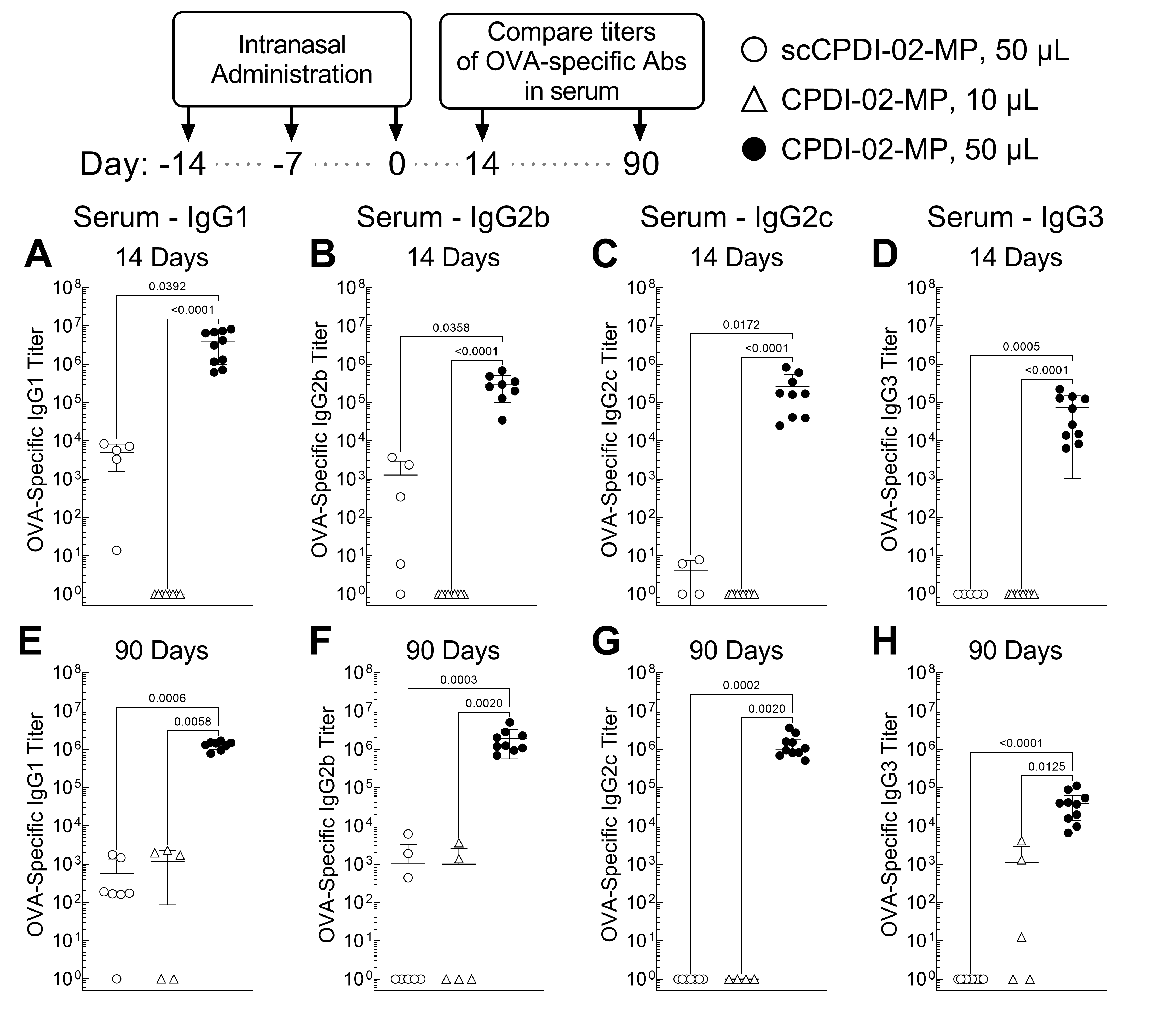
3.5. Surface Modification of ~1 μm Biodegradable Microparticles with CPDI-02 and Increased Pulmonary Delivery Greatly Increase Short-Term and Long-Term Systemic IgG Antibody Subclasses against Encapsulated Protein Antigen in the Sera of Young, Naïve Mice
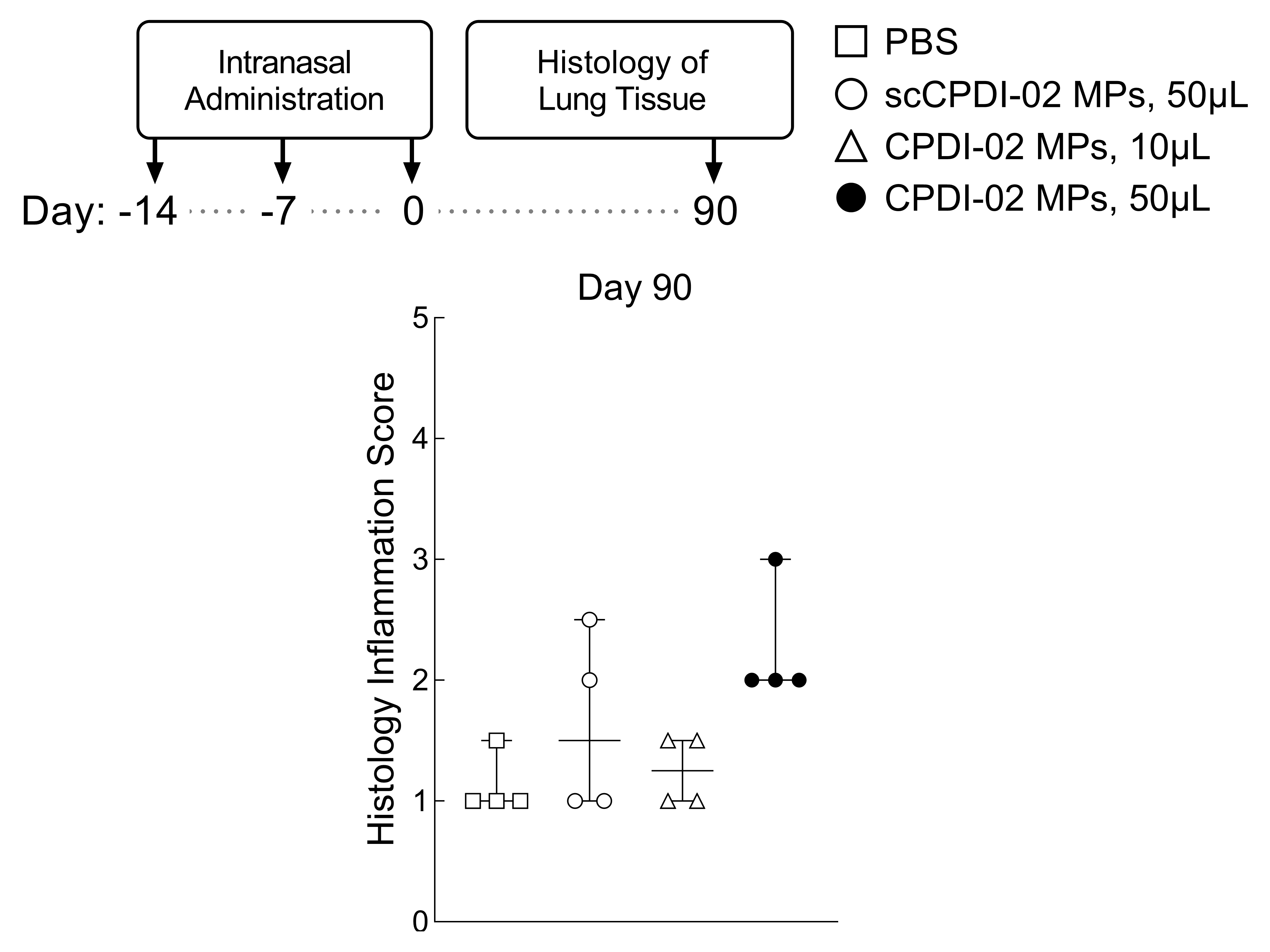
3.6. Preliminary Assessment of Long-Term Lung Inflammation in Healthy Young Mice after Respiratory Immunization with Surface-Modified MP
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nguipdop Djomo, P.; Thomas, S.L.; Fine, P.E.M. Correlates of Vaccine-Induced Protection: Methods and Implications; WHO Document Production Services: Geneva, Switzerland, 2013; Volume 1, pp. 1–49. [Google Scholar]
- Plotkin, S.A. Complex correlates of protection after vaccination. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2013, 56, 1458–1465. [Google Scholar] [CrossRef]
- Tallapaka, S.B.; Karuturi, B.V.K.; Yeapuri, P.; Curran, S.M.; Sonawane, Y.A.; Phillips, J.A.; Smith, D.D.; Sanderson, S.D.; Vetro, J.A. Surface conjugation of EP67 to biodegradable nanoparticles increases the generation of long-lived mucosal and systemic memory T-cells by encapsulated protein vaccine after respiratory immunization and subsequent T-cell-mediated protection against respiratory infection. Int. J. Pharm. 2019, 565, 242–257. [Google Scholar] [CrossRef]
- Siegrist, C.-A. Vaccine Immunology. In Vaccines, 6th ed.; Plotkin, S., Orenstein, W., Offit, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 14–33. [Google Scholar]
- Ionescu, L.; Urschel, S. Memory B Cells and Long-lived Plasma Cells. Transplantation 2019, 103, 890–898. [Google Scholar] [CrossRef]
- Thakur, A.; Pedersen, L.E.; Jungersen, G. Immune markers and correlates of protection for vaccine induced immune responses. Vaccine 2012, 30, 4907–4920. [Google Scholar] [CrossRef]
- Rappuoli, R. Bridging the knowledge gaps in vaccine design. Nat. Biotechnol. 2007, 25, 1361–1366. [Google Scholar] [CrossRef]
- Renegar, K.B.; Small, P.A., Jr.; Boykins, L.G.; Wright, P.F. Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J. Immunol. 2004, 173, 1978–1986. [Google Scholar] [CrossRef]
- Lavelle, E.C.; Ward, R.W. Mucosal vaccines—Fortifying the frontiers. Nat. Reviews. Immunol. 2022, 22, 236–250. [Google Scholar] [CrossRef]
- Lycke, N. Recent progress in mucosal vaccine development: Potential and limitations. Nat. Reviews. Immunol. 2012, 12, 592–605. [Google Scholar] [CrossRef]
