

Anticancer Drug Conjugates Incorporating Estrogen Receptor Ligands


Abstract
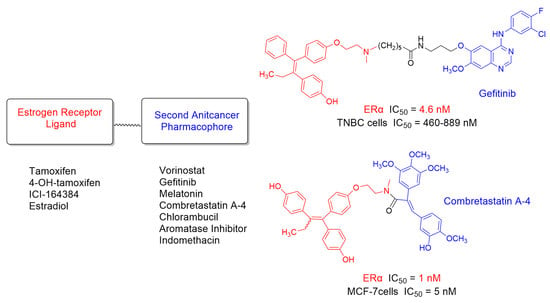
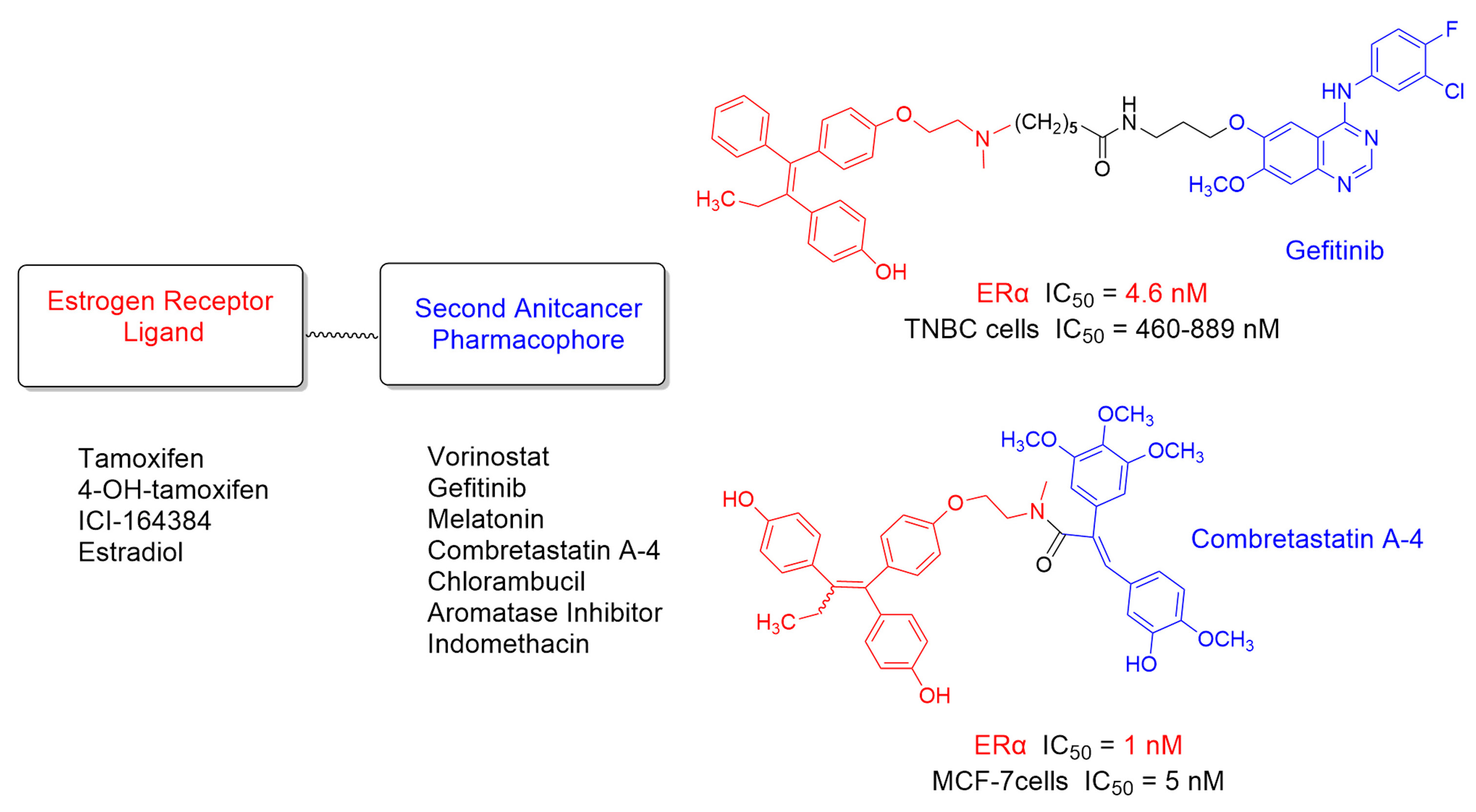
:
1. Introduction
2. Hybrid Ligands Incorporating Tamoxifen
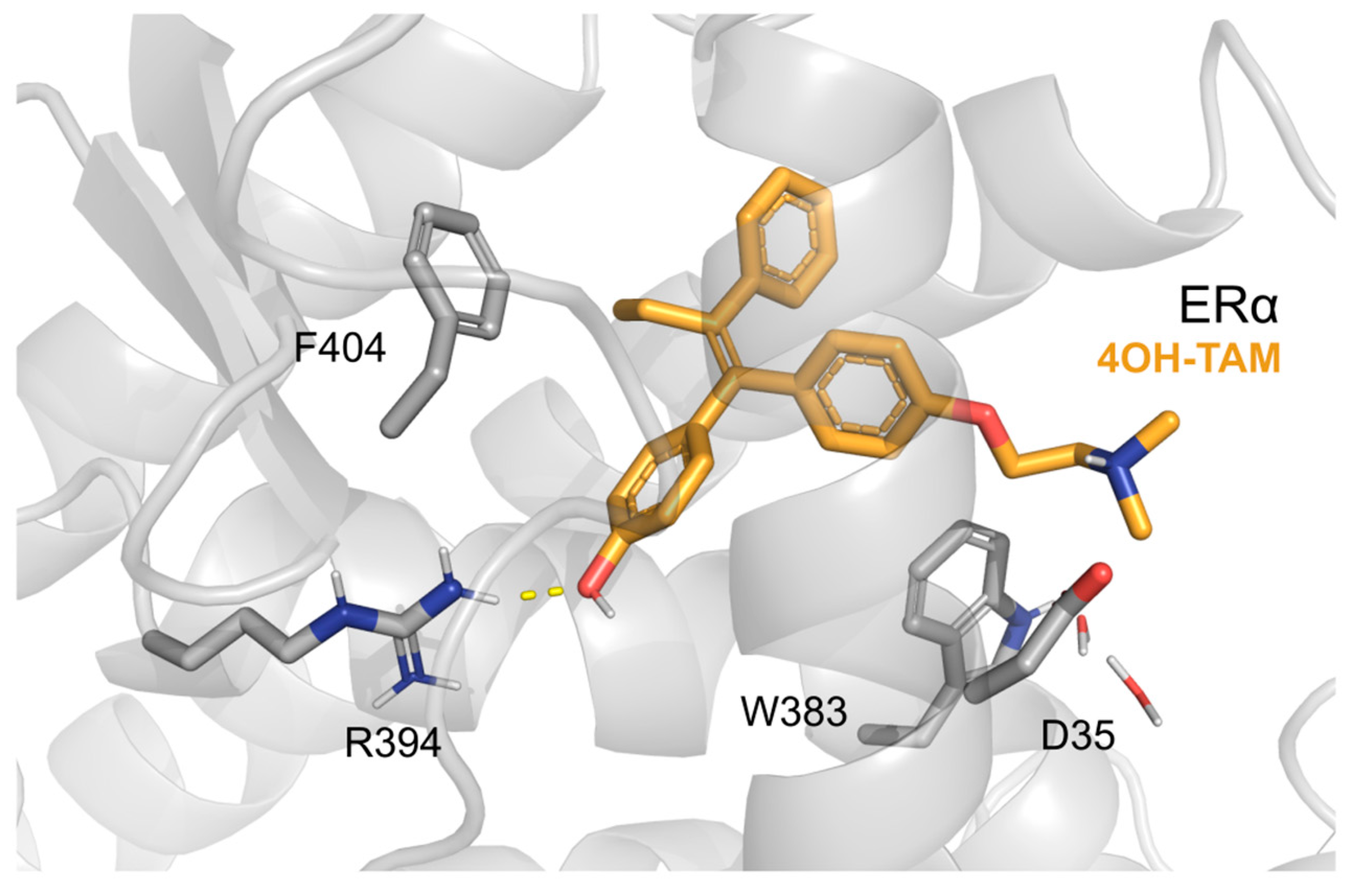
3. Hybrid Ligands Incorporating 4-Hydroxytamoxifen
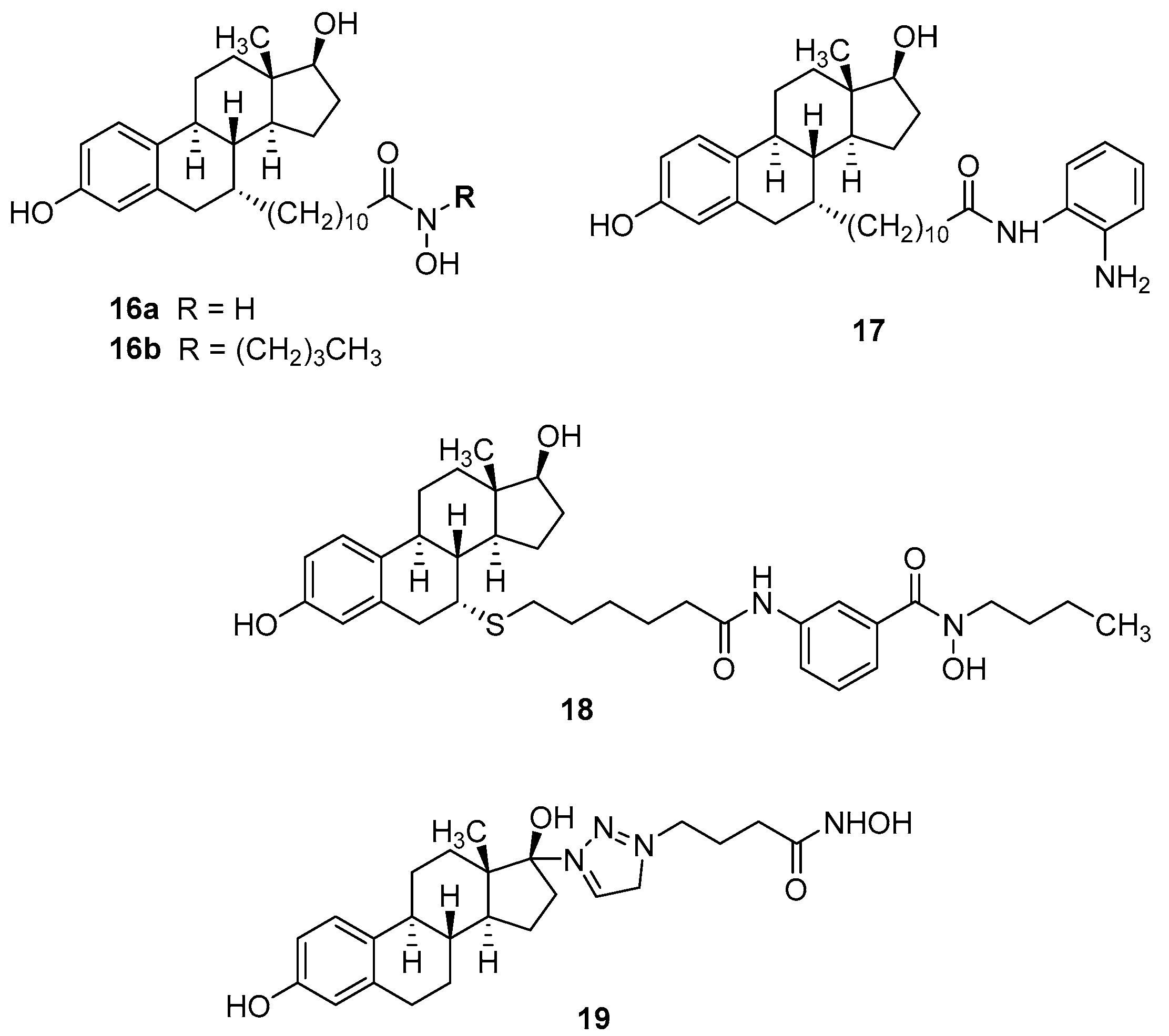
4. Hybrid Ligands Incorporating Steroids
5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fortin, S.; Bérubé, G. Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2013, 8, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Singh, P.; Koorbanally, N.; Raj, R.; Kumur, V. Recent advances (2015–2016) in anticancer hybrids. Eur. J. Med. Chem. 2017, 142, 179–212. [Google Scholar] [CrossRef] [PubMed]
- Ivasiv, V.; Albertini, C.; Gonzalves, A.E.; Rossi, M.; Bolognesi, M.L. Molecular Hybridization as a Tool for Designing Multitarget Drug Candidates for Complex Diseases. Curr. Top. Med. Chem. 2019, 19, 1694–1711. [Google Scholar] [CrossRef] [PubMed]
- Kucuksayan, E.; Ozben, T. Hybrid Compounds as Multitarget Directed Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 907–918. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://pubmed.ncbi.nlm.nih.gov/?term=anticancer+hybrids (accessed on 22 December 2022).
