
Advances in Antibody-Based Therapeutics for Cerebral Ischemia

 , , ,
, , ,
Abstract
:1. Introduction
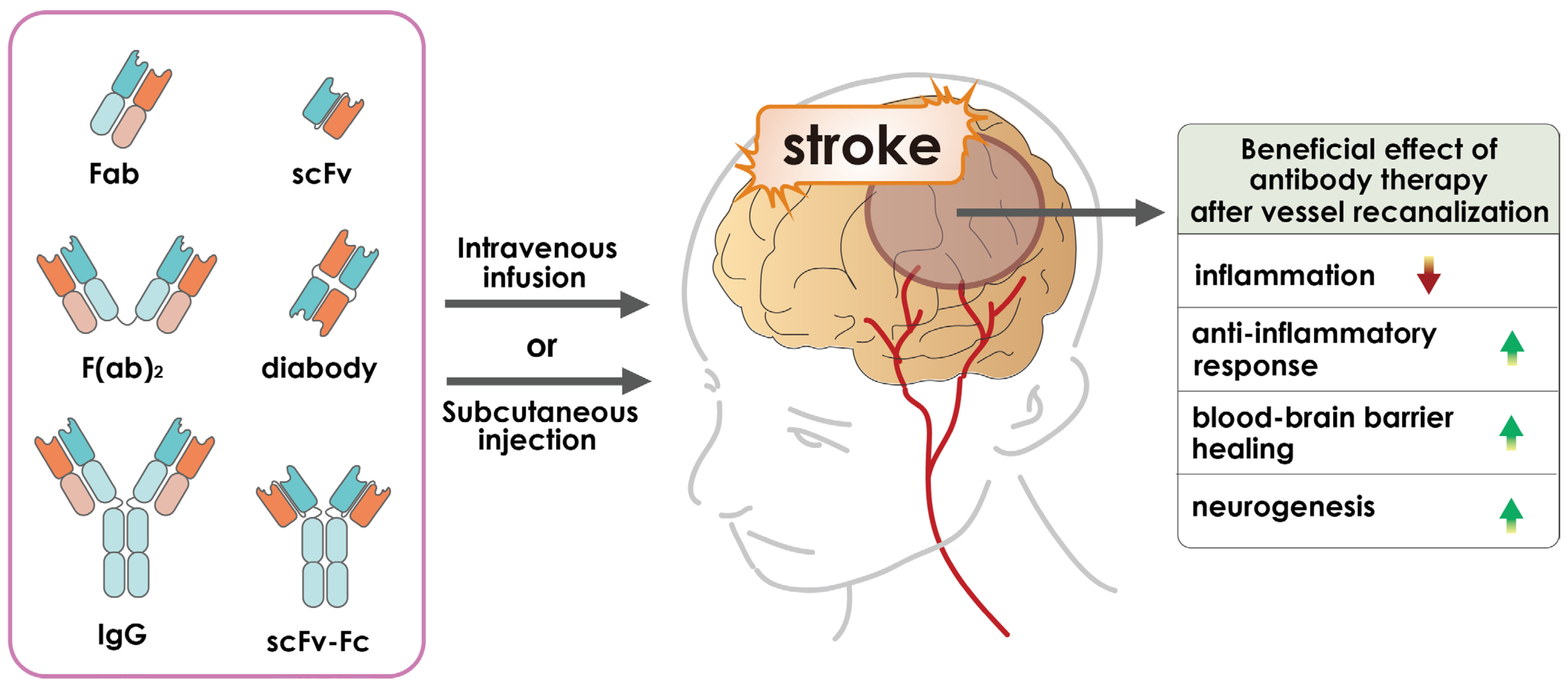
2. Antibody-Based Therapeutics
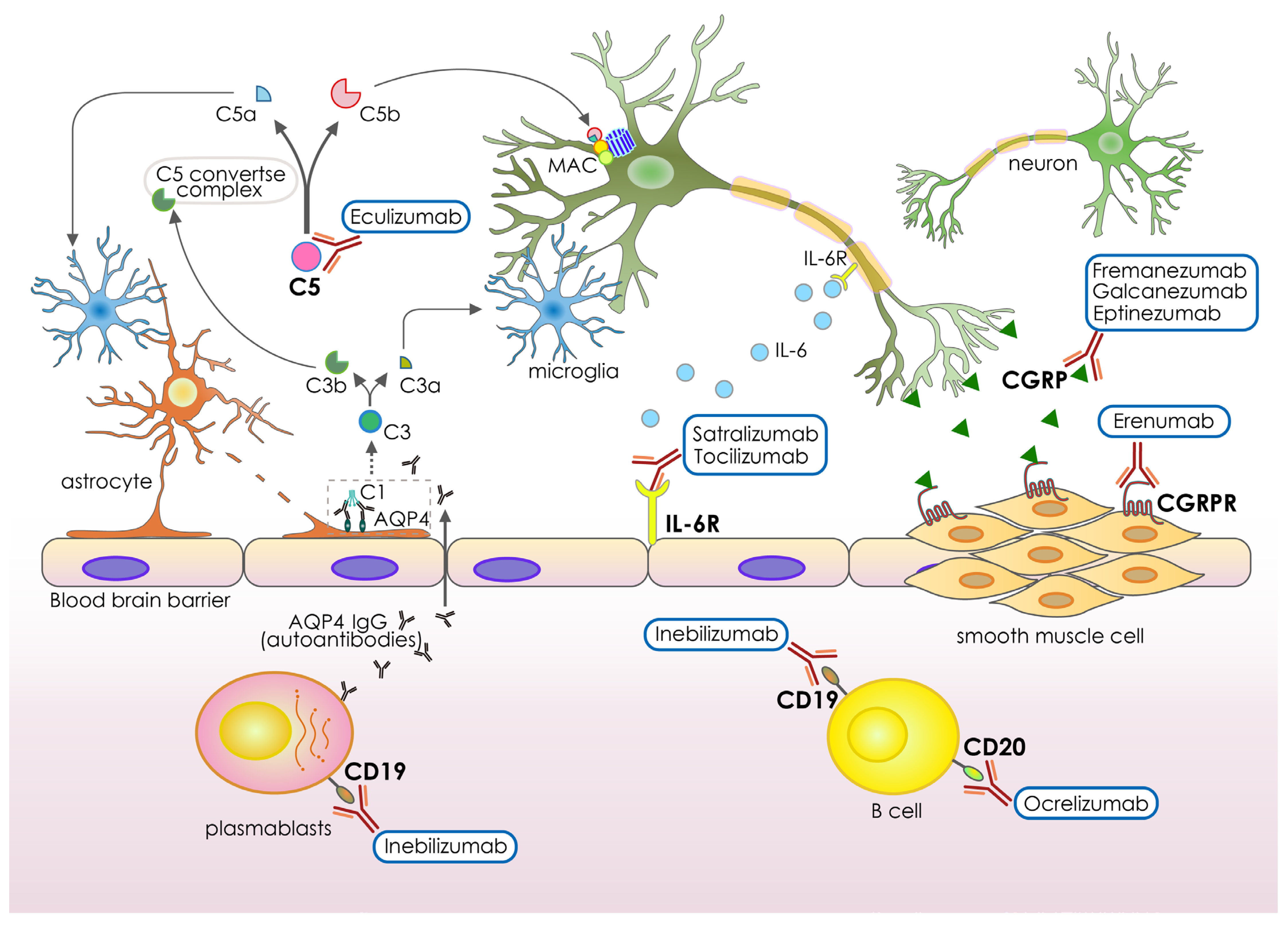
3. Recent Antibody-Based Drugs for Neurological Disorders
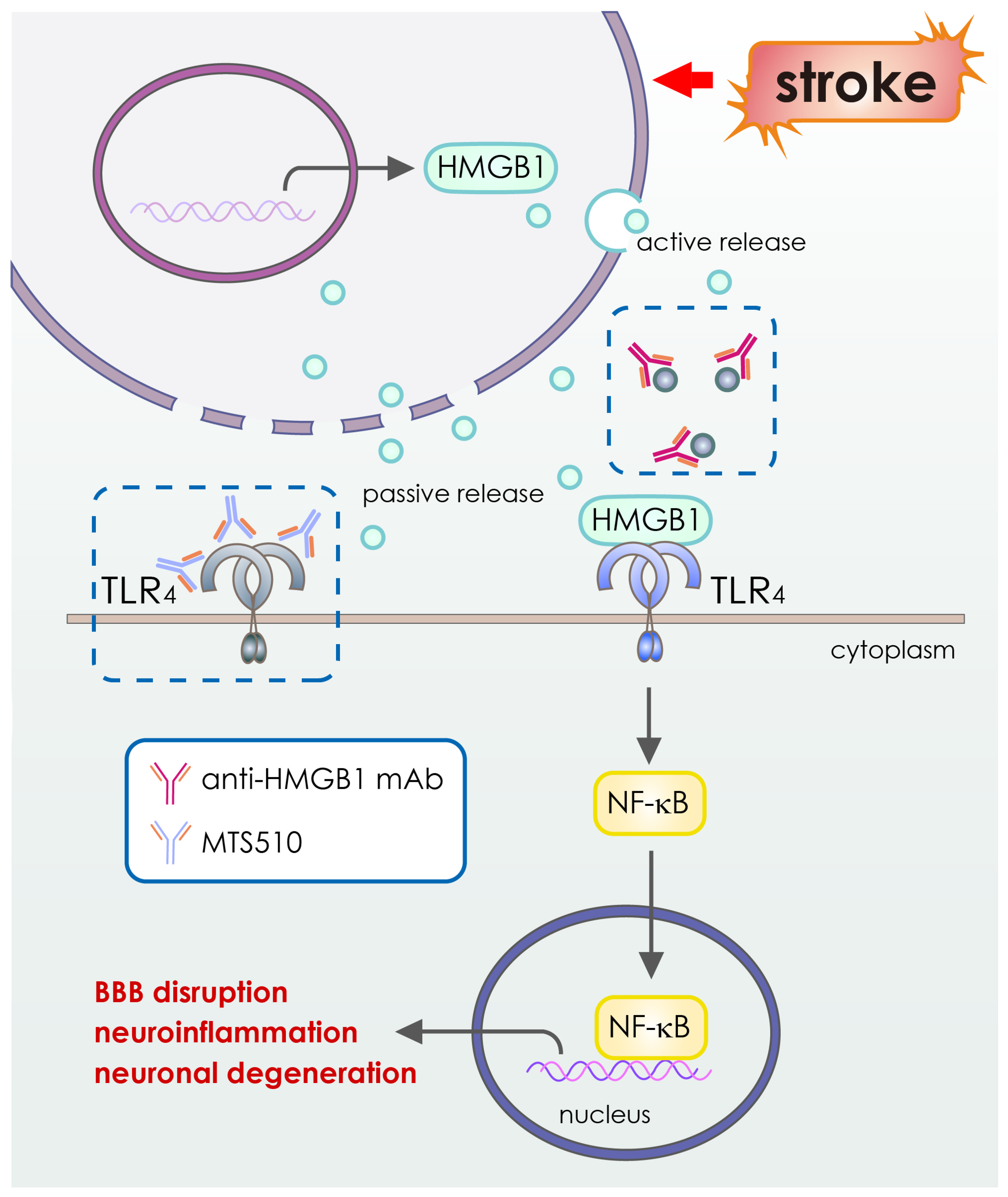
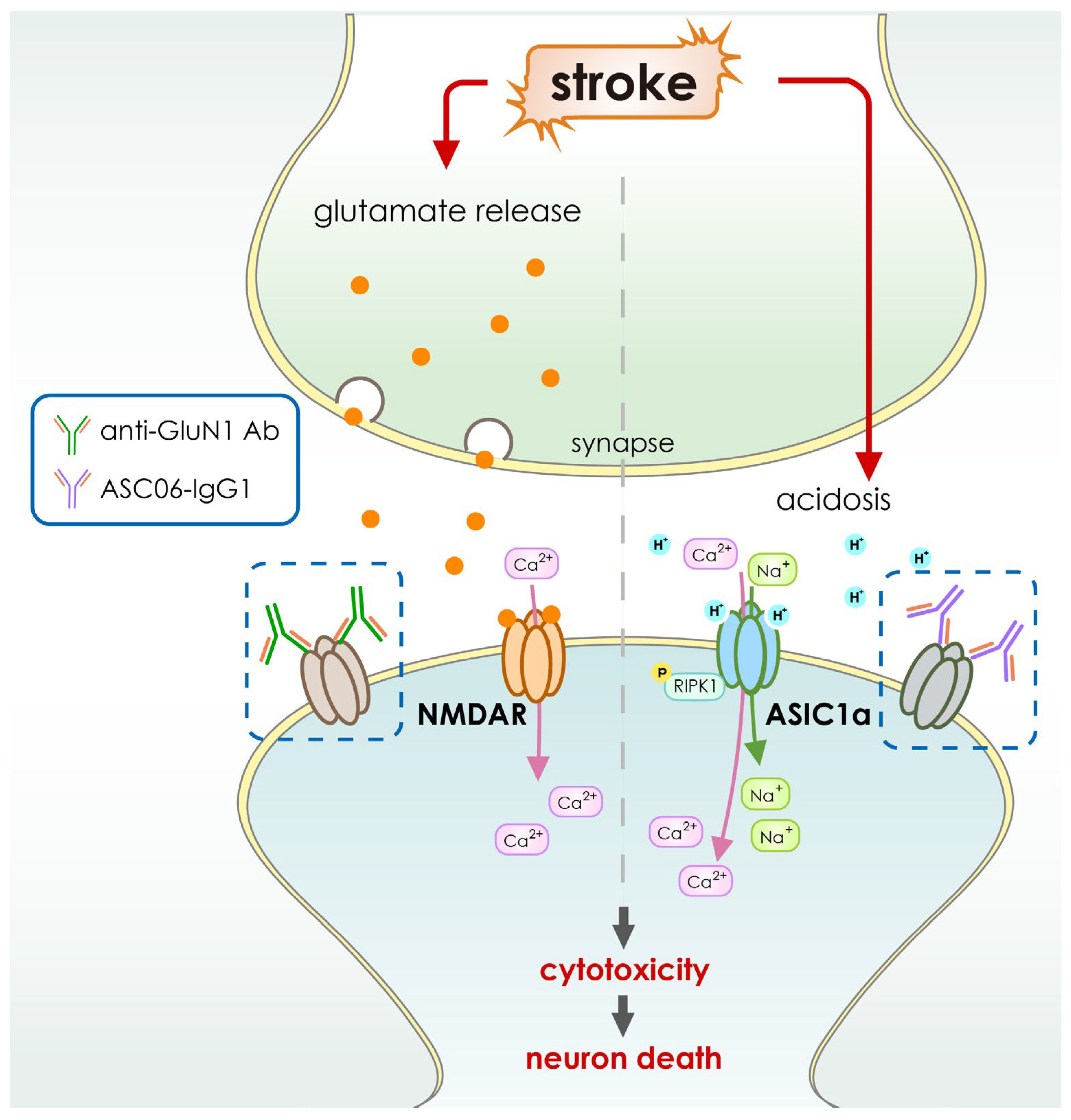
4. Application of Antibody-Based Drugs for Cerebral Ischemia
5. Pros and Cons of Antibody-Based Drugs for Cerebral Ischemia
6. Strategies to Improve the Efficacy of Antibody-Based Drugs for Cerebral Ischemia
7. Clinical Translation of Antibody-Based Drugs for Cerebral Ischemia
8. Limitation of Antibody-Based Drug for Cerebral Ischemia
9. Conclusions and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnson, W.; Onuma, O.; Owolabi, M.; Sachdev, S. Stroke: A global response is needed. Bull. World Health Organ. 2016, 94, 634–634A. [Google Scholar] [CrossRef] [PubMed]
- Minhas, J.; Robinson, T. Latest Developments in Clinical Stroke Care. J. R. Coll. Physicians Edinb. 2017, 47, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Owolabi, M.O.; Akarolo-Anthony, S.; Akinyemi, R.; Arnett, D.; Gebregziabher, M.; Jenkins, C.; Tiwari, H.; Arulogun, O.; Akpalu, A.; Sarfo, F.S.; et al. The burden of stroke in Africa: A glance at the present and a glimpse into the future. Cardiovasc. J. Afr. 2015, 26, S27–S38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakroun-Walha, O.; Samet, A.; Ben Abdallah, M.; Benmansour, S.; Issaoui, F.; Rebai, M.; Ben Messaoud, K.; Benali, C.; Mokni, W.; Nasri, A.; et al. Stroke knowledge among emergency centre visitors: A cross-sectional multicenter survey. Afr. J. Emerg. Med. 2021, 11, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Sarikaya, H.; Ferro, J.; Arnold, M. Stroke Prevention—Medical and Lifestyle Measures. Eur. Neurol. 2015, 73, 150–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.C.; Mendis, S.; Mathers, C.D. Global variation in stroke burden and mortality: Estimates from monitoring, surveillance, and modelling. Lancet Neurol. 2009, 8, 345–354. [Google Scholar] [CrossRef]
- O’Donnell, M.J.; Xavier, D.; Liu, L.; Zhang, H.; Chin, S.L.; Rao-Melacini, P.; Rangarajan, S.; Islam, S.; Pais, P.; McQueen, M.J.; et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): A case-control study. Lancet 2010, 376, 112–123. [Google Scholar] [CrossRef]
- Mayosi, B.M.; Lawn, J.E.; van Niekerk, A.; Bradshaw, D.; Karim, S.S.A.; Coovadia, H.M. Health in South Africa: Changes and challenges since 2009. Lancet 2012, 380, 2029–2043. [Google Scholar] [CrossRef]
- Gibson, C.L. Cerebral ischemic stroke: Is gender important? J. Cereb. Blood Flow 2013, 33, 1355–1361. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, Y.; Sun, H.; Xing, Y. Hemorrhagic transformation after cerebral infarction: Current concepts and challenges. Ann. Transl. Med. 2014, 2, 81. [Google Scholar] [CrossRef]
- Seet, R.C.S.; Rabinstein, A.A. Symptomatic Intracranial Hemorrhage following Intravenous Thrombolysis for Acute Ischemic Stroke: A Critical Review of Case Definitions. Cerebrovasc. Dis. 2012, 34, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Grysiewicz, R.A.; Thomas, K.; Pandey, D.K. Epidemiology of Ischemic and Hemorrhagic Stroke: Incidence, Prevalence, Mortality, and Risk Factors. Neurol. Clin. 2008, 26, 871–895. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Dávalos, A.; Rogalewski, A.; Schneider, A.; Ringelstein, E.B.; Schäbitz, W.-R. Toward a Multimodal Neuroprotective Treatment of Stroke. Stroke 2006, 37, 1129–1136. [Google Scholar] [CrossRef]
- Kuriakose, D.; Xiao, Z. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 7609. [Google Scholar] [CrossRef]
- Zhou, Z.; Lu, J.; Liu, W.-W.; Manaenko, A.; Hou, X.; Mei, Q.; Huang, J.-L.; Tang, J.; Zhang, J.H.; Yao, H.; et al. Advances in stroke pharmacology. Pharmacol. Ther. 2018, 191, 23–42. [Google Scholar] [CrossRef]
- Gomez, C.R. Time is brain: The stroke theory of relativity. J. Stroke Cerebrovasc. Dis. 2018, 27, 2214–2227. [Google Scholar] [CrossRef]
- French, B.R.; Boddepalli, R.S.; Govindarajan, R. Acute Ischemic Stroke: Current Status and Future Directions. Mo. Med. 2016, 113, 480–486. [Google Scholar]
- Bevers, M.B.; Kimberly, W.T. Critical Care Management of Acute Ischemic Stroke. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 41. [Google Scholar] [CrossRef]
- Hasan, T.F.; Rabinstein, A.A.; Middlebrooks, E.H.; Haranhalli, N.; Silliman, S.L.; Meschia, J.F.; Tawk, R.G. Diagnosis and Management of Acute Ischemic Stroke. Mayo Clin. Proc. 2018, 93, 523–538. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C.; Anrather, J.J.N.M. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Lyden, P.; Buchan, A.; Boltze, J.; Fisher, M.; Ansari, S.; Broderick, J.P.; Campbell, B.C.; Chaisinanunkul, N.; Chen, C.; Grotta, J.C.; et al. Top Priorities for Cerebroprotective Studies—A Paradigm Shift: Report from STAIR XI. Stroke 2021, 52, 3063–3071. [Google Scholar] [CrossRef] [PubMed]
- Savitz, S.I.; Baron, J.-C.; Fisher, M.; Stroke, S.X.C.J. Stroke treatment academic industry roundtable X: Brain cytoprotection therapies in the reperfusion era. Stroke 2019, 50, 1026–1031. [Google Scholar] [CrossRef]
- Siniscalchi, A.; Iannacchero, R.; Anticoli, S.; Pezzella, F.R.; De Sarro, G.; Gallelli, L. Anti-inflammatory strategies in stroke: A potential therapeutic target. Curr. Vasc. Pharmacol. 2016, 14, 98–105. [Google Scholar] [CrossRef]
- Zafar, M.; Memon, R.S.; Mussa, M.; Merchant, R.; Khurshid, A.; Khosa, F. Does the administration of sonothrombolysis along with tissue plasminogen activator improve outcomes in acute ischemic stroke? A systematic review and meta-analysis. J. Thromb. Thrombolysis 2019, 48, 203–208. [Google Scholar] [CrossRef]