- Czerkinsky, C.; Holmgren, J. Mucosal Delivery Routes for Optimal Immunization: Targeting Immunity to the Right Tissues. In Mucosal Vaccines: Modern Concepts, Strategies, and Challenges; Kozlowski, P.A., Ed.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 1–18. [Google Scholar]
- Ebensen, T.; Guzman, C.A. Immune modulators with defined molecular targets: Cornerstone to optimize rational vaccine design. Hum Vaccin 2008, 4, 13–22. [Google Scholar] [CrossRef]
- Kraehenbuhl, J.P.; Neutra, M.R. Mucosal vaccines: Where do we stand? Curr. Top. Med. Chem. 2013, 13, 2609–2628. [Google Scholar]
- Woodrow, K.A.; Bennett, K.M.; Lo, D.D. Mucosal vaccine design and delivery. Annu. Rev. Biomed. Eng. 2012, 14, 17–46. [Google Scholar] [CrossRef] [Green Version]
- Delany, I.; Rappuoli, R.; De Gregorio, E. Vaccines for the 21st century. EMBO Mol. Med. 2014, 6, 708–720. [Google Scholar] [CrossRef]
- Vetter, V.; Denizer, G.; Friedland, L.R.; Krishnan, J.; Shapiro, M. Understanding modern-day vaccines: What you need to know. Ann. Med. 2018, 50, 110–120. [Google Scholar] [CrossRef]
- Santos, E.; Levitz, S.M. Fungal vaccines and immunotherapeutics. Cold Spring Harb. Perspect. Med. 2014, 4, a019711. [Google Scholar] [CrossRef]
- Nami, S.; Mohammadi, R.; Vakili, M.; Khezripour, K.; Mirzaei, H.; Morovati, H. Fungal vaccines, mechanism of actions and immunology: A comprehensive review. Biomed. Pharm. 2019, 109, 333–344. [Google Scholar] [CrossRef]
- McAllister, M.M. Successful vaccines for naturally occurring protozoal diseases of animals should guide human vaccine research. A review of protozoal vaccines and their designs. Parasitology 2014, 141, 624–640. [Google Scholar] [CrossRef]
- Eldridge, J.H.; Meulbroek, J.A.; Staas, J.K.; Tice, T.R.; Gilley, R.M. Vaccine-containing biodegradable microspheres specifically enter the gut-associated lymphoid tissue following oral administration and induce a disseminated mucosal immune response. Adv. Exp. Med. Biol. 1989, 251, 191–202. [Google Scholar]
- Demento, S.L.; Cui, W.; Criscione, J.M.; Stern, E.; Tulipan, J.; Kaech, S.M.; Fahmy, T.M. Role of sustained antigen release from nanoparticle vaccines in shaping the T cell memory phenotype. Biomaterials 2012, 33, 4957–4964. [Google Scholar] [CrossRef]
- Shen, H.; Ackerman, A.L.; Cody, V.; Giodini, A.; Hinson, E.R.; Cresswell, P.; Edelson, R.L.; Saltzman, W.M.; Hanlon, D.J. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology 2006, 117, 78–88. [Google Scholar] [CrossRef]
