


Drug Repurposing at the Interface of Melanoma Immunotherapy and Autoimmune Disease
 ,
, 
Abstract
:1. Introduction
2. Drivers of Metastatic Melanoma as Hallmarks of Cancer Progression
2.1. E2F1 as Driver and Therapeutic Target of Melanoma Metastasis
2.2. Role of the TME for Metastasis
3. Immunotherapeutic Strategies against Metastatic Melanoma
3.1. Immune Checkpoint Inhibitors and Other Immunotherapeutic Approaches
3.2. Shortfalls of Immunotherapies—irAEs and Autoimmune Disease
4. A Look at the Interface of Cancer and AD
4.1. Melanoma and Autoimmune Disease
4.2. AD, Cancer, and Immunotherapy—An Underinvestigated Conncetion: Hurdles or Chances?
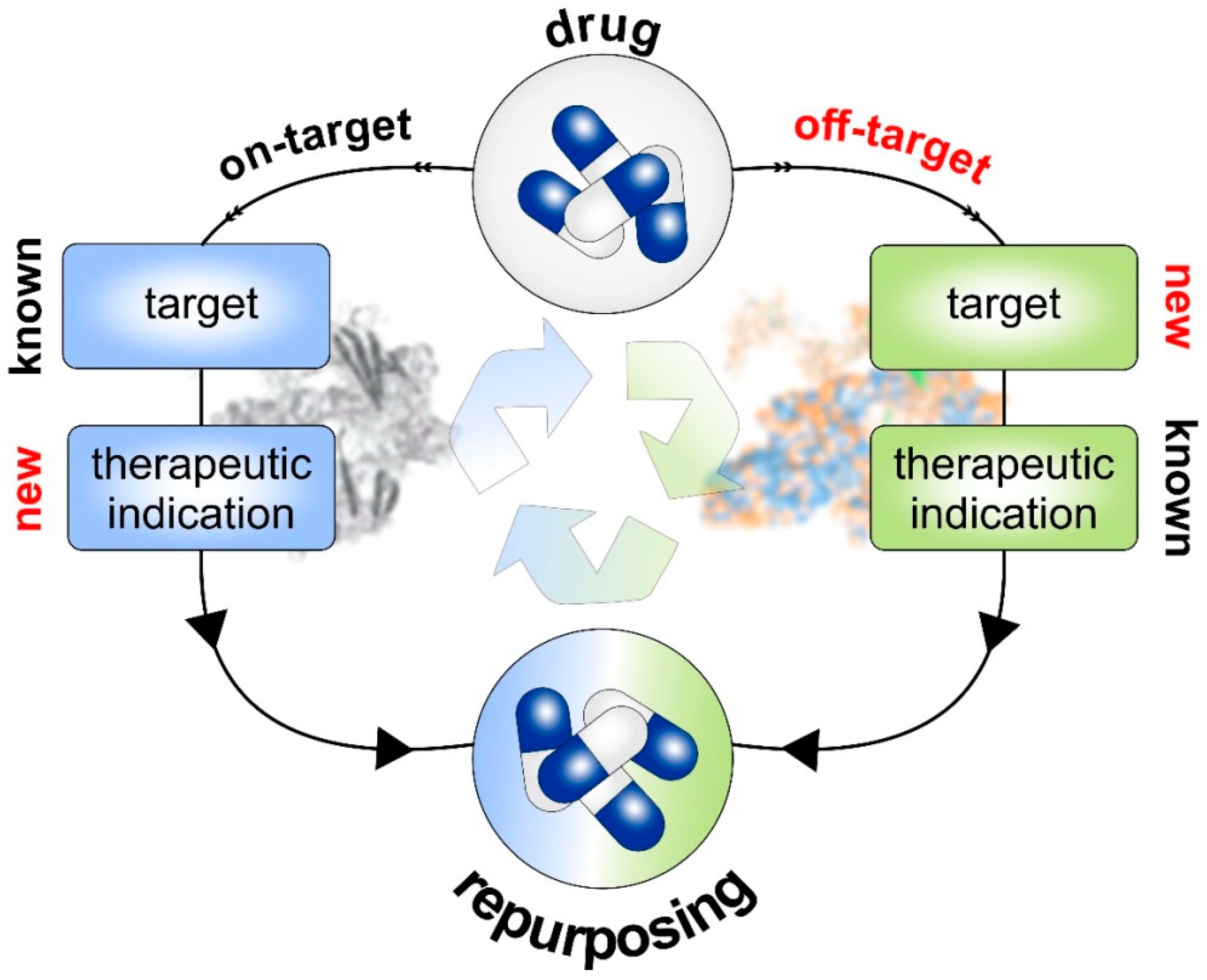
5. Prospects of Drug Repurposing
5.1. Repurposed Drugs in the Context of Anti-Metastatic Treatment and Cancer Immunotherapy
5.1.1. Niclosamide
5.1.2. Aspirin
5.1.3. Denosumab
5.1.4. Metformin
5.2. Perspectives of Drug Repurposing at the Interface of Cancer, AD and Immunotherapy
6. Computational Approaches for Drug-Repurposing
6.1. Traditional Drug Discovery versus Drug Repurposing
6.2. Strategies of Drug Repurposing
6.2.1. Drug-Based Strategies
6.2.2. Target-Based Strategies
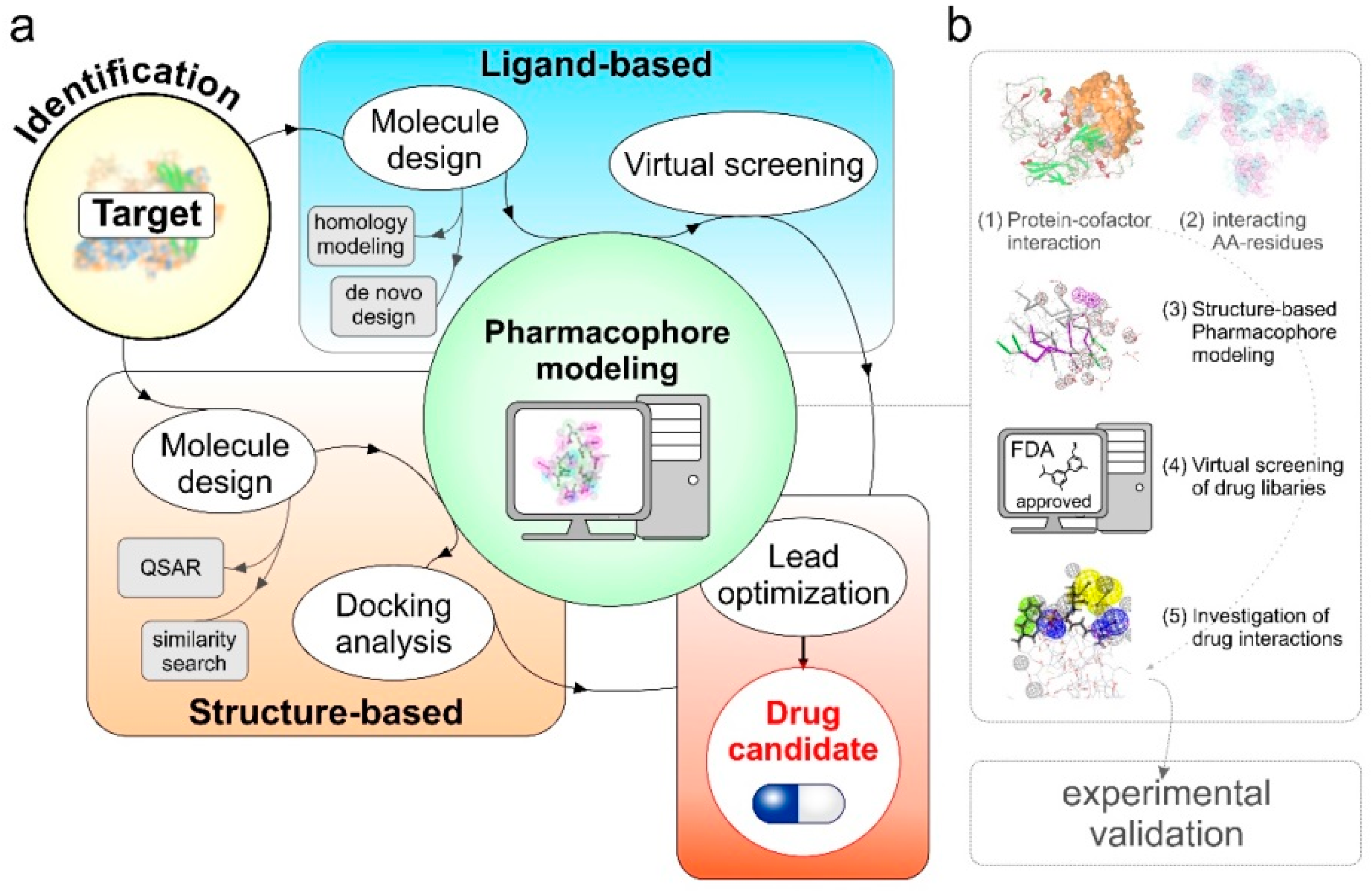
7. Pharmacophore Modeling towards Therapy Personalization in Melanoma
7.1. Pharmacophore Modeling
- Predictive-pharmacophore: when the list of drug molecules benefiting a clinical phenotype is known but the related target information is missing. These methods learn from the chemical features present in active and inactive compounds and quickly screen a virtual library of similar compounds for drug repositioning [158,159].
- Receptor–ligand pharmacophore: when the information about the drug and target is known, these methods generate pharmacophore models using the features from receptor-ligand interactions [160].
- Structure-based pharmacophore: these methods are suitable for the cases where the information about the potential therapeutic targets, protein–protein complexes, protein-cofactors driving this phenotype in known. The main goal is to find potential inhibitors that may fit to the active site or interfere in complex formation to benefit the clinical phenotype [161].
7.2. Argatroban as a Case Study
7.2.1. Structure Modeling and Quality Assessment of E2F1 and MTA1 Proteins [9,11]
7.2.2. In Silico PPI Docking Studies Suggest MTA1-E2F1 Physical Interaction
7.2.3. Screening of Non-Peptidic Small Molecule Inhibitors Disrupting E2F1-MTA1 Interaction in Metastatic Tumor Cells Using Pharmacophore Modeling