- Cai, X.; Zhai, H.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.; Bao, R.; Qian, C. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptan amide (CUDC-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef]
- Available online: https://adisinsight.springer.com/drugs/800028795 (accessed on 22 December 2022).
- Younes, A.; Berdeja, J.G.; Patel, M.R.; Flinn, I.; Gerecitano, J.F.; Neelapu, S.S.; Kelly, K.R.; Copeland, A.R.; Akins, A.; Clancy, M.S.; et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: An open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2016, 17, 622–631. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.; Leak, R.K.; Stradfort, R.; Zlotos, D.P.; Witt-Enderby, P.A. Drug conjugates—An emerging approach to treat breast cancer. Pharmacol. Res. Perspect. 2018, 6, e00417. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Nilsson, S.; Gustafsson, J.A. Biological role of estrogen and estrogen receptors. Crit. Rev. Biochem. Mol. Biol. 2002, 37, 1–28. [Google Scholar] [CrossRef]
- Sanchez, R.; Nguyen, D.; Rocha, W.; White, J.H.; Mader, S. Diversity in the mechanisms of gene regulation by estrogen receptors. Bioessays 2002, 24, 244–254. [Google Scholar] [CrossRef]
- Ricketts, D.; Turnbull, L.; Ryall, G.; Bakhshi, R.; Rawson, N.S.; Gazet, J.C.; Nolan, C.; Coombes, R.C. Estrogen and progesterone receptors in the normal female breast. Cancer Res. 1991, 51, 1817–1822. [Google Scholar] [PubMed]
- Ariazi, E.A.; Ariazi, J.L.; Cordera, F.; Jordan, V.C. Estrogen receptors as therapeutic targets in breast cancer. Curr. Top. Med. Chem. 2006, 6, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Wijayaratne, A.L.; McDonnell, D.P. The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J. Biol. Chem. 2001, 276, 35684–35692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wall, M.E.; Abernethy, G.S., Jr.; Carroll, F.I.; Taylor, D.J. The effects of some steroidal alkylating agents on experimental animal mammary tumor and leukemia systems. J. Med. Chem. 1969, 12, 810–818. [Google Scholar] [CrossRef]
- Dao, K.L.; Hanson, R.N. Targeting the estrogen receptor using steroid-therapeutic drug conjugates (hybrids). Bioconjug. Chem. 2012, 23, 2139–2158. [Google Scholar] [CrossRef]
- Keely, N.O.; Meegan, M.J. Targeting tumors using estrogen receptor ligand conjugates. Curr. Cancer Drug Targets 2009, 9, 370–380. [Google Scholar] [CrossRef]
- Jones, M.E.; van Leeuwen, F.E.; Hoogendoorn, W.E.; Mourits, M.J.E.; Hollema, H.; van Boven, H.; Swerdlow, A.J. Endometrial cancer survival after breast cancer in relation to tamoxifen treatment: Pooled results from three countries. Breast Cancer Res. 2012, 14, R91. [Google Scholar] [CrossRef] [Green Version]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, B.; Liu, J.; Liu, J.; Li, C.; Dong, W.; Fang, S.; Li, M.; Song, B.; Tang, B.; et al. Mechanisms of Gefitinib-mediated reversal of tamoxifen resistance in MCF-7 breast cancer cells by inducing ERα re-expression. Sci. Rep. 2015, 5, 7835. [Google Scholar] [CrossRef]