- Dennis, M.S.; Burn, J.P.; Sandercock, P.A.; Bamford, J.M.; Wade, D.T.; Warlow, C.P. Long-term survival after first-ever stroke: The Oxfordshire Community Stroke Project. Stroke 1993, 24, 796–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augusto, D.E.; Álvarez, L.M.; Costa, F.T. Actualización en hemorragia cerebral espontánea. Med. Intensiv. 2008, 32, 282–295. [Google Scholar] [CrossRef] [Green Version]
- Kolias, A.G.; Viaroli, E.; Rubiano, A.M.; Adams, H.; Khan, T.; Gupta, D.; Adeleye, A.; Iaccarino, C.; Servadei, F.; Devi, B.I.; et al. The Current Status of Decompressive Craniectomy in Traumatic Brain Injury. Curr. Trauma Rep. 2018, 4, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Moradiya, Y.; Murthy, S.B.; Newman-Toker, D.E.; Hanley, D.F.; Ziai, W.C. Intraventricular Thrombolysis in Intracerebral Hemorrhage Requiring Ventriculostomy: A decade-long real-world experience. Stroke 2014, 45, 2629–2635. [Google Scholar] [CrossRef]
- Ruan, C.; Long, H.; Sun, H.; He, M.; Yang, K.; Zhang, H.; Mao, B. Endovascular coiling vs. surgical clipping for unruptured intracranial aneurysm: A meta-analysis. Br. J. Neurosurg. 2015, 29, 485–492. [Google Scholar] [CrossRef]
- Starke, R.M.; Kano, H.; Ding, D.; Lee, J.Y.K.; Mathieu, D.; Whitesell, J.; Pierce, J.T.; Huang, P.P.; Kondziolka, D.; Yen, C.-P.; et al. Stereotactic radiosurgery for cerebral arteriovenous malformations: Evaluation of long-term outcomes in a multicenter cohort. J. Neurosurg. 2017, 126, 36–44. [Google Scholar] [CrossRef]
- Urdaneta, A.E.; Bhalla, P. Cutting Edge Acute Ischemic Stroke Management. Emerg. Med. Clin. North Am. 2019, 37, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Watts, R.J. Developing Therapeutic Antibodies for Neurodegenerative Disease. Neurotherapeutics 2013, 10, 459–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadkar, K.; Yadav, D.B.; Zuchero, J.Y.; Couch, J.A.; Kanodia, J.; Kenrick, M.K.; Atwal, J.K.; Dennis, M.S.; Prabhu, S.; Watts, R.J.; et al. Mathematical PKPD and safety model of bispecific TfR/BACE1 antibodies for the optimization of antibody uptake in brain. Eur. J. Pharm. Biopharm. 2016, 101, 53–61. [Google Scholar] [CrossRef]
- Kanodia, J.S.; Gadkar, K.; Bumbaca, D.; Zhang, Y.; Tong, R.K.; Luk, W.; Hoyte, K.; Lu, Y.; Wildsmith, K.R.; Couch, J.A.; et al. Prospective Design of Anti-Transferrin Receptor Bispecific Antibodies for Optimal Delivery into the Human Brain. CPT Pharmacometrics Syst. Pharmacol. 2016, 5, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, Z. CNS Drug Design: Balancing Physicochemical Properties for Optimal Brain Exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef]
- Khawli, L.A.; Prabhu, S. Drug delivery across the blood–brain barrier. Mol. Pharm. 2013, 10, 1471–1472. [Google Scholar] [CrossRef]
- Deo, A.K.; Theil, F.-P.; Nicolas, J.-M. Confounding Parameters in Preclinical Assessment of Blood–Brain Barrier Permeation: An Overview with Emphasis on Species Differences and Effect of Disease States. Mol. Pharm. 2013, 10, 1581–1595. [Google Scholar] [CrossRef]
- Borsook, D. Neurological diseases and pain. Brain 2012, 135, 320–344. [Google Scholar] [CrossRef] [Green Version]
- Dumurgier, J.; Tzourio, C. Epidemiology of neurological diseases in older adults. Rev. Neurol. 2020, 176, 642–648. [Google Scholar] [CrossRef]
- Wraith, D.C.; Nicholson, L.B. The adaptive immune system in diseases of the central nervous system. J. Clin. Investig. 2012, 122, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Medina, K.L. Overview of the immune system. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 133, pp. 61–76. [Google Scholar]
- Mamik, M.K.; Power, C. Inflammasomes in neurological diseases: Emerging pathogenic and therapeutic concepts. Brain 2017, 140, 2273–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedlack, R.S.; Cudkowicz, M.E. Clinical Trials in Progressive Neurological Diseases. In Clinical Trials in the Neurosciences; Karger Publishers: Basel, Switzerland, 2009; Volume 25, pp. 144–151. [Google Scholar]
- Mitchell, A.J.; Kemp, S.; Benito-León, J.; Reuber, M. The influence of cognitive impairment on health-related quality of life in neurological disease. Acta Neuropsychiatr. 2010, 22, 2–13. [Google Scholar] [CrossRef]
- Amantea, D.; Petrelli, F.; Greco, R.; Tassorelli, C.; Corasaniti, M.T.; Tonin, P.; Bagetta, G. Azithromycin Affords Neuroprotection in Rat Undergone Transient Focal Cerebral Ischemia. Front. Neurosci. 2019, 13, 1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, Y.C.; Kriz, J. Differential neuroprotective effects of a minocycline-based drug cocktail in transient and permanent focal cerebral ischemia. Exp. Neurol. 2007, 204, 433–442. [Google Scholar] [CrossRef]
- Yrjänheikki, J.; Tikka, T.; Keinänen, R.; Goldsteins, G.; Chan, P.H.; Koistinaho, J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc. Natl. Acad. Sci. USA 1999, 96, 13496–13500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2021. In MAbs; Taylor & Francis: New York, NY, USA, 2021; p. 1860476. [Google Scholar]
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to watch in 2022. In MAbs; Taylor & Francis: New York, NY, USA, 2022; p. 2014296. [Google Scholar]
- Dhillon, S. Eptinezumab: First Approval. Drugs 2020, 80, 733–739. [Google Scholar] [CrossRef]
- Hoy, S.M. Fremanezumab: First Global Approval. Drugs 2018, 78, 1829–1834. [Google Scholar] [CrossRef]
- Belin, A.C.; Ran, C.; Edvinsson, L. Calcitonin Gene-Related Peptide (CGRP) and Cluster Headache. Brain Sci. 2020, 10, 30. [Google Scholar] [CrossRef] [Green Version]
- Markham, A. Erenumab: First Global Approval. Drugs 2018, 78, 1157–1161. [Google Scholar] [CrossRef]