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nosengo, N. Can you teach old drugs new tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkbak, N.J.; McGranahan, N. Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 2020, 37, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Alla, V.; Engelmann, D.; Niemetz, A.; Pahnke, J.; Schmidt, A.; Kunz, M.; Emmrich, S.; Steder, M.; Koczan, D.; Pützer, B.M. E2F1 in melanoma progression and metastasis. J. Natl. Cancer Inst. 2010, 102, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pützer, B.M.; Steder, M.; Alla, V. Predicting and preventing melanoma invasiveness: Advances in clarifying E2F1 function. Expert Rev. Anticancer Ther. 2010, 10, 1707–1720. [Google Scholar] [CrossRef] [PubMed]
- Andrechek, E.R. HER2/Neu tumorigenesis and metastasis is regulated by E2F activator transcription factors. Oncogene 2015, 34, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-S.; Leem, S.-H.; Lee, S.-Y.; Kim, S.-C.; Park, E.-S.; Kim, S.-B.; Kim, S.-K.; Kim, Y.-J.; Kim, W.-J.; Chu, I.-S. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J. Clin. Oncol. 2010, 28, 2660–2667. [Google Scholar] [CrossRef]
- Khan, F.M.; Marquardt, S.; Gupta, S.K.; Knoll, S.; Schmitz, U.; Spitschak, A.; Engelmann, D.; Vera, J.; Wolkenhauer, O.; Pützer, B.M. Unraveling a tumor type-specific regulatory core underlying E2F1-mediated epithelial-mesenchymal transition to predict receptor protein signatures. Nat. Commun. 2017, 8, 198. [Google Scholar] [CrossRef] [Green Version]
- Vera, J.; Schmitz, U.; Lai, X.; Engelmann, D.; Khan, F.M.; Wolkenhauer, O.; Pützer, B.M. Kinetic modeling-based detection of genetic signatures that provide chemoresistance via the E2F1-p73/DNp73-miR-205 network. Cancer Res. 2013, 73, 3511–3524. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Alla, V.; Goody, D.; Gupta, S.K.; Spitschak, A.; Wolkenhauer, O.; Pützer, B.M.; Engelmann, D. Epigenetic factor EPC1 is a master regulator of DNA damage response by interacting with E2F1 to silence death and activate metastasis-related gene signatures. Nucleic Acids Res. 2016, 44, 117–133. [Google Scholar] [CrossRef] [Green Version]
- Engelmann, D.; Mayoli-Nüssle, D.; Mayrhofer, C.; Fürst, K.; Alla, V.; Stoll, A.; Spitschak, A.; Abshagen, K.; Vollmar, B.; Ran, S.; et al. E2F1 promotes angiogenesis through the VEGF-C/VEGFR-3 axis in a feedback loop for cooperative induction of PDGF-B. J. Mol. Cell Biol. 2013, 5, 391–403. [Google Scholar] [CrossRef]
- Meier, C.; Spitschak, A.; Abshagen, K.; Gupta, S.; Mor, J.M.; Wolkenhauer, O.; Haier, J.; Vollmar, B.; Alla, V.; Pützer, B.M. Association of RHAMM with E2F1 promotes tumour cell extravasation by transcriptional up-regulation of fibronectin. J. Pathol. 2014, 234, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Knoll, S.; Fürst, K.; Kowtharapu, B.; Schmitz, U.; Marquardt, S.; Wolkenhauer, O.; Martin, H.; Pützer, B.M. E2F1 induces miR-224/452 expression to drive EMT through TXNIP downregulation. EMBO Rep. 2014, 15, 1315–1329. [Google Scholar] [CrossRef] [Green Version]