- Munster, P.N.; Lacevic, M.; Thomas, S.; Ismail-Khan, R.; Rugo, H.; Melisko, M.; Minton, S.E. Clinical Phase II Study of Vorinostat, a Hydroxamic Type Histone Deacetylase Inhibitor, in Combination with Tamoxifen to Reverse Acquired Hormone Resistance in Breast Cancer Patients Who Progressed on Hormone Therapy. Cancer Res. 2009, 69, 856S. [Google Scholar] [CrossRef]
- Wilson, S.T.; Blask, D.E.; Lemus-Wilson, A.M. Melatonin augments the sensitivity of MCF-7 human breast cancer cells to tamoxifen in vitro. J. Clin. Endocrinol. Metab. 1992, 75, 669–670. [Google Scholar] [PubMed]
- Lissoni, P.; Barni, S.; Meregalli, S.; Fossati, V.; Cazzaniga, M.; Esposti, D.; Tancini, G. Modulation of cancer endocrine therapy by melatonin: A phase II study of tamoxifen plus melatonin in metastatic breast cancer patients progressing under tamoxifen alone. Br. J. Cancer 1995, 71, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Rato, A.G.; Pedrero, J.G.; Martinez, M.A.; del Rio, B.; Lazo, P.S.; Ramos, S. Melatonin blocks the activation of estrogen receptor for DNA binding. FASEB J. 1999, 13, 857–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.M.; Spriggs, L.L.; Simon, M.A.; Muraoka, H.; Blask, D.E. The growth inhibitory action of melatonin on human breast cancer cells is linked to the estrogen response system. Cancer Lett. 1992, 64, 249–256. [Google Scholar] [CrossRef]
- Gryder, B.E.; Rood, M.K.; Johnson, K.A.; Vishal, P.; Raftery, E.D.; Yao, L.-P.D.; Rice, M.; Azizi, B.; Doyle, D.F.; Oyelere, A.K. Histone deacetylase inhibitors equipped with estrogen receptor modulation activity. J. Med. Chem. 2013, 56, 5782–5796. [Google Scholar] [CrossRef] [Green Version]
- Abdelmalek, C.M.; Hu, Z.; Kronenberger, T.; Küblbeck, J.; Kinnen, F.J.M.; Hesse, S.S.; Malik, A.; Kudolo, M.; Niess, R.; Gehringer, M.; et al. Gefitinib-Tamoxifen Hybrid Ligands as Potent Agents against Triple-Negative Breast Cancer. J. Med. Chem. 2022, 65, 4616–4632. [Google Scholar] [CrossRef]
- Witt-Enderby, P.A.; Davis, V.L.; Lapinsky, D. Anti-Cancer Tamoxifen-Melatonin Hybrid Ligand. U.S. Patent 8,785,501, 22 July 2014. [Google Scholar]
- Kelly, P.M.; Keely, N.O.; Bright, S.A.; Yassin, B.; Ana, G.; Fayne, D.; Zisterer, D.M.; Meegan, M.J. Novel Selective Estrogen Receptor Ligand Conjugates Incorporating Endoxifen-Combretastatin and Cyclofenil-Combretastatin Hybrid Scaffolds: Synthesis and Biochemical Evaluation. Molecules 2017, 22, 1440. [Google Scholar] [CrossRef]