- Audran, R.; Peter, K.; Dannull, J.; Men, Y.; Scandella, E.; Groettrup, M.; Gander, B.; Corradin, G. Encapsulation of peptides in biodegradable microspheres prolongs their MHC class-I presentation by dendritic cells and macrophages in vitro. Vaccine 2003, 21, 1250–1255. [Google Scholar]
- Eyles, J.E.; Bramwell, V.W.; Williamson, E.D.; Alpar, H.O. Microsphere translocation and immunopotentiation in systemic tissues following intranasal administration. Vaccine 2001, 19, 4732–4742. [Google Scholar]
- Eyles, J.E.; Spiers, I.D.; Williamson, E.D.; Alpar, H.O. Tissue distribution of radioactivity following intranasal administration of radioactive microspheres. J. Pharm. Pharm. 2001, 53, 601–607. [Google Scholar]
- Fischer, S.; Schlosser, E.; Mueller, M.; Csaba, N.; Merkle, H.P.; Groettrup, M.; Gander, B. Concomitant delivery of a CTL-restricted peptide antigen and CpG ODN by PLGA microparticles induces cellular immune response. J. Drug Target. 2009, 17, 652–661. [Google Scholar] [CrossRef]
- Silva, A.L.; Rosalia, R.A.; Sazak, A.; Carstens, M.G.; Ossendorp, F.; Oostendorp, J.; Jiskoot, W. Optimization of encapsulation of a synthetic long peptide in PLGA nanoparticles: Low-burst release is crucial for efficient CD8(+) T cell activation. Eur. J. Pharm. Biopharm. 2013, 83, 338–345. [Google Scholar] [CrossRef]
- Ma, W.; Chen, M.; Kaushal, S.; McElroy, M.; Zhang, Y.; Ozkan, C.; Bouvet, M.; Kruse, C.; Grotjahn, D.; Ichim, T.; et al. PLGA nanoparticle-mediated delivery of tumor antigenic peptides elicits effective immune responses. Int. J. Nanomed. 2012, 7, 1475–1487. [Google Scholar] [CrossRef] [Green Version]
- Mohan, T.; Sharma, C.; Bhat, A.A.; Rao, D.N. Modulation of HIV peptide antigen specific cellular immune response by synthetic alpha- and beta-defensin peptides. Vaccine 2013, 31, 1707–1716. [Google Scholar] [CrossRef]
- Zhang, Z.; Tongchusak, S.; Mizukami, Y.; Kang, Y.J.; Ioji, T.; Touma, M.; Reinhold, B.; Keskin, D.B.; Reinherz, E.L.; Sasada, T. Induction of anti-tumor cytotoxic T cell responses through PLGA-nanoparticle mediated antigen delivery. Biomaterials 2011, 32, 3666–3678. [Google Scholar] [CrossRef]
- Rodrigues, C.M.C.; Pinto, M.V.; Sadarangani, M.; Plotkin, S.A. Whither vaccines? J. Infect. 2017, 74 (Suppl. S1), S2–S9. [Google Scholar] [CrossRef]
- Longet, S.; Lundahl, M.L.E.; Lavelle, E.C. Targeted Strategies for Mucosal Vaccination. Bioconjug. Chem. 2018, 29, 613–623. [Google Scholar] [CrossRef]
- Czerkinsky, C.; Holmgren, J. Enteric vaccines for the developing world: A challenge for mucosal immunology. Mucosal Immunol. 2009, 2, 284–287. [Google Scholar] [CrossRef]