- Logotheti, S.; Marquardt, S.; Gupta, S.K.; Richter, C.; Edelhäuser, B.A.H.; Engelmann, D.; Brenmoehl, J.; Söhnchen, C.; Murr, N.; Alpers, M.; et al. LncRNA-SLC16A1-AS1 induces metabolic reprogramming during Bladder Cancer progression as target and co-activator of E2F1. Theranostics 2020, 10, 9620–9643. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Marquardt, S.; Li, F.; Spitschak, A.; Murr, N.; Edelhäuser, B.A.H.; Iliakis, G.; Pützer, B.M.; Logotheti, S. Rewiring E2F1 with classical NHEJ via APLF suppression promotes bladder cancer invasiveness. J. Exp. Clin. Cancer Res. 2019, 38, 292. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, S.; Solanki, M.; Spitschak, A.; Vera, J.; Pützer, B.M. Emerging functional markers for cancer stem cell-based therapies: Understanding signaling networks for targeting metastasis. Semin. Cancer Biol. 2018, 53, 90–109. [Google Scholar] [CrossRef]
- Goody, D.; Gupta, S.K.; Engelmann, D.; Spitschak, A.; Marquardt, S.; Mikkat, S.; Meier, C.; Hauser, C.; Gundlach, J.-P.; Egberts, J.-H.; et al. Drug Repositioning Inferred from E2F1-Coregulator Interactions Studies for the Prevention and Treatment of Metastatic Cancers. Theranostics 2019, 9, 1490–1509. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Quail, D.F.; Siegers, G.M.; Jewer, M.; Postovit, L.-M. Nodal signalling in embryogenesis and tumourigenesis. Int. J. Biochem. Cell Biol. 2013, 45, 885–898. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Lee, H.-Y.; Park, K.-K.; Choi, Y.-K.; Nam, J.-S.; Hong, I.-S. CWP232228 targets liver cancer stem cells through Wnt/β-catenin signaling: A novel therapeutic approach for liver cancer treatment. Oncotarget 2016, 7, 20395–20409. [Google Scholar] [CrossRef] [Green Version]
- Kuol, N.; Stojanovska, L.; Apostolopoulos, V.; Nurgali, K. Crosstalk between cancer and the neuro-immune system. J. Neuroimmunol. 2018, 315, 15–23. [Google Scholar] [CrossRef]
- Kuol, N.; Stojanovska, L.; Apostolopoulos, V.; Nurgali, K. Role of the nervous system in cancer metastasis. J. Exp. Clin. Cancer Res. 2018, 37, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shurin, M.R.; Shurin, G.V.; Zlotnikov, S.B.; Bunimovich, Y.L. The Neuroimmune Axis in the Tumor Microenvironment. J. Immunol. 2020, 204, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Dora, D.; Rivard, C.; Yu, H.; Bunn, P.; Suda, K.; Ren, S.; Lueke Pickard, S.; Laszlo, V.; Harko, T.; Megyesfalvi, Z.; et al. Neuroendocrine subtypes of small cell lung cancer differ in terms of immune microenvironment and checkpoint molecule distribution. Mol. Oncol. 2020, 14, 1947–1965. [Google Scholar] [CrossRef] [PubMed]