- Fröhlich, T.; Mai, C.; Bogautdinov, R.P.; Morozkina, S.N.; Shavva, A.G.; Friedrich, O.; Gilbert, D.F.; Tsogoeva, S.B. Synthesis of Tamoxifen-Artemisinin and Estrogen-Artemisinin Hybrids Highly Potent against Breast and Prostate Cancer. ChemMedChem 2020, 15, 1473–1479. [Google Scholar] [CrossRef]
- Jockers, R.; Delagrange, P.; Dubocovich, M.L.; Markus, R.P.; Renault, N.; Tosini, G.; Cecon, E.; Zlotos, D.P. Update on melatonin receptors: IUPHAR Review 20. Br. J. Pharmacol. 2016, 173, 2702–2725. [Google Scholar] [CrossRef]
- Zlotos, D.P.; Jockers, R.; Cecon, E.; Rivara, S.; Witt-Enderby, P.A. MT1 and MT2 melatonin receptors: Ligands, models, oligomers, and therapeutic potential. J. Med. Chem. 2014, 57, 3161–3185. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Marzouk, M.A.; Adhikari, S.; Wright, T.D.; Miller, B.P.; Matossian, M.D.; Elliot, S.; Wright, M.; Alzoubi, M.; Collins-Burow, B.M.; et al. Pharmacological, Mechanistic and Pharmacokinetic Assessment of Novel Melatonin-Tamoxifen Drug Conjugates as Breast Cancer Drugs. Mol. Pharmacol. 2019, 96, 272–296. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Desta, Z.; Stearns, V.; Ward, B.; Ho, H.; Lee, K.H.; Skaar, T.; Storniolo, A.M.; Li, L.; Araba, A.; et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J. Natl. Cancer Inst. 2005, 97, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, V.C. Metabolites of tamoxifen in animals and man: Identification, pharmacology, and significance. Breast Cancer Res. Treat. 1982, 2, 123–138. [Google Scholar] [CrossRef]
- Coezy, E.; Borgna, J.L.; Rochefort, H. Tamoxifen and metabolites in MCF7 cells: Correlation between binding to estrogen receptor and inhibition of cell growth. Cancer Res. 1982, 42, 317–323. [Google Scholar]
- Malet, C.; Gompel, A.; Spritzer, P.; Bricout, N.; Yaneva, H.; Mowszowicz, I.; Kuttenn, F.; Mauvais-Jarvis, P. Tamoxifen and hydroxytamoxifen isomers versus estradiol effects on normal human breast cells in culture. Cancer Res. 1988, 48 Pt 1, 7193–71999. [Google Scholar]
- Katzenellenbogen, B.S.; Norman, M.J.; Eckert, R.L.; Peltz, S.W.; Mangel, W.F. Bioactivities, estrogen receptor interactions, and plasminogen activator-inducing activities of tamoxifen and hydroxy-tamoxifen isomers in MCF-7 human breast cancer cells. Cancer Res. 1984, 44, 112–119. [Google Scholar]
- Katzenellenbogen, J.A.; Carlson, K.E.; Katzenellenbogen, B.S. Facile geometric isomerization of phenolic non-steroidal estrogens and antiestrogens: Limitations to the interpretation of experiments characterizing the activity of individual isomers. J. Steroid Biochem. 1985, 22, 589–596. [Google Scholar] [CrossRef]