- Frampton, J.E. Ocrelizumab: First Global Approval. Drugs 2017, 77, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, C.A.; Budisteanu, M.; Falup-Pecurariu, C. Monoclonal antibodies—A revolutionary therapy in multiple sclerosis. Neurol. i Neurochir. Polska 2020, 54, 21–27. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef] [Green Version]
- Frampton, J.E. Inebilizumab: First Approval. Drugs 2020, 80, 1259–1264. [Google Scholar] [CrossRef]
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayzenberg, I.; Kleiter, I.; Schröder, A.; Hellwig, K.; Chan, A.; Yamamura, T.; Gold, R. Interleukin 6 Receptor Blockade in Patients with Neuromyelitis Optica Nonresponsive to Anti-CD20 Therapy. JAMA Neurol. 2013, 70, 394–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieseier, B.C.; Stüve, O.; Dehmel, T.; Goebels, N.; Leussink, V.I.; Mausberg, A.K.; Ringelstein, M.; Turowski, B.; Aktas, O.; Antoch, G.; et al. Disease Amelioration with Tocilizumab in a Treatment-Resistant Patient with Neuromyelitis Optica: Implication for cellular immune responses. JAMA Neurol. 2013, 70, 390–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, M.; Aranami, T.; Matsuoka, T.; Nakamura, M.; Miyake, S.; Yamamura, T. Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod. Rheumatol. 2013, 23, 827–831. [Google Scholar] [CrossRef]
- Yamamura, T.; Kleiter, I.; Fujihara, K.; Palace, J.; Greenberg, B.; Zakrzewska-Pniewska, B.; Patti, F.; Tsai, C.-P.; Saiz, A.; Yamazaki, H.; et al. Trial of Satralizumab in Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 2114–2124. [Google Scholar] [CrossRef]
- Traboulsee, A.; Greenberg, B.M.; Bennett, J.L.; Szczechowski, L.; Fox, E.; Shkrobot, S.; Yamamura, T.; Terada, Y.; Kawata, Y.; Wright, P.; et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: A randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020, 19, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in Aquaporin-4–Positive Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 614–625. [Google Scholar] [CrossRef]
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakamura, M.; Watanabe, S.; Takahashi, T.; Nakashima, I.; Takahashi, H.; Itoyama, Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007, 130, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Saadoun, S.; Waters, P.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010, 133, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Zador, Z.; Stiver, S.; Wang, V.; Manley, G.T. Role of Aquaporin-4 in Cerebral Edema and Stroke. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2009; pp. 159–170. [Google Scholar] [CrossRef] [Green Version]
- Saadoun, S.; Papadopoulos, M. Aquaporin-4 in brain and spinal cord oedema. Neuroscience 2010, 168, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.; Yang, Y.; Lu, Q.; Gao, B.; Chen, Z.; Wang, Z. Efficacy and Safety of Monoclonal Antibody Therapy in Neuromyelitis Optica Spectrum Disorders: Evidence from Randomized Controlled Trials. Mult. Scler. Relat. Disord. 2020, 43, 102166. [Google Scholar] [CrossRef]
- Ashina, M.; Saper, J.; Cady, R.; Schaeffler, B.A.; Biondi, D.M.; Hirman, J.; Pederson, S.; Allan, B.; Smith, J. Eptinezumab in episodic migraine: A randomized, double-blind, placebo-controlled study (PROMISE-1). Cephalalgia 2020, 40, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Goadsby, P.J.; Reuter, U.; Hallström, Y.; Broessner, G.; Bonner, J.H.; Zhang, F.; Sapra, S.; Picard, H.; Mikol, D.D.; Lenz, R.A. A Controlled Trial of Erenumab for Episodic Migraine. N. Engl. J. Med. 2017, 377, 2123–2132. [Google Scholar] [CrossRef]
- Dodick, D.W.; Silberstein, S.D.; Bigal, M.E.; Yeung, P.P.; Goadsby, P.J.; Blankenbiller, T.; Grozinski-Wolff, M.; Yang, R.; Ma, Y.; Aycardi, E. Effect of Fremanezumab Compared with Placebo for Prevention of Episodic Migraine: A Randomized Clinical Trial. JAMA 2018, 319, 1999–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauffer, V.L.; Dodick, D.W.; Zhang, Q.; Carter, J.N.; Ailani, J.; Conley, R.R. Evaluation of Galcanezumab for the Prevention of Episodic Migraine: The EVOLVE-1 Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1080–1088. [Google Scholar] [CrossRef] [Green Version]
- Skljarevski, V.; Matharu, M.; Millen, B.A.; Ossipov, M.H.; Kim, B.-K.; Yang, J.Y. Efficacy and safety of galcanezumab for the prevention of episodic migraine: Results of the EVOLVE-2 Phase 3 randomized controlled clinical trial. Cephalalgia 2018, 38, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Detke, H.C.; Goadsby, P.J.; Wang, S.; Friedman, D.I.; Selzler, K.J.; Aurora, S.K. Galcanezumab in chronic migraine: The randomized, double-blind, placebo-controlled REGAIN study. Neurology 2018, 91, e2211. [Google Scholar] [CrossRef] [Green Version]
- Gklinos, P.; Mitsikostas, D.D. Galcanezumab in migraine prevention: A systematic review and meta-analysis of randomized controlled trials. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420918088. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J.; Dodick, D.W.; Leone, M.; Bardos, J.N.; Oakes, T.M.; Millen, B.A.; Zhou, C.; Dowsett, S.A.; Aurora, S.K.; Ahn, A.H.; et al. Trial of Galcanezumab in Prevention of Episodic Cluster Headache. N. Engl. J. Med. 2019, 381, 132–141. [Google Scholar] [CrossRef]
- Kokoti, L.; Drellia, K.; Papadopoulos, D.; Mitsikostas, D.D. Placebo and nocebo phenomena in anti- CGRP monoclonal antibody trials for migraine prevention: A meta-analysis. J. Neurol. 2020, 267, 1158–1170. [Google Scholar] [CrossRef]
- Drellia, K.; Kokoti, L.; Deligianni, C.I.; Papadopoulos, D.; Mitsikostas, D.D. Anti-CGRP monoclonal antibodies for migraine prevention: A systematic review and likelihood to help or harm analysis. Cephalalgia 2021, 41, 851–864. [Google Scholar] [CrossRef]