- Logotheti, S.; Pützer, B.M. STAT3 and STAT5 Targeting for Simultaneous Management of Melanoma and Autoimmune Diseases. Cancers 2019, 11, 1448. [Google Scholar] [CrossRef] [Green Version]
- Pützer, B.M.; Hitt, M.; Muller, W.J.; Emtage, P.; Gauldie, J.; Graham, F.L. Interleukin 12 and B7-1 costimulatory molecule expressed by an adenovirus vector act synergistically to facilitate tumor regression. Proc. Natl. Acad. Sci. USA 1997, 94, 10889–10894. [Google Scholar] [CrossRef] [Green Version]
- Pützer, B.M.; Stiewe, T.; Rödicker, F.; Schildgen, O.; Rühm, S.; Dirsch, O.; Fiedler, M.; Damen, U.; Tennant, B.; Scherer, C.; et al. Large nontransplanted hepatocellular carcinoma in woodchucks: Treatment with adenovirus-mediated delivery of interleukin 12/B7.1 genes. J. Natl. Cancer Inst. 2001, 93, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Zitvogel, L. Immune checkpoint inhibitors. J. Exp. Med. 2021, 218, e20201979. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Janssen, L.M.E.; Ramsay, E.E.; Logsdon, C.D.; Overwijk, W.W. The immune system in cancer metastasis: Friend or foe? J. Immunother. Cancer 2017, 5, 79. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Cancer Immunotherapy, Part 1: Current Strategies and Agents. Pharm. Ther. 2017, 42, 375–383. [Google Scholar]
- Ma, Q.; Shilkrut, M.; Zhao, Z.; Li, M.; Batty, N.; Barber, B. Autoimmune comorbidities in patients with metastatic melanoma: A retrospective analysis of us claims data. BMC Cancer 2018, 18, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.-J.; Kuen, D.-S.; Chung, Y. Future prospects of immune checkpoint blockade in cancer: From response prediction to overcoming resistance. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Taylor, N.A.; Vick, S.C.; Iglesia, M.D.; Brickey, W.J.; Midkiff, B.R.; McKinnon, K.P.; Reisdorf, S.; Anders, C.K.; Carey, L.A.; Parker, J.S.; et al. Treg depletion potentiates checkpoint inhibition in claudin-low breast cancer. J. Clin. Investig. 2017, 127, 3472–3483. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Blake, S.J.; Harjunpää, H.; Fairfax, K.A.; Yong, M.C.R.; Allen, S.; Kohrt, H.E.; Takeda, K.; Smyth, M.J.; Teng, M.W.L. Assessing Immune-Related Adverse Events of Efficacious Combination Immunotherapies in Preclinical Models of Cancer. Cancer Res. 2016, 76, 5288–5301. [Google Scholar] [CrossRef] [Green Version]