- Keely, N.O.; Carr, M.; Yassin, B.; Ana, G.; Lloyd, D.G.; Zisterer, D.; Meegan, M.J. Design, Synthesis and Biochemical Evaluation of Novel Selective Estrogen Receptor Ligand Conjugates Incorporating an Endoxifen-Combretastatin Hybrid Scaffold. Biomedicines 2016, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Keely, N.O.; Zisterer, D.M.; Meegan, M.J. Design, Synthesis and Biochemical Evaluation of Estrogen Receptor Ligand Conjugates as Tumor Targeting Agents. Lett. Drug Des. Discov. 2012, 9, 295–304. [Google Scholar] [CrossRef]
- Palermo, A.F.; Diennet, M.; El Ezzy, M.; Williams, B.M.; Cotnoir-White, D.; Mader, S.; Gleason, J.L. Incorporation of histone deacetylase inhibitory activity into the core of tamoxifen—A new hybrid design paradigm. Bioorg. Med. Chem. 2018, 26, 4428–4440. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Sanchez, R.; Cotnoir-White, D.; Kulpa, J.; Jutras, I.; Pottel, J.; Moitessier, N.; Mader, S.; Gleason, J.L. Design, synthesis and evaluation of antiestrogen and histone deacetylase inhibitor molecular hybrids. Bioorg. Med. Chem. 2015, 23, 7597–7606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Mehrling, T.; Chen, Y. The Alkylating-HDAC Inhibition Fusion Principle: Taking Chemotherapy to the Next Level with the First in Class Molecule EDO-S101. Anti-Cancer Agents Med. Chem. 2016, 16, 20–28. [Google Scholar] [CrossRef]
- Kumar, S.; Kumar, V. Have molecular hybrids delivered effective anti-cancer treatments and what should future drug discovery focus on? Expert Opin. Drug Discov. 2021, 16, 335–363. [Google Scholar]






{kind=link}
{kind=link}
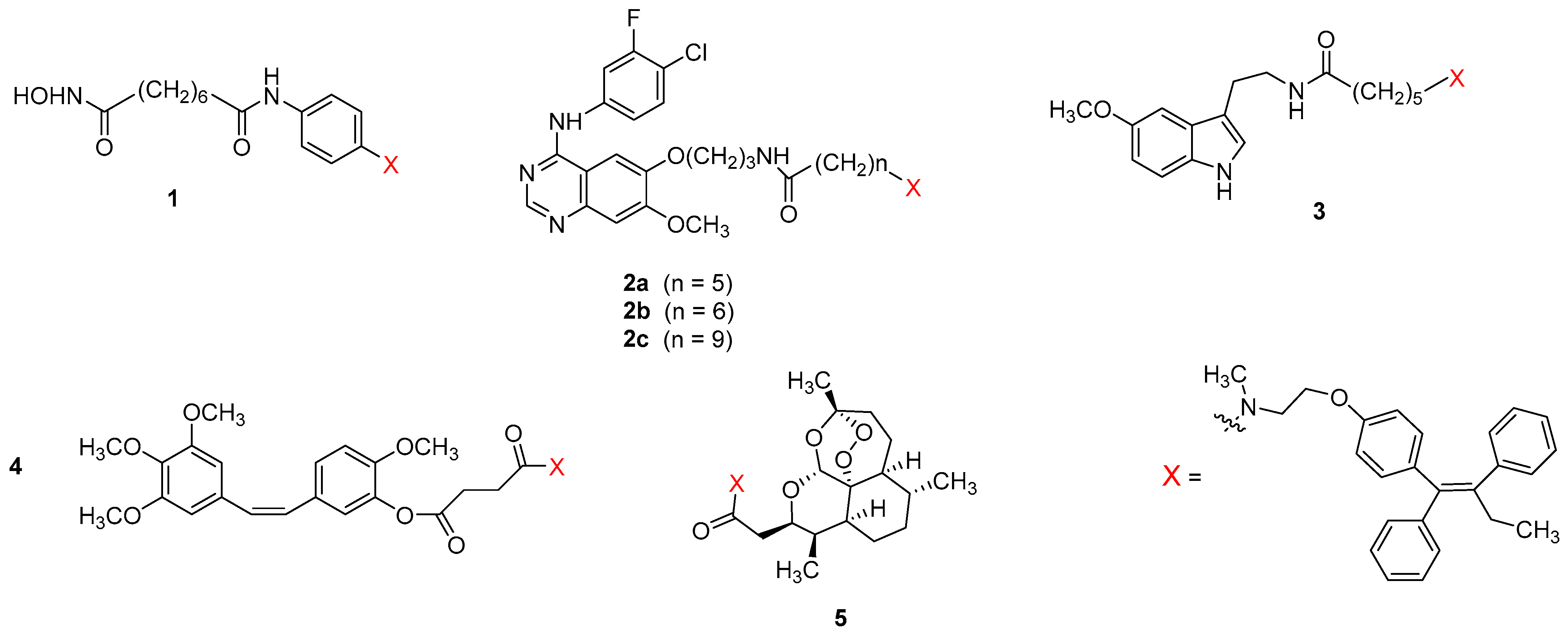{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activity at ERα | Antiproliferative Activity | Activity at Second Target | Lit. | ||
|---|---|---|---|---|---|
| IC50 | MCF-7 | MDA-MB-231 | |||
| 1 | 127 nM a | 3.8 µM | 8.1 µM | HDAC1 2.7 µM | [28] |
| HDAC6 221 nM | |||||
| tamoxifen | 39 nM a | 16 µM | 17 µM | - | |
| vorinostat | - | 4.4 µM | 3.4 µM | HDAC1 42 nM | |
| HDAC6 34 nM | |||||
| 2a | 101 nM a | 1.2 µM | 780 nM | - | [29] |
| 2b | 11 nM a | 1.5 µM | 850 nM | EGFR 1.1 nM | |
| 2c | 232 nM a | - | - | - | |
| tamoxifen | 49 nM a | ||||
| gefitinib | - | - | - | EGFR < 0.1 nM | |
| 3 | 2 nM b | - | - | MT1 2.8 nM c | [30] |
| tamoxifen | 10 nM b | - | - | - | |
| melatonin | - | - | - | MT1 8.6 nM c | |
| 4 | 80 nM d | 90 nM | - | - | [31] |
| tamoxifen | - | 2.1 µM | - | - | |
| combretastatin | - | 8 nM | - | ||
| 5 | - | 3.9 µM | - | - | [32] |
| artesunic acid | - | 32 µM | - | ||
 | ||||
| plasma concentration | 363 nM | 655 nM | 9 nM | 63 nM |
| ERα antagonist action from luciferase reporter assay IC50 | 49 nM | - | 0.21 nM | 0.14 nM |