- Holmgren, J. Actions of cholera toxin and the prevention and treatment of cholera. Nature 1981, 292, 413–417. [Google Scholar]
- Gluck, U.; Gebbers, J.O.; Gluck, R. Phase 1 evaluation of intranasal virosomal influenza vaccine with and without Escherichia coli heat-labile toxin in adult volunteers. J. Virol. 1999, 73, 7780–7786. [Google Scholar]
- Mutsch, M.; Zhou, W.; Rhodes, P.; Bopp, M.; Chen, R.T.; Linder, T.; Spyr, C.; Steffen, R. Use of the inactivated intranasal influenza vaccine and the risk of Bell’s palsy in Switzerland. N. Engl. J. Med. 2004, 350, 896–903. [Google Scholar] [CrossRef]
- Lawson, L.B.; Norton, E.B.; Clements, J.D. Defending the mucosa: Adjuvant and carrier formulations for mucosal immunity. Curr. Opin. Immunol. 2011, 23, 414–420. [Google Scholar] [CrossRef]
- Chadwick, S.; Kriegel, C.; Amiji, M. Nanotechnology solutions for mucosal immunization. Adv. Drug. Deliv. Rev. 2010, 62, 394–407. [Google Scholar] [CrossRef]
- Rhee, J.H.; Lee, S.E.; Kim, S.Y. Mucosal vaccine adjuvants update. Clin. Exp. Vaccine Res. 2012, 1, 50–63. [Google Scholar] [CrossRef]
- Newsted, D.; Fallahi, F.; Golshani, A.; Azizi, A. Advances and challenges in mucosal adjuvant technology. Vaccine 2015, 33, 2399–2405. [Google Scholar] [CrossRef]
- Borges, O.; Lebre, F.; Bento, D.; Borchard, G.; Junginger, H.E. Mucosal vaccines: Recent progress in understanding the natural barriers. Pharm. Res. 2010, 27, 211–223. [Google Scholar] [CrossRef]
- Morgan, E.L.; Morgan, B.N.; Stein, E.A.; Vitrs, E.L.; Thoman, M.L.; Sanderson, S.D.; Phillips, J.A. Enhancement of in vivo and in vitro immune functions by a conformationally biased, response-selective agonist of human C5a: Implications for a novel adjuvant in vaccine design. Vaccine 2009, 28, 463–469. [Google Scholar] [CrossRef]
- Sheen, T.R.; Cavaco, C.K.; Ebrahimi, C.M.; Thoman, M.L.; Sanderson, S.D.; Morgan, E.L.; Doran, K.S. Control of methicillin resistant Staphylococcus aureus infection utilizing a novel immunostimulatory peptide. Vaccine 2011, 30, 9–13. [Google Scholar] [CrossRef]
- Hung, C.Y.; Hurtgen, B.J.; Bellecourt, M.; Sanderson, S.D.; Morgan, E.L.; Cole, G.T. An agonist of human complement fragment C5a enhances vaccine immunity against Coccidioides infection. Vaccine 2012, 30, 4681–4690. [Google Scholar] [CrossRef]
- Sanderson, S.D.; Thoman, M.L.; Kis, K.; Virts, E.L.; Herrera, E.B.; Widmann, S.; Sepulveda, H.; Phillips, J.A. Innate immune induction and influenza protection elicited by a response-selective agonist of human C5a. PLoS ONE 2012, 7, e40303. [Google Scholar] [CrossRef]