- Ashina, M.; Goadsby, P.J.; Reuter, U.; Silberstein, S.; Dodick, D.W.; Xue, F.; Zhang, F.; Lima, G.P.d.S.; Cheng, S.; Mikol, D.D. Long-term efficacy and safety of erenumab in migraine prevention: Results from a 5-year, open-label treatment phase of a randomized clinical trial. Eur. J. Neurol. 2021, 28, 1716–1725. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Silberstein, S.D.; Yeung, P.P.; Cohen, J.M.; Ning, X.; Yang, R.; Dodick, D.W. Long-term safety, tolerability, and efficacy of fremanezumab in migraine: A randomized study. Neurology 2020, 95, e2487–e2499. [Google Scholar] [CrossRef]
- Camporeale, A.; Kudrow, D.; Sides, R.; Wang, S.; Van Dycke, A.; Selzler, K.J.; Stauffer, V.L. A phase 3, long-term, open-label safety study of Galcanezumab in patients with migraine. BMC Neurol. 2018, 18, 188. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Ogihara, S.; Harada, S.; Tokuyama, S. Activation of cerebral sodium-glucose transporter type 1 function mediated by post-ischemic hyperglycemia exacerbates the development of cerebral ischemia. Neuroscience 2015, 310, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, Q.; Cai, H.; Xu, C.; Liu, G.; Li, Z. Calcitonin gene-related peptide prevents blood–brain barrier injury and brain edema induced by focal cerebral ischemia reperfusion. Regul. Pept. 2011, 171, 19–25. [Google Scholar] [CrossRef]
- Shao, B.; Zhou, Y.L.; Wang, H.; Lin, Y.S. The role of calcitonin gene-related peptide in post-stroke depression in chronic mild stress-treated ischemic rats. Physiol. Behav. 2015, 139, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Gertz, K.; Kronenberg, G.; Kälin, R.E.; Baldinger, T.; Werner, C.; Balkaya, M.; Eom, G.D.; Hellmann-Regen, J.; Kröber, J.; Miller, K.R.; et al. Essential role of interleukin-6 in post-stroke angiogenesis. Brain 2012, 135, 1964–1980. [Google Scholar] [CrossRef] [Green Version]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a Major Cytokine in the Central Nervous System. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Pawluk, H.; Woźniak, A.; Grześk, G.; Kołodziejska, R.; Kozakiewicz, M.; Kopkowska, E.; Grzechowiak, E.; Kozera, G. The Role of Selected Pro-Inflammatory Cytokines in Pathogenesis of Ischemic Stroke. Clin. Interv. Aging 2020, 15, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Mantani, P.T.; Ljungcrantz, I.; Andersson, L.; Alm, R.; Hedblad, B.; Björkbacka, H.; Nilsson, J.; Fredrikson, G.N. Circulating CD40 + and CD86 + B Cell Subsets Demonstrate Opposing Associations with Risk of Stroke. Arter. Thromb. Vasc. Biol. 2014, 34, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Pawluk, H.; Kołodziejska, R.; Grześk, G.; Kozakiewicz, M.; Woźniak, A.; Pawluk, M.; Kosinska, A.; Grześk, M.; Wojtasik, J.; Kozera, G. Selected Mediators of Inflammation in Patients with Acute Ischemic Stroke. Int. J. Mol. Sci. 2022, 23, 10614. [Google Scholar] [CrossRef]
- van Dijk, B.J.; Meijers, J.C.; Kloek, A.T.; Knaup, V.L.; Rinkel, G.J.; Morgan, B.P.; van der Kamp, M.J.; Osuka, K.; Aronica, E.; Ruigrok, Y.M.; et al. Complement C5 Contributes to Brain Injury After Subarachnoid Hemorrhage. Transl. Stroke Res. 2020, 11, 678–688. [Google Scholar] [CrossRef]
- Du, Z.; Zhang, H.; Chen, Q.; Gao, Y.; Sun, B. Intranasal Calcitonin Gene-Related Peptide Protects Against Focal Cerebral Ischemic Injury in Rats Through the Wnt/β-Catenin Pathway. Med. Sci. Monit. 2018, 24, 8860–8869. [Google Scholar] [CrossRef]
- Kimura, M.; Fujiwara, S.; Tanaka, A.; Omura, Y.; Yamashita, D.; Hinoda, T.; Sakai, N.; Kohara, N. Multiple Cerebral Hemorrhages with Microbleeds in Intravascular Large B-Cell Lymphoma. J. Stroke Cerebrovasc. Dis. 2020, 29, 104798. [Google Scholar] [CrossRef]
- Yu, C.Y.; Ng, G.; Liao, P. Therapeutic Antibodies in Stroke. Transl. Stroke Res. 2013, 4, 477–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, P.I.; Siedlak, S.L.; Aliev, G.; Zhu, X.; Cash, A.D.; Smith, M.A.; Perry, G. Oxidative stress mechanisms and potential therapeutics in Alzheimer disease. J. Neural Transm. 2005, 112, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; LaFerla, F.M. The role of nicotinic acetylcholine receptors in Alzheimer’s disease. J. Physiol. 2006, 99, 172–179. [Google Scholar] [CrossRef]
- Wenk, G.L.; Rosi, S.; McGann, K.; Hauss-Wegrzyniak, B. A nitric oxide-donating flurbiprofen derivative reduces neuroinflammation without interacting with galantamine in the rat. Eur. J. Pharmacol. 2002, 453, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Asai, T.; Oyama, D.; Agato, Y.; Yasuda, N.; Fukuta, T.; Shimizu, K.; Minamino, T.; Oku, N. Treatment of cerebral ischemia-reperfusion injury with PEGylated liposomes encapsulating FK506. FASEB J. 2013, 27, 1362–1370. [Google Scholar] [CrossRef]
- Peng, T.; Britton, G.L.; Kim, H.; Cattano, D.; Aronowski, J.; Grotta, J.; McPherson, D.D.; Huang, S.-L. Therapeutic Time Window and Dose Dependence of Xenon Delivered via Echogenic Liposomes for Neuroprotection in Stroke. CNS Neurosci. Ther. 2013, 19, 773–784. [Google Scholar] [CrossRef]
- Qu, C.; Ni, J.; Yang, P.; Zhang, H.; Lin, L.; Li, J.; Tian, J.; Fu, J.; Yin, X.; Kong, H. Altered plasma and brain disposition of isopropylidene shikimic acid liposome in rats and the brain protection in cerebral ischemia–reperfusion. Drug Dev. Ind. Pharm. 2013, 39, 1291–1295. [Google Scholar] [CrossRef]
- Migliore, M.; Ortiz, R.; Dye, S.; Campbell, R.; Amiji, M.; Waszczak, B. Neurotrophic and neuroprotective efficacy of intranasal GDNF in a rat model of Parkinson’s disease. Neuroscience 2014, 274, 11–23. [Google Scholar] [CrossRef]