- Shitara, K.; Nishikawa, H. Regulatory T cells: A potential target in cancer immunotherapy. Ann. New York Acad. Sci. 2018, 1417, 104–115. [Google Scholar] [CrossRef]
- Han, S.; Toker, A.; Liu, Z.Q.; Ohashi, P.S. Turning the Tide Against Regulatory T Cells. Front. Oncol. 2019, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.-C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Voron, T.; Marcheteau, E.; Pernot, S.; Colussi, O.; Tartour, E.; Taieb, J.; Terme, M. Control of the immune response by pro-angiogenic factors. Front. Oncol. 2014, 4, 70. [Google Scholar] [CrossRef] [Green Version]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Horvat, T.Z.; Adel, N.G.; Dang, T.-O.; Momtaz, P.; Postow, M.A.; Callahan, M.K.; Carvajal, R.D.; Dickson, M.A.; D’Angelo, S.P.; Woo, K.M.; et al. Immune-Related Adverse Events, Need for Systemic Immunosuppression, and Effects on Survival and Time to Treatment Failure in Patients with Melanoma Treated with Ipilimumab at Memorial Sloan Kettering Cancer Center. J. Clin. Oncol. 2015, 33, 3193–3198. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Sullivan, R.J.; Ott, P.A.; Carlino, M.S.; Khushalani, N.I.; Ye, F.; Guminski, A.; Puzanov, I.; Lawrence, D.P.; Buchbinder, E.I.; et al. Ipilimumab Therapy in Patients with Advanced Melanoma and Preexisting Autoimmune Disorders. JAMA Oncol. 2016, 2, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Menzies, A.M.; Johnson, D.B.; Ramanujam, S.; Atkinson, V.G.; Wong, A.N.M.; Park, J.J.; McQuade, J.L.; Shoushtari, A.N.; Tsai, K.K.; Eroglu, Z.; et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann. Oncol. 2017, 28, 368–376. [Google Scholar] [CrossRef]
- Karin, M.; Lawrence, T.; Nizet, V. Innate immunity gone awry: Linking microbial infections to chronic inflammation and cancer. Cell 2006, 124, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.W.; Reeve, B.B.; Bellizzi, K.M.; Harlan, L.C.; Klabunde, C.N.; Amsellem, M.; Bierman, A.S.; Hays, R.D. Cancer, comorbidities, and health-related quality of life of older adults. Health Care Financ. Rev. 2008, 29, 41–56. [Google Scholar] [PubMed]
- Wogan, G.N.; Dedon, P.C.; Tannenbaum, S.R.; Fox, J.G. Infection, inflammation and colon carcinogenesis. Oncotarget 2012, 3, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Franks, A.L.; Slansky, J.E. Multiple associations between a broad spectrum of autoimmune diseases, chronic inflammatory diseases and cancer. Anticancer Res. 2012, 32, 1119–1136. [Google Scholar] [PubMed]
- Zhang, K.; Kong, X.; Li, Y.; Wang, Z.; Zhang, L.; Xuan, L. PD-1/PD-L1 Inhibitors in Patients With Preexisting Autoimmune Diseases. Front. Pharmacol. 2022, 13, 854967. [Google Scholar] [CrossRef]