- Phillips, J.A.; Morgan, E.L.; Dong, Y.; Cole, G.T.; McMahan, C.; Hung, C.Y.; Sanderson, S.D. Single-step conjugation of bioactive peptides to proteins via a self-contained succinimidyl bis-arylhydrazone. Bioconjug. Chem. 2009, 20, 1950–1957. [Google Scholar] [CrossRef]
- Morgan, E.L.; Thoman, M.L.; Sanderson, S.D.; Phillips, J.A. A novel adjuvant for vaccine development in the aged. Vaccine 2010, 28, 8275–8279. [Google Scholar] [CrossRef]
- Karuturi, B.V.K.; Tallapaka, S.B.; Yeapuri, P.; Curran, S.M.; Sanderson, S.D.; Vetro, J.A. Encapsulation of an EP67-Conjugated CTL Peptide Vaccine in Nanoscale Biodegradable Particles Increases the Efficacy of Respiratory Immunization and Affects the Magnitude and Memory Subsets of Vaccine-Generated Mucosal and Systemic CD8+ T Cells in a Diameter-Dependent Manner. Mol. Pharm. 2017, 14, 1469–1481. [Google Scholar] [CrossRef]
- Karuturi, B.V.; Tallapaka, S.B.; Phillips, J.A.; Sanderson, S.D.; Vetro, J.A. Preliminary evidence that the novel host-derived immunostimulant EP67 can act as a mucosal adjuvant. Clin. Immunol. 2015, 161, 251–259. [Google Scholar] [CrossRef]
- Alshammari, A.M.; Smith, D.D.; Parriott, J.; Stewart, J.P.; Curran, S.M.; McCulloh, R.J.; Barry, P.A.; Iyer, S.S.; Palermo, N.; Phillips, J.A.; et al. Targeted Amino Acid Substitution Overcomes Scale-Up Challenges with the Human C5a-Derived Decapeptide Immunostimulant EP67. ACS Infect. Dis. 2020, 6, 1169–1181. [Google Scholar] [CrossRef]
- Kollessery, G.; Nordgren, T.M.; Mittal, A.K.; Joshi, S.S.; Sanderson, S.D. Tumor-specific peptide-based vaccines containing the conformationally biased, response-selective C5a agonists EP54 and EP67 protect against aggressive large B cell lymphoma in a syngeneic murine model. Vaccine 2011, 29, 5904–5910. [Google Scholar] [CrossRef]
- Tifrea, D.F.; Pal, S.; Le Bon, C.; Giusti, F.; Popot, J.L.; Cocco, M.J.; Zoonens, M.; de la Maza, L.M. Co-delivery of amphipol-conjugated adjuvant with antigen, and adjuvant combinations, enhance immune protection elicited by a membrane protein-based vaccine against a mucosal challenge with Chlamydia. Vaccine 2018, 36, 6640–6649. [Google Scholar] [CrossRef]
- Zasowski, E.J.; Blackford, M. Lower Respiratory Tract Infections. In Pharmacotherapy: A Pathophysiologic Approach, 11th ed.; DiPiro, J.T., Yee, G.C., Posey, L.M., Haines, S.T., Nolin, T.D., Ellingrod, V., Eds.; McGraw-Hill Education: New York, NY, USA, 2020. [Google Scholar]
- Murphy, K.M.; Weaver, C.; Berg, L.J. Janeway’s Immunobiology, 10th ed.; W.W. Norton & Company, Inc.: New York, NY, USA, 2022. [Google Scholar]