- Babcock, A.A.; Wirenfeldt, M.; Holm, T.; Nielsen, H.H.; Dissing-Olesen, L.; Toft-Hansen, H.; Millward, J.M.; Landmann, R.; Rivest, S.; Finsen, B.; et al. Toll-Like Receptor 2 Signaling in Response to Brain Injury: An Innate Bridge to Neuroinflammation. J. Neurosci. 2006, 26, 12826–12837. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.-C.; Arumugam, T.V.; Xu, X.; Cheng, A.; Mughal, M.R.; Jo, D.-G.; Lathia, J.D.; Siler, D.A.; Chigurupati, S.; Ouyang, X.; et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 13798–13803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caso, J.; Pradillo, J.; Hurtado, O.; Lorenzo, P.; Moro, M.A.; Lizasoain, I. Toll-Like Receptor 4 Is Involved in Brain Damage and Inflammation After Experimental Stroke. Circulation 2007, 115, 1599–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, C.-X.; Yang, Q.-W.; Lv, F.-L.; Cui, J.; Fu, H.-B.; Wang, J.-Z. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem. Biophys. Res. Commun. 2007, 353, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.T.; Puskas, F.; Agoston, V.A.; Cleveland, J.C., Jr.; Freeman, K.A.; Gamboni, F.; Herson, P.S.; Meng, X.; Smith, P.D.; Weyant, M.J.; et al. Toll-Like Receptor 4–Dependent Microglial Activation Mediates Spinal Cord Ischemia–Reperfusion Injury. Circulation 2013, 128, S152–S156. [Google Scholar] [CrossRef] [Green Version]
- Chekhonin, V.P.; Lebedev, S.V.; Ryabukhin, I.A.; Petrov, S.V.; Gurina, O.I.; Dmitrieva, T.B.; Volkov, A.I.; Kashparov, I.A.; Skoblov, Y.S. Selective accumulation of monoclonal antibodies against neurospecific enolase in brain tissue of rats with middle cerebral artery occlusion. Bull. Exp. Biol. Med. 2004, 138, 343–347. [Google Scholar] [CrossRef]
- Andresen, L.; Theodorou, K.; Grünewald, S.; Czech-Zechmeister, B.; Könnecke, B.; Lühder, F.; Trendelenburg, G. Evaluation of the Therapeutic Potential of Anti-TLR4-Antibody MTS510 in Experimental Stroke and Significance of Different Routes of Application. PLoS ONE 2016, 11, e0148428. [Google Scholar] [CrossRef]
- Qiang, M.; Dong, X.; Zha, Z.; Zuo, X.-K.; Song, X.-L.; Zhao, L.; Yuan, C.; Huang, C.; Tao, P.; Hu, Q.; et al. Selection of an ASIC1a-blocking combinatorial antibody that protects cells from ischemic death. Proc. Natl. Acad. Sci. USA 2018, 115, E7469–E7477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, T.N.; Hamilton, J.R.; Morel-Kopp, M.-C.; Zheng, Z.; Chen, T.-Y.T.; Hearn, J.I.; Sun, P.P.; Flanagan, J.U.; Young, D.; Barber, P.A.; et al. Inhibition of NMDA receptor function with an anti-GluN1-S2 antibody impairs human platelet function and thrombosis. Platelets 2017, 28, 799–811. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Liu, K.; Wake, H.; Teshigawara, K.; Mori, S.; Nishibori, M. Anti-high mobility group box-1 (HMGB1) antibody inhibits hemorrhage-induced brain injury and improved neurological deficits in rats. Sci. Rep. 2017, 7, srep46243. [Google Scholar] [CrossRef]
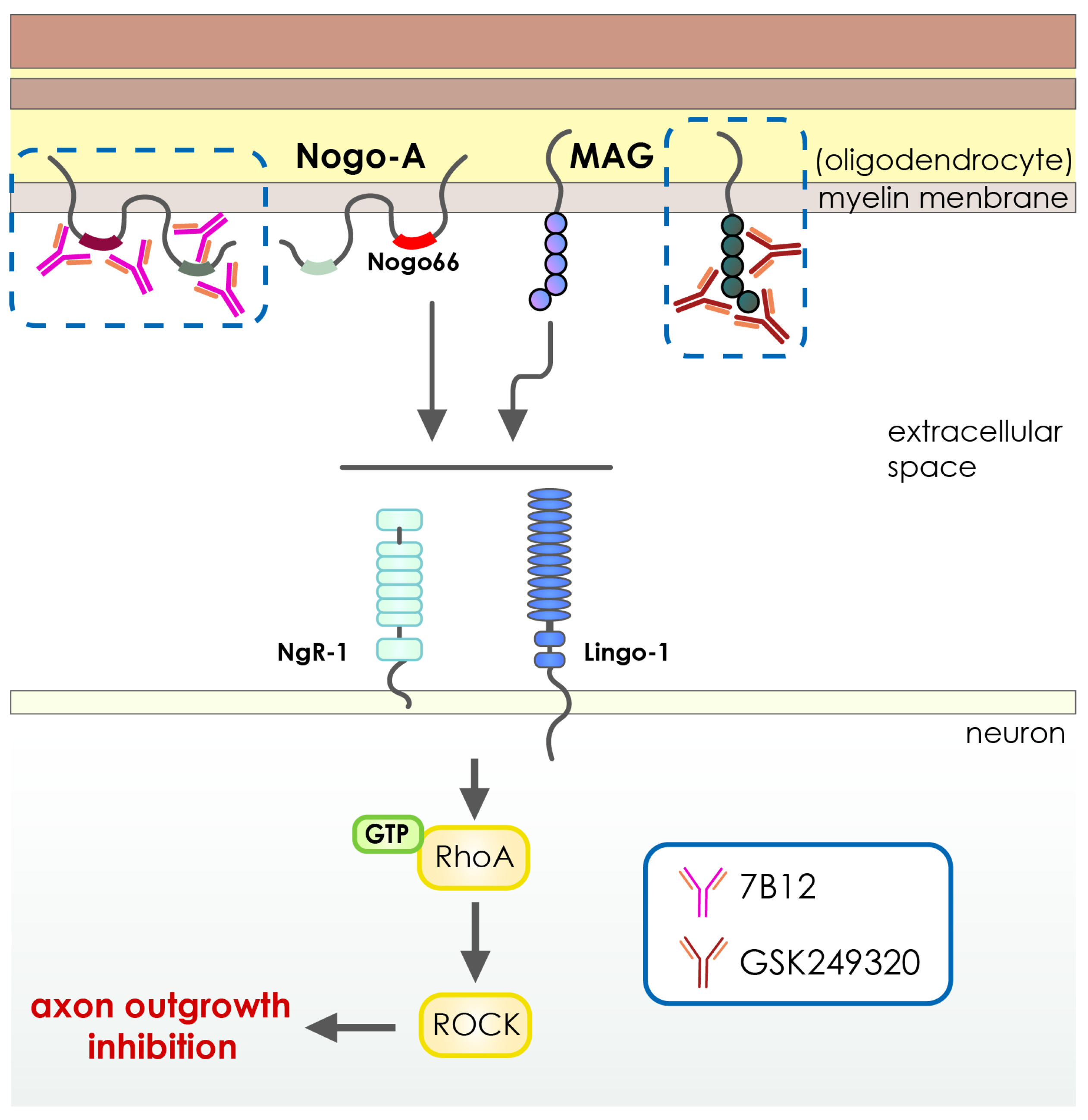
- Cash, D.; Easton, A.C.; Mesquita, M.; Beech, J.; Williams, S.; Lloyd, A.; Irving, E.; Cramer, S.C. GSK249320, A Monoclonal Antibody Against the Axon Outgrowth Inhibition Molecule Myelin-Associated Glycoprotein, Improves Outcome of Rodents with Experimental Stroke. J. Neurol. Exp. Neurosci. 2016, 2, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Wiessner, C.; Bareyre, F.M.; Allegrini, P.R.; Mir, A.K.; Frentzel, S.; Zurini, M.; Schnell, L.; Oertle, T.; Schwab, M.E. Anti—Nogo-A Antibody Infusion 24 Hours after Experimental Stroke Improved Behavioral Outcome and Corticospinal Plasticity in Normotensive and Spontaneously Hypertensive Rats. J. Cereb. Blood Flow Metab. 2003, 23, 154–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilmes, M.; Espinoza, C.P.; Ludewig, P.; Stabernack, J.; Liesz, A.; Nicke, A.; Gelderblom, M.; Gerloff, C.; Falzoni, S.; Tolosa, E.; et al. Blocking P2X7 by intracerebroventricular injection of P2X7-specific nanobodies reduces stroke lesions. J. Neuroinflammation 2022, 19, 256. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nature 1997, 386, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Gründer, S.; Chen, X. Structure, function, and pharmacology of acid-sensing ion channels (ASICs): Focus on ASIC1a. Int. J. Physiol. Pathophysiol. Pharmacol. 2010, 2, 73–94. [Google Scholar] [PubMed]
- Xiong, Z.-G.; Zhu, X.-M.; Chu, X.-P.; Minami, M.; Hey, J.; Wei, W.-L.; MacDonald, J.F.; Wemmie, J.A.; Price, M.P.; Welsh, M.J.; et al. Neuroprotection in Ischemia: Blocking Calcium-Permeable Acid-Sensing Ion Channels. Cell 2004, 118, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Duan, B.; Wang, D.-G.; Deng, X.-H.; Zhang, G.-Y.; Xu, L.; Xu, T.-L. Coupling between NMDA Receptor and Acid-Sensing Ion Channel Contributes to Ischemic Neuronal Death. Neuron 2005, 48, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-Z.; Wang, J.-J.; Huang, Y.; Liu, F.; Zeng, W.-Z.; Li, Y.; Xiong, Z.-G.; Zhu, M.X.; Xu, T.-L. Tissue acidosis induces neuronal necroptosis via ASIC1a channel independent of its ionic conduction. eLife 2015, 4, e05682. [Google Scholar] [CrossRef]
- Petroff, E.; Leonard, A.S.; Schnizler, M.K.; Abboud, F.M.; Welsh, M.J. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. USA 2004, 101, 6752–6757. [Google Scholar] [CrossRef] [Green Version]
- Baron, A.; Lingueglia, E. Pharmacology of acid-sensing ion channels—Physiological and therapeutical perspectives. Neuropharmacology 2015, 94, 19–35. [Google Scholar] [CrossRef]
- Li, M.-H.; Inoue, K.; Si, H.-F.; Xiong, Z.-G. Calcium-permeable ion channels involved in glutamate receptor-independent ischemic brain injury. Acta Pharmacol. Sin. 2011, 32, 734–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-J.; Liu, F.; Yang, F.; Wang, Y.-Z.; Qi, X.; Li, Y.; Hu, Q.; Zhu, M.X.; Xu, T.-L. Disruption of auto-inhibition underlies conformational signaling of ASIC1a to induce neuronal necroptosis. Nat. Commun. 2020, 11, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusser, S.A.; Pless, S.A. Acid-sensing ion channels as potential therapeutic targets. Trends Pharmacol. Sci. 2021, 42, 1035–1050. [Google Scholar] [CrossRef]