| Inhibition of [3H]-estradiol binding to rat uterine ERs IC50 | ~100 nM | ~200 nM | ~3 nM | - |
| Activity at ERα | Antiproliferative Activity | Activity at Second Target | Lit. | ||
|---|---|---|---|---|---|
| IC50 | MCF-7 | MDA-MB-231 | |||
| 6a | 4.6 nM a | 1.4 µM | 890 nM | EGFR 2.5 nM | [29] |
| 6b | 4.4 nM a | 2.0 µM | 970 nM | EGFR 260 nM | |
| Z-4-OH-tamoxifen | 0.21 nM a | - | - | - | |
| Z-endoxifen | 0.14 nM a | - | - | - | |
| gefitinib | - | EGFR < 0.1 nM | |||
| 7 | 52 nM b | 5.7 nM | - | [31] | |
| 8 | - | 33 nM | 2.7 µM | [42] | |
| 9 | 1 nM b | 5 nM | 2.5 µM | ||
| E/Z-endoxifen | 47 nM b | 29 nM | - | ||
| 4-OH-tamoxifen | 30 nM b | - | 18 µM | ||
| tamoxifen | - | - | 20 µM | ||
| combretastatin | - | 8 nM | 43 nM | ||
| 10 | 524 nM | 1.6 µM | [43] | ||
| 11 | 490 nM | 30 µM | |||
| 12 | 51 nM | 13 µM | |||
| 13 | 79 nM | 12 µM | |||
| 14 | 36 nM | >50 µM | |||
| tamoxifen | 70 nM | 4 µM | |||
| 15a | 800 nM c | 1.2 µM | HDAC6 1.78 µM | [44] | |
| 15b | 820 nM c | 790 nM | 1.1 µM | HDAC3 2.10 µM | |
| HDAC6 0.30 µM | |||||
| HDAC3 0.73 µM | |||||
| 4-OH-tamoxifen | 500 nM c | 3.3 µM | 2.5 µM | HDAC6 0.06 µM | |
| vorinostat | - | 450 nM | 610 nM | ||
| HDAC3 0.11 µM | |||||
| Activity at ERα IC50 a | Antiproliferative Activity MCF-7 Cells | HDAC6 Inhibition | HDAC3 Inhibition | Lit | |
|---|---|---|---|---|---|
| 16a | 1.06 µM | 2.93 µM | 1.15 µM | 960 nM | [45] |
| 16b | 2.10 µM | 9.11 µM | >50 µM | >5 µM | |
| 17 | 720 nM | 1.90 µM | >50 µM | 3.18 µM | |
| 18 | 180 nM | 340 nM | 43.7 µM | 1.55 µM | |
| Z-4-OH-tamoxifen | 10 nM | 150 nM | - | - | |
| ICI-164384 | 50 nM | 930 nM | - | - | |
| vorinostat | - | 320 nM | 350 nM | 170 nM | |
| entinostat | - | 350 nM | - | 310 nM | |
| 19 | 22 µM | 22 µM | 8 nM | - | [28] |
| estradiol | - | - | - | - | |
| vorinostat | 4.4 µM | 4.4 µM | 34 nM | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zlotos, D.P.; Kronenberger, T.; Laufer, S.A. Anticancer Drug Conjugates Incorporating Estrogen Receptor Ligands. Pharmaceutics 2023, 15, 67. https://doi.org/10.3390/pharmaceutics15010067
Zlotos DP, Kronenberger T, Laufer SA. Anticancer Drug Conjugates Incorporating Estrogen Receptor Ligands. Pharmaceutics. 2023; 15(1):67. https://doi.org/10.3390/pharmaceutics15010067
Chicago/Turabian StyleZlotos, Darius P., Thales Kronenberger, and Stefan A. Laufer. 2023. "Anticancer Drug Conjugates Incorporating Estrogen Receptor Ligands" Pharmaceutics 15, no. 1: 67. https://doi.org/10.3390/pharmaceutics15010067
APA StyleZlotos, D. P., Kronenberger, T., & Laufer, S. A. (2023). Anticancer Drug Conjugates Incorporating Estrogen Receptor Ligands. Pharmaceutics, 15(1), 67. https://doi.org/10.3390/pharmaceutics15010067