- Beyaert, R.; Beaugerie, L.; van Assche, G.; Brochez, L.; Renauld, J.-C.; Viguier, M.; Cocquyt, V.; Jerusalem, G.; Machiels, J.-P.; Prenen, H.; et al. Cancer risk in immune-mediated inflammatory diseases (IMID). Mol. Cancer 2013, 12, 98. [Google Scholar] [CrossRef] [Green Version]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef]
- Amos, S.M.; Duong, C.P.M.; Westwood, J.A.; Ritchie, D.S.; Junghans, R.P.; Darcy, P.K.; Kershaw, M.H. Autoimmunity associated with immunotherapy of cancer. Blood 2011, 118, 499–509. [Google Scholar] [CrossRef]
- Cohen, I.R. Activation of benign autoimmunity as both tumor and autoimmune disease immunotherapy: A comprehensive review. J. Autoimmun. 2014, 54, 112–117. [Google Scholar] [CrossRef]
- Teulings, H.E.; Overkamp, M.; Ceylan, E.; Nieuweboer-Krobotova, L.; Bos, J.D.; Nijsten, T.; Wolkerstorfer, A.W.; Luiten, R.M.; van der Veen, J.P.W. Decreased risk of melanoma and nonmelanoma skin cancer in patients with vitiligo: A survey among 1307 patients and their partners. Br. J. Dermatol. 2013, 168, 162–171. [Google Scholar] [CrossRef]
- Teulings, H.-E.; Limpens, J.; Jansen, S.N.; Zwinderman, A.H.; Reitsma, J.B.; Spuls, P.I.; Luiten, R.M. Vitiligo-like depigmentation in patients with stage III-IV melanoma receiving immunotherapy and its association with survival: A systematic review and meta-analysis. J. Clin. Oncol. 2015, 33, 773–781. [Google Scholar] [CrossRef]
- Cho, E.A.; Lee, M.A.; Kang, H.; Lee, S.D.; Kim, H.O.; Park, Y.M. Vitiligo-like Depigmentation Associated with Metastatic Melanoma of an Unknown Origin. Ann. Dermatol. 2009, 21, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Fürst, K.; Steder, M.; Logotheti, S.; Angerilli, A.; Spitschak, A.; Marquardt, S.; Schumacher, T.; Engelmann, D.; Herchenröder, O.; Rupp, R.A.W.; et al. DNp73-induced degradation of tyrosinase links depigmentation with EMT-driven melanoma progression. Cancer Lett. 2019, 442, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Hölzel, M.; Tüting, T. Inflammation-Induced Plasticity in Melanoma Therapy and Metastasis. Trends Immunol. 2016, 37, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Bottoni, U.; Paolino, G.; Ambrifi, M.; Didona, D.; Albanesi, M.; Clerico, R.; Lido, P.; Brachini, A.; Corsetti, P.; Richetta, A.G.; et al. Association between autoimmune disease and cutaneous melanoma with regard to melanoma prognosis. Clin. Exp. Dermatol. 2015, 40, 254–259. [Google Scholar] [CrossRef]
- Reichert, J.M. Antibodies to watch in 2014. MAbs 2014, 6, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Reichert, J.M. Antibodies to watch in 2015. MAbs 2015, 7, 1–8. [Google Scholar] [CrossRef]