- Li, S.; Carmichael, S.T. Growth-associated gene and protein expression in the region of axonal sprouting in the aged brain after stroke. Neurobiol. Dis. 2006, 23, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Cheatwood, J.L.; Emerick, A.J.; Schwab, M.E.; Kartje, G.L. Nogo-A Expression After Focal Ischemic Stroke in the Adult Rat. Stroke 2008, 39, 2091–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, M.N.; Ma, H.K.; Arakawa, S.; Howells, D.W.; Markus, R.; Rowe, C.C.; Donnan, G.A. Inflammation following stroke. J. Clin. Neurosci. 2006, 13, 1–8. [Google Scholar] [CrossRef]
- Malone, K.; Amu, S.; Moore, A.C.; Waeber, C. Immunomodulatory Therapeutic Strategies in Stroke. Front. Pharmacol. 2019, 10, 630. [Google Scholar] [CrossRef] [Green Version]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Huang, J.; Upadhyay, U.M.; Tamargo, R.J. Inflammation in stroke and focal cerebral ischemia. Surg. Neurol. 2006, 66, 232–245. [Google Scholar] [CrossRef]
- Gelderblom, M.; Sobey, C.G.; Kleinschnitz, C.; Magnus, T. Danger signals in stroke. Ageing Res. Rev. 2015, 24, 77–82. [Google Scholar] [CrossRef]
- Hattori, M.; Gouaux, E. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 2012, 485, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Llovera, G.; Hofmann, K.; Roth, S.; Salas-Pérdomo, A.; Ferrer-Ferrer, M.; Perego, C.; Zanier, E.R.; Mamrak, U.; Rex, A.; Party, H.J.S.t.m. Results of a preclinical randomized controlled multicenter trial (pRCT): Anti-CD49d treatment for acute brain ischemia. Sci. Transl. Med. 2015, 7, 299ra121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, B.; Masliah, E. Immunotherapy for Alzheimer’s disease: Past, present and future. Front. Aging Neurosci. 2014, 6, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajes, M.; Ramos-Fernández, E.; Weng-Jiang, X.; Bosch-Morató, M.; Guivernau, B.; Eraso-Pichot, A.; Salvador, B.; Fernàndez-Busquets, X.; Roquer, J.; Muñoz, F.J. The blood-brain barrier: Structure, function and therapeutic approaches to cross it. Mol. Membr. Biol. 2014, 31, 152–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
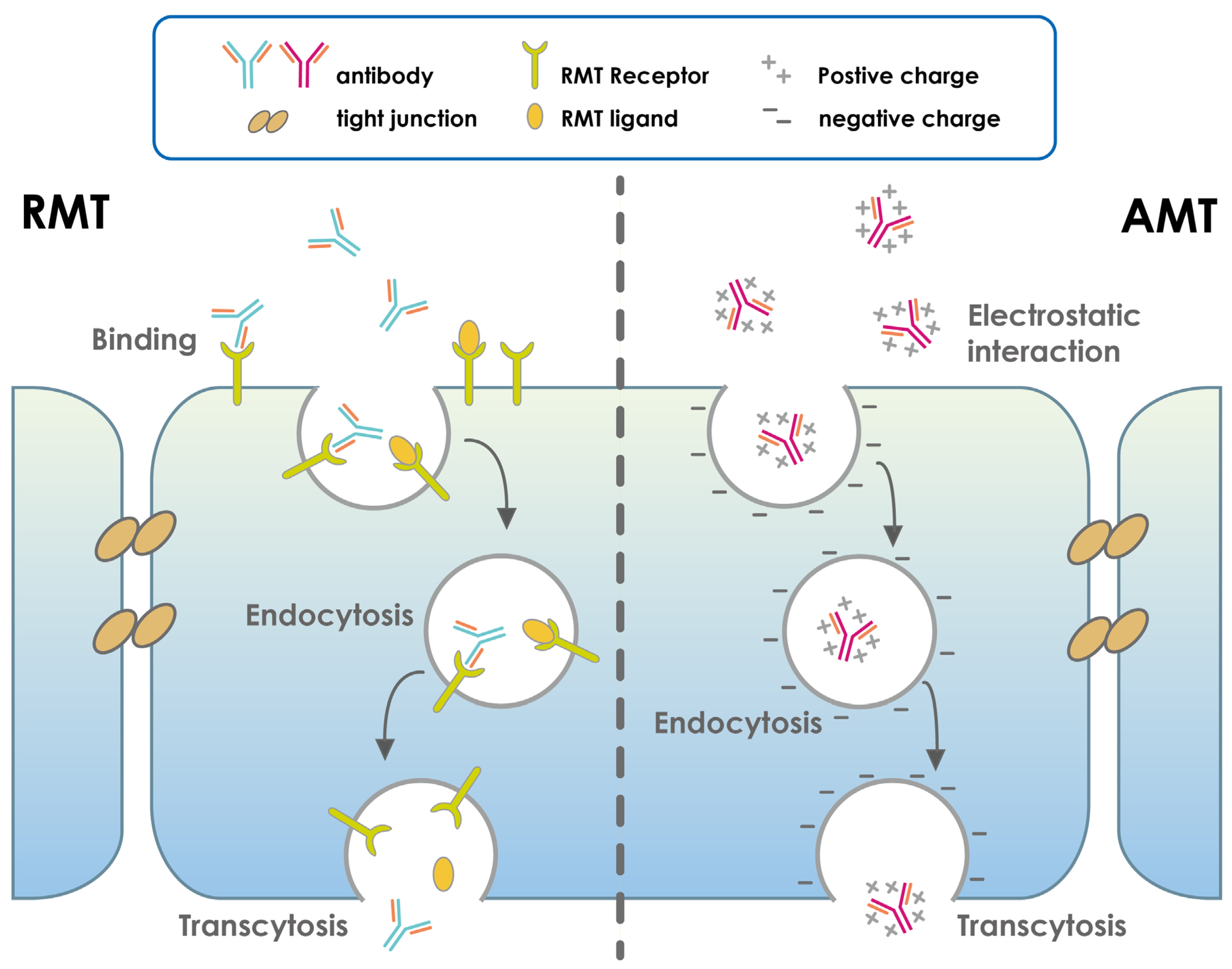
- Pulgar, V.M. Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front. Neurosci. 2018, 12, 1019. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Boado, R.J. Reengineering Biopharmaceuticals for Targeted Delivery Across the Blood–Brain Barrier. Methods Enzymol. 2012, 503, 269–292. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug Transport across the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Coloma, M.J.; Lee, H.J.; Kurihara, A.; Landaw, E.M.; Boado, R.J.; Morrison, S.L.; Pardridge, W.M. Transport across the primate blood-brain barrier of a genetically engineered chimeric monoclonal antibody to the human insulin receptor. Pharm. Res. 2000, 17, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-brain barrier drug targeting: The future of brain drug development. Mol. Interv. 2003, 3, 90–105. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pardridge, W.M. Blood–brain barrier targeting of BDNF improves motor function in rats with middle cerebral artery occlusion. Brain Res. 2006, 1111, 227–229. [Google Scholar] [CrossRef]
- Bien-Ly, N.; Yu, Y.J.; Bumbaca, D.; Elstrott, J.; Boswell, C.A.; Zhang, Y.; Luk, W.; Lu, Y.; Dennis, M.S.; Weimer, R.M.; et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 2014, 211, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.J.; Zhang, Y.; Kenrick, M.; Hoyte, K.; Luk, W.; Lu, Y.; Atwal, J.; Elliott, J.M.; Prabhu, S.; Watts, R.J.; et al. Boosting Brain Uptake of a Therapeutic Antibody by Reducing Its Affinity for a Transcytosis Target. Sci. Transl. Med. 2011, 3, 84ra44. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Wu, D.; Sakane, T. Combined use of carboxyl-directed protein pegylation and vector-mediated blood-brain barrier drug delivery system optimizes brain uptake of brain-derived neurotrophic factor following intravenous administration. Pharm. Res. 1998, 15, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yang, J.; Pardridge, W.M. Drug targeting of a peptide radiopharmaceutical through the primate blood-brain barrier in vivo with a monoclonal antibody to the human insulin receptor. J. Clin. Investig. 1997, 100, 1804–1812. [Google Scholar] [CrossRef]
- Patel, M.M.; Goyal, B.R.; Bhadada, S.V.; Bhatt, J.S.; Amin, A.F. Getting into the Brain. CNS Drugs 2009, 23, 35–58. [Google Scholar] [CrossRef]
- Qian, Z.M.; Li, H.; Sun, H.; Ho, K. Targeted Drug Delivery via the Transferrin Receptor-Mediated Endocytosis Pathway. Pharmacol. Rev. 2002, 54, 561–587. [Google Scholar] [CrossRef]
- Cannon, R.E.; Peart, J.C.; Hawkins, B.T.; Campos, C.R.; Miller, D.S. Targeting blood-brain barrier sphingolipid signaling reduces basal P-glycoprotein activity and improves drug delivery to the brain. Proc. Natl. Acad. Sci. USA 2012, 109, 15930–15935. [Google Scholar] [CrossRef] [Green Version]
- Gorin, F.; Harley, W.; Schnier, J.; Lyeth, B.; Jue, T. Perinecrotic glioma proliferation and metabolic profile within an intracerebral tumor xenograft. Acta Neuropathol. 2004, 107, 235–244. [Google Scholar] [CrossRef]
- Guo, L.; Ren, J.; Jiang, X. Perspectives on brain-targeting drug delivery systems. Curr. Pharm. Biotechnol. 2012, 13, 2310–2318. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood–brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- Xin, H.; Sha, X.; Jiang, X.; Zhang, W.; Chen, L.; Fang, X. Anti-glioblastoma efficacy and safety of paclitaxel-loading Angiopep-conjugated dual targeting PEG-PCL nanoparticles. Biomaterials 2012, 33, 8167–8176. [Google Scholar] [CrossRef]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.-E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-H.; Wen, C.-J.; Al-Suwayeh, S.; Chang, H.-W.; Yen, T.-C.; Fang, J.-Y. Physicochemical characterization and in vivo bioluminescence imaging of nanostructured lipid carriers for targeting the brain: Apomorphine as a model drug. Nanotechnology 2010, 21, 405101. [Google Scholar] [CrossRef] [Green Version]