- Mizuno, K.; Nakamura, T.; Ohshima, T.; Tanaka, S.; Matsuo, H. Characterization of KEX2-encoded endopeptidase from yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1989, 159, 305–311. [Google Scholar] [CrossRef]
- Nakayama, K. Furin: A mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem. J. 1997, 327 Pt 3, 625–635. [Google Scholar] [CrossRef]
- Eyles, J.E.; Williamson, E.D.; Alpar, H.O. Immunological responses to nasal delivery of free and encapsulated tetanus toxoid: Studies on the effect of vehicle volume. Int. J. Pharm. 1999, 189, 75–79. [Google Scholar]
- Iorns, E.; Drews-Elger, K.; Ward, T.M.; Dean, S.; Clarke, J.; Berry, D.; El Ashry, D.; Lippman, M. A new mouse model for the study of human breast cancer metastasis. PLoS ONE 2012, 7, e47995. [Google Scholar] [CrossRef]
- Southam, D.S.; Dolovich, M.; O’Byrne, P.M.; Inman, M.D. Distribution of intranasal instillations in mice: Effects of volume, time, body position, and anesthesia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L833–L839. [Google Scholar] [CrossRef]
- Golde, W.T.; Gollobin, P.; Rodriguez, L.L. A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab. Anim. 2005, 34, 39–43. [Google Scholar] [CrossRef]
- McCaskill, M.L.; Romberger, D.J.; DeVasure, J.; Boten, J.; Sisson, J.H.; Bailey, K.L.; Poole, J.A.; Wyatt, T.A. Alcohol exposure alters mouse lung inflammation in response to inhaled dust. Nutrients 2012, 4, 695–710. [Google Scholar] [CrossRef]
- Takahashi, Y.; Kagawa, Y.; Izawa, K.; Ono, R.; Akagi, M.; Kamei, C. Effect of histamine H4 receptor antagonist on allergic rhinitis in mice. Int. Immunopharmacol. 2009, 9, 734–738. [Google Scholar] [CrossRef]
- Frey, A.; Di Canzio, J.; Zurakowski, D. A statistically defined endpoint titer determination method for immunoassays. J. Immunol. Methods 1998, 221, 35–41. [Google Scholar] [CrossRef]
- Zrein, M.; De Marcillac, G.; Van Regenmortel, M.H. Quantitation of rheumatoid factors by enzyme immunoassay using biotinylated human IgG. J. Immunol. Methods 1986, 87, 229–237. [Google Scholar] [CrossRef]
- Gross, E.A.; Swenberg, J.A.; Fields, S.; Popp, J.A. Comparative morphometry of the nasal cavity in rats and mice. J. Anat. 1982, 135, 83–88. [Google Scholar]
- Poole, J.A.; Romberger, D.J.; Bauer, C.; Gleason, A.M.; Sisson, J.H.; Oldenburg, P.J.; West, W.W.; Wyatt, T.A. Protein kinase C epsilon is important in modulating organic-dust-induced airway inflammation. Exp. Lung. Res. 2012, 38, 383–395. [Google Scholar] [CrossRef]