- Geldenhuys, W.J.; Allen, D.D. The blood-brain barrier choline transporter. Cent. Nerv. Syst. Agents Med. Chem. 2012, 12, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-T.; Li, W.; Meng, G.; Wang, P.; Liao, W. Strategies for transporting nanoparticles across the blood–brain barrier. Biomater. Sci. 2016, 4, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Gabathuler, R. Approaches to transport therapeutic drugs across the blood–brain barrier to treat brain diseases. Neurobiol. Dis. 2010, 37, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M.; Patel, B.M. Crossing the Blood–Brain Barrier: Recent Advances in Drug Delivery to the Brain. CNS Drugs 2017, 31, 109–133. [Google Scholar] [CrossRef]
- Zhu, X.; Jin, K.; Huang, Y.; Pang, Z. Brain drug delivery by adsorption-mediated transcytosis. In Brain Targeted Drug Delivery System; Elsevier: Amsterdam, The Netherlands, 2019; pp. 159–183. [Google Scholar]
- Soni, V.; Jain, A.; Khare, P.; Gulbake, A.; Jain, S.K. Potential approaches for drug delivery to the brain: Past, present, and future. Crit. Rev. Ther. Drug Carr. Syst. 2010, 27, 187–236. [Google Scholar] [CrossRef]
- Pardridge, W.M. Re-Engineering Biopharmaceuticals for Delivery to Brain with Molecular Trojan Horses. Bioconjugate Chem. 2008, 19, 1327–1338. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug and gene targeting to the brain with molecular trojan horses. Nat. Rev. Drug Discov. 2002, 1, 131–139. [Google Scholar] [CrossRef]
- Pardridge, W.M. Molecular Trojan horses for blood-brain barrier drug delivery. Discov. Med. 2009, 6, 139–143. [Google Scholar] [CrossRef] [PubMed]
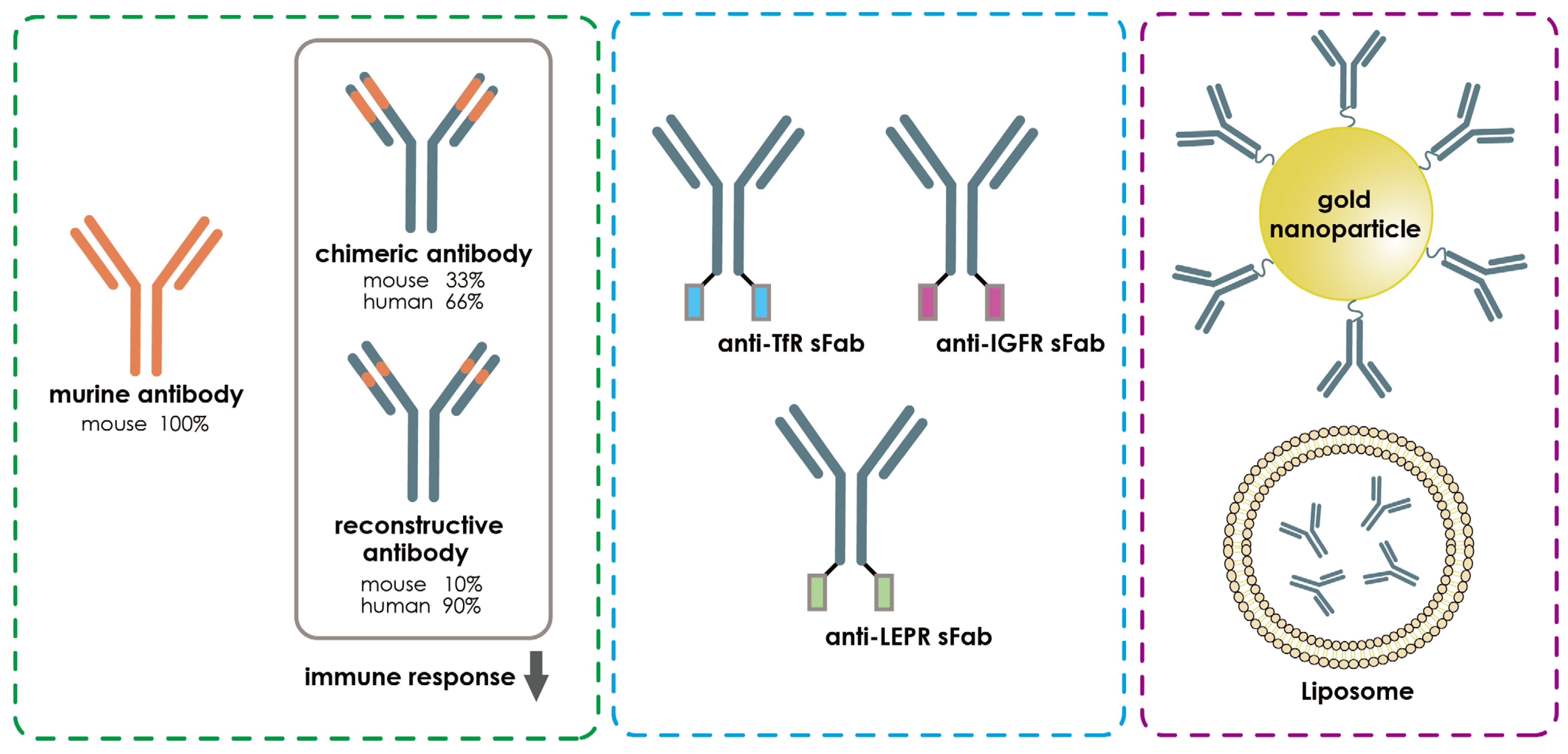
- Da Silva, F.A.; Corte-Real, S.; Goncalves, J. Recombinant Antibodies as Therapeutic Agents. BioDrugs 2008, 22, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Thangudu, S.; Cheng, F.-Y.; Su, C.-H. Advancements in the Blood–Brain Barrier Penetrating Nanoplatforms for Brain Related Disease Diagnostics and Therapeutic Applications. Polymers 2020, 12, 3055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mazouzi, Y.; Salmain, M.; Liedberg, B.; Boujday, S. Antibody-Gold Nanoparticle Bioconjugates for Biosensors: Synthesis, Characterization and Selected Applications. Biosens. Bioelectron. 2020, 165, 112370. [Google Scholar] [CrossRef]
- Sun, Z.-T.; Ma, C.; Li, G.-J.; Zheng, X.-Y.; Hao, Y.-T.; Yang, Y.; Wang, X. Application of Antibody Fragments Against Aβ With Emphasis on Combined Application with Nanoparticles in Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 654611. [Google Scholar] [CrossRef] [PubMed]
- Gopalan, D.; Pandey, A.; Alex, A.T.; Kalthur, G.; Pandey, S.; Udupa, N.; Mutalik, S. Nanoconstructs as a versatile tool for detection and diagnosis of Alzheimer biomarkers. Nanotechnology 2021, 32, 142002. [Google Scholar] [CrossRef] [PubMed]
- Neves, V.; Aires-Da-Silva, F.; Corte-Real, S.; Castanho, M.A.R.B. Antibody Approaches to Treat Brain Diseases. Trends Biotechnol. 2016, 34, 36–48. [Google Scholar] [CrossRef]
- Bai, B.; Yan, Z.; Hao, Y.; Zhang, Z.; Li, G.; Dekker, J.; Qiu, C. A randomised controlled multimodal intervention trial in patients with ischaemic stroke in Shandong, China: Design and rationale. Lancet 2017, 390, S13. [Google Scholar] [CrossRef] [Green Version]
- Cramer, S.C.; Enney, L.A.; Russell, C.K.; Simeoni, M.; Thompson, T.R. Proof-of-Concept Randomized Trial of the Monoclonal Antibody GSK249320 Versus Placebo in Stroke Patients. Stroke 2017, 48, 692–698. [Google Scholar] [CrossRef] [Green Version]
- Ruck, T.; Nimmerjahn, F.; Wiendl, H.; Lünemann, J.D. Next-generation antibody-based therapies in neurology. Brain 2022, 145, 1229–1241. [Google Scholar] [CrossRef]
- Kumar, A.; Aakriti; Gupta, V. A review on animal models of stroke: An update. Brain Res. Bull. 2016, 122, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Wairkar, S. Biotechnology-based therapeutics for management of cerebral stroke. Eur. J. Pharmacol. 2021, 913, 174638. [Google Scholar] [CrossRef] [PubMed]
- West, F.D.; Kaiser, E.E. Large animal ischemic stroke models: Replicating human stroke pathophysiology. Neural Regen. Res. 2020, 15, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Fluri, F.; Schuhmann, M. Animal models of ischemic stroke and their application in clinical research. Drug Des. Dev. Ther. 2015, 9, 3445–3454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, T.; Tagashira, Y.; Egashira, S.; Morimoto, M.; Irie, K.; Hosokawa, M.; Hayashi, T.; Egawa, T.; Hayakawa, K.; Mishima, K. Therapeutic effect of anti-HMGB1 antibody in a mouse model of 4-h middle cerebral artery occlusion: Comparison with tissue plasminogen activator. Neuroreport 2022, 33, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Koopman, I.; Tack, R.W.; Rinkel, G.J.; Vergouwen, M.D. CLASH study group Abstract 96: CompLement C5 Antibodies for Decreasing Brain Injury After Aneurysmal Subarachnoid Hemorrhage (CLASH): A Randomized Controlled Phase II Clinical Trial. Stroke 2022, 53, A96. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generic Names | Antigens | Designs | Neurological Conditions | Administration | FDA Approval | Funders |
|---|---|---|---|---|---|---|
| Inebilizumab-cdon | CD19 | Humanized IgG1 | Neuromyelitis optica and neuromyelitis optica spectrum disorders | Intravenous infusion | 2021 | Viela Bio |
| Satralizumab-mwge | IL-6R | Humanized IgG2 | Neuromyelitis optica spectrum disorders | Subcutaneous injection | 2020 | Genentech Inc. |
| Tocilizumab | IL-6R | Humanized IgG1 | Neuromyelitis optica spectrum disorder | Intravenous infusion | 2019 | Chugai/Roche |
| Eculizumab | C5 complement protein | Humanized IgG2 | Neuromyelitis optica spectrum disorder, Myasthenia gravis | Intravenous infusion | 2019, 2018, respectively | Alexion Pharmaceuticals |
| Eptinezumab | CGRP ligand | Humanized IgG1 | Episodic or chronic migraine | Intravenous infusion | 2020 | Lundbeck Seattle Biopharmaceuticals, Inc. |
| Galcanezumab-gnlm | CGRP ligand | Humanized IgG4 | Cluster headache or chronic migraine | Subcutaneous injection | 2019 | Eli Lilly and Company |
| Fremanezumabvfrm | CGRP ligand | Humanized IgG2 | Episodic or chronic migraine | Subcutaneous injection | 2018 | Teva Pharmaceuticals |
| Erenumab-aooe | CGRP receptor | Human IgG2 | Episodic or chronic migraine | Subcutaneous injection | 2018 | Amgen and Novartis |
| Ocrelizumab | CD20 | Humanized IgG1 | Relapsing-remitting Multiple sclerosis | Intravenous infusion | 2017 | Hoffmann-La Roche |
| Antibody | Antigen | Design | Route | Main Findings | Citation |
|---|---|---|---|---|---|
| mABs clone MTS510 | TLR4 | Recombinant | Intravenous infusion |
| [112] |
| ASC06-IgG1 | ASIC blocker | Recombinant IgG1 | Intravenous infusion |
| [113] |
| Anti-GluN1 antibodies | Blocks NMDA receptor associated calcium influx | Recombinant | Subcutaneous injection |
| [114] |
| Anti-High Mobility Group Box-1 mAbs | HMGB1, inflammatory proteins | Recombinant | Intravenous infusion |
| [115] |
| GSK249320 | MAG | Humanized recombinant IgG1 | Intravenous infusion |
| [116] |
| Anti-Nogo-A antibody 7B12 | NAM-associated neurite outgrowth protein | Recombinant IgG1 | Intravenous infusion |
| [117] |
| Anti-P2X7 antibodies | Adenosine triphosphate (ATP) | Recombinant nanobodies | Intravenous and icv injections |
| [118] |
| Pros | Cons | |
|---|---|---|
| 1 | Monoclonal antibodies are extremely sensitive and specific for antigens. | Injection-related reactions (IRRs). They occur most frequently in a period of 10 min to 4 h after the start of antibody administration. However, anaphylactic reactions are not expected to occur during the first mAbs infusion, except in the rare case that pre-existing immunoglobulins E (IgEs) cross-react with the infused mAb. |
| 2 | Without alteration, nanobodies such as VHH can cross the blood–brain barrier. | Cytokine release syndrome (CRS) depends largely on the cell type targeted by the mAb rather than its allergenic properties. mAbs that activate T cells are most likely to cause CRS, when large amounts of proinflammatory cytokines are released by activated astrocytes and white blood cells, including B cells, T cells, natural killer cells, macrophages, dendritic cells, and monocytes. |
| 3 | Antibodies are stable, especially nanobodies in extreme conditions such as temperature and pH. | The more immunogenic the mAbs, the more likely they are to produce anti-drug antibodies (ADA). This explains why ADAs are more likely to be produced against chimeric than human mAbs, and ADA production can lead to neutralization of mAbs, rapid elimination, and loss of efficacy. |
| 4 | Nanobodies can penetrate hard-to-reach epitopes. | Opportunistic infections occur when mAbs affect immune function by depleting cell populations (e.g., ocrelizumab) or blocking immune cell migration through endothelial barriers; dizziness. |
| 5 | Bispecific antibodies (BsAbs) can facilitate transport across the BBB. They are designed to contain one arm with specificity against a BBB RMT receptor that drives their migration across the BBB and a second arm with a therapeutic function that produces the pharmacological effect when the BsAb encounters the target. | MAb-associated malignancy. In a phase III study of the treatment of primary progressive multiple sclerosis with ocrelizumab, an anti-CD20, B-cell-depleting mAb, 11 cases of malignancy were reported in the active treatment group, 4 of which were adenocarcinomas of the breast [57]; back pain; diarrhea. |
| 6 | Antibodies can be easily cleared from the system without causing damage to the tissues as compared to chemotherapy. | Autoimmune disorders; nasopharyngitis |
| Receptor-Mediated Targets (RMT) | Citations | |
|---|---|---|
| 1 | Transferrin receptor (TfR) | [151,152,153] |
| 2 | Insulin receptor (IR) and Diphtheria toxin receptor (DTR) | [154,155] |
| 3 | Low-density lipoprotein-related protein (LDLRP) | [154,156] |
| 4 | Insulin-like growth factor receptor (IGFR) | [154] |
| 5 | Nicotinic acetylcholine receptor (NACR) | [154,155,157] |
| 6 | Leptin receptor (LEPR) and Scavenger receptor, Class B | [154,155,158,159] |
| 7 | Fc fragment of IgG receptor transports alpha (FCGRT) | [155,158,160] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.-M.; Yen, T.-L.; Jan, J.-S.; Mwale, P.F.; Teng, R.-D.; Taliyan, R.; Hsieh, C.-T.; Yang, C.-H. Advances in Antibody-Based Therapeutics for Cerebral Ischemia. Pharmaceutics 2023, 15, 145. https://doi.org/10.3390/pharmaceutics15010145
Sun J-M, Yen T-L, Jan J-S, Mwale PF, Teng R-D, Taliyan R, Hsieh C-T, Yang C-H. Advances in Antibody-Based Therapeutics for Cerebral Ischemia. Pharmaceutics. 2023; 15(1):145. https://doi.org/10.3390/pharmaceutics15010145
Chicago/Turabian StyleSun, Jui-Ming, Ting-Lin Yen, Jing-Shiun Jan, Pharaoh Fellow Mwale, Ruei-Dun Teng, Rajeev Taliyan, Cheng-Ta Hsieh, and Chih-Hao Yang. 2023. "Advances in Antibody-Based Therapeutics for Cerebral Ischemia" Pharmaceutics 15, no. 1: 145. https://doi.org/10.3390/pharmaceutics15010145
APA StyleSun, J. -M., Yen, T. -L., Jan, J. -S., Mwale, P. F., Teng, R. -D., Taliyan, R., Hsieh, C. -T., & Yang, C. -H. (2023). Advances in Antibody-Based Therapeutics for Cerebral Ischemia. Pharmaceutics, 15(1), 145. https://doi.org/10.3390/pharmaceutics15010145