- Gutierro, I.; Hernandez, R.M.; Igartua, M.; Gascon, A.R.; Pedraz, J.L. Size dependent immune response after subcutaneous, oral and intranasal administration of BSA loaded nanospheres. Vaccine 2002, 21, 67–77. [Google Scholar]
- Patil, Y.B.; Toti, U.S.; Khdair, A.; Ma, L.; Panyam, J. Single-step surface functionalization of polymeric nanoparticles for targeted drug delivery. Biomaterials 2009, 30, 859–866. [Google Scholar] [CrossRef]
- Toti, U.S.; Guru, B.R.; Grill, A.E.; Panyam, J. Interfacial activity assisted surface functionalization: A novel approach to incorporate maleimide functional groups and cRGD peptide on polymeric nanoparticles for targeted drug delivery. Mol. Pharm. 2010, 7, 1108–1117. [Google Scholar] [CrossRef]
- Lee, F.E.; Halliley, J.L.; Walsh, E.E.; Moscatiello, A.P.; Kmush, B.L.; Falsey, A.R.; Randall, T.D.; Kaminiski, D.A.; Miller, R.K.; Sanz, I. Circulating human antibody-secreting cells during vaccinations and respiratory viral infections are characterized by high specificity and lack of bystander effect. J. Immunol. 2011, 186, 5514–5521. [Google Scholar] [CrossRef]
- Halliley, J.L.; Kyu, S.; Kobie, J.J.; Walsh, E.E.; Falsey, A.R.; Randall, T.D.; Treanor, J.; Feng, C.; Sanz, I.; Lee, F.E. Peak frequencies of circulating human influenza-specific antibody secreting cells correlate with serum antibody response after immunization. Vaccine 2010, 28, 3582–3587. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Hellfritzsch, M.; Scherliess, R. Mucosal Vaccination via the Respiratory Tract. Pharmaceutics 2019, 11, 375. [Google Scholar] [CrossRef]
- Mettelman, R.C.; Allen, E.K.; Thomas, P.G. Mucosal immune responses to infection and vaccination in the respiratory tract. Immunity 2022, 55, 749–780. [Google Scholar] [CrossRef]
- Collins, A.M. IgG subclass co-expression brings harmony to the quartet model of murine IgG function. Immunol. Cell. Biol. 2016, 94, 949–954. [Google Scholar] [CrossRef]
- Kim, S.H.; Jung, D.I.; Yang, I.Y.; Kim, J.; Lee, K.Y.; Nochi, T.; Kiyono, H.; Jang, Y.S. M cells expressing the complement C5a receptor are efficient targets for mucosal vaccine delivery. Eur. J. Immunol. 2011, 41, 3219–3229. [Google Scholar] [CrossRef]
- Wang, M.; Gao, Z.; Zhang, Z.; Pan, L.; Zhang, Y. Roles of M cells in infection and mucosal vaccines. Hum. Vaccin. Immunother. 2014, 10, 3544–3551. [Google Scholar] [CrossRef]
- Islam, M.A.; Firdous, J.; Badruddoza, A.Z.M.; Reesor, E.; Azad, M.; Hasan, A.; Lim, M.; Cao, W.; Guillemette, S.; Cho, C.S. M cell targeting engineered biomaterials for effective vaccination. Biomaterials 2019, 192, 75–94. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | OVA Loading 1 (μg/mg MP ± SD) | Burst Release 1 (% Loaded OVA) | CPDI-02 Conjugation 2 (μg/mg MP ± SD) | Diameter 3 (μm ± SD) | Polydispersity Index 3 (PDI ± SD) | Zeta Potential 3 (mv ± SD) |
|---|---|---|---|---|---|---|
| CPDI-02-MP | 62 ± 13 | 0.8 ± 0.2 | 4.0 ± 0.6 | 1.1 ± 0.2 | 0.3 ± 0.1 | −22 ± 3 |
| scCPDI-02-MP | 52 ± 13 | 0.4 ± 0.1 | 3.9 ± 0.2 | 1.21 ± 0.02 | 0.36 ± 0.08 | −24 ± 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parriott, J.E.; Stewart, J.P.; Smith, D.D.; Curran, S.M.; Bauer, C.D.; Wyatt, T.A.; Phillips, J.A.; Lyden, E.; Thiele, G.M.; Vetro, J.A. Surface Modification of Biodegradable Microparticles with the Novel Host-Derived Immunostimulant CPDI-02 Significantly Increases Short-Term and Long-Term Mucosal and Systemic Antibodies against Encapsulated Protein Antigen in Young Naïve Mice after Respiratory Immunization. Pharmaceutics 2022, 14, 1843. https://doi.org/10.3390/pharmaceutics14091843
Parriott JE, Stewart JP, Smith DD, Curran SM, Bauer CD, Wyatt TA, Phillips JA, Lyden E, Thiele GM, Vetro JA. Surface Modification of Biodegradable Microparticles with the Novel Host-Derived Immunostimulant CPDI-02 Significantly Increases Short-Term and Long-Term Mucosal and Systemic Antibodies against Encapsulated Protein Antigen in Young Naïve Mice after Respiratory Immunization. Pharmaceutics. 2022; 14(9):1843. https://doi.org/10.3390/pharmaceutics14091843
Chicago/Turabian StyleParriott, Jacob E., Jason P. Stewart, D. David Smith, Stephen M. Curran, Christopher D. Bauer, Todd A. Wyatt, Joy A. Phillips, Elizabeth Lyden, Geoffrey M. Thiele, and Joseph A. Vetro. 2022. "Surface Modification of Biodegradable Microparticles with the Novel Host-Derived Immunostimulant CPDI-02 Significantly Increases Short-Term and Long-Term Mucosal and Systemic Antibodies against Encapsulated Protein Antigen in Young Naïve Mice after Respiratory Immunization" Pharmaceutics 14, no. 9: 1843. https://doi.org/10.3390/pharmaceutics14091843
APA StyleParriott, J. E., Stewart, J. P., Smith, D. D., Curran, S. M., Bauer, C. D., Wyatt, T. A., Phillips, J. A., Lyden, E., Thiele, G. M., & Vetro, J. A. (2022). Surface Modification of Biodegradable Microparticles with the Novel Host-Derived Immunostimulant CPDI-02 Significantly Increases Short-Term and Long-Term Mucosal and Systemic Antibodies against Encapsulated Protein Antigen in Young Naïve Mice after Respiratory Immunization. Pharmaceutics, 14(9), 1843. https://doi.org/10.3390/pharmaceutics14091843