- Reichert, J.M. Antibodies to watch in 2016. MAbs 2016, 8, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Reichert, J.M. Antibodies to watch in 2017. MAbs 2017, 9, 167–181. [Google Scholar] [CrossRef] [Green Version]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2018. MAbs 2018, 10, 183–203. [Google Scholar] [CrossRef] [Green Version]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. MAbs 2020, 12, 1703531. [Google Scholar] [CrossRef] [Green Version]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2021. MAbs 2021, 13, 1860476. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.C.; Carter, P.J. Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol. 2010, 10, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.L.; Dhimolea, E.; Reichert, J.M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 2010, 9, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Schwab, N.; Schneider-Hohendorf, T.; Wiendl, H. Therapeutic uses of anti-α4-integrin (anti-VLA-4) antibodies in multiple sclerosis. Int. Immunol. 2015, 27, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabol, R.A.; Noxon, V.; Sartor, O.; Berger, J.R.; Qureshi, Z.; Raisch, D.W.; Norris, L.B.; Yarnold, P.R.; Georgantopoulos, P.; Hrushesky, W.J.; et al. Melanoma complicating treatment with natalizumab for multiple sclerosis: A report from the Southern Network on Adverse Reactions (SONAR). Cancer Med. 2017, 6, 1541–1551. [Google Scholar] [CrossRef] [Green Version]
- Havrdova, E.; Cohen, J.A.; Horakova, D.; Kovarova, I.; Meluzinova, E. Understanding the positive benefit:risk profile of alemtuzumab in relapsing multiple sclerosis: Perspectives from the Alemtuzumab Clinical Development Program. Ther. Clin. Risk Manag. 2017, 13, 1423–1437. [Google Scholar] [CrossRef] [Green Version]
- Guarnera, C.; Bramanti, P.; Mazzon, E. Alemtuzumab: A review of efficacy and risks in the treatment of relapsing remitting multiple sclerosis. Ther. Clin. Risk Manag. 2017, 13, 871–879. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Zheng, Y.; Chen, X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharmacol. 2017, 8, 460. [Google Scholar] [CrossRef]
- Bongartz, T.; Sutton, A.J.; Sweeting, M.J.; Buchan, I.; Matteson, E.L.; Montori, V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: Systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 2006, 295, 2275–2285. [Google Scholar] [CrossRef]
- Siegel, C.A.; Marden, S.M.; Persing, S.M.; Larson, R.J.; Sands, B.E. Risk of lymphoma associated with combination anti-tumor necrosis factor and immunomodulator therapy for the treatment of Crohn’s disease: A meta-analysis. Clin. Gastroenterol. Hepatol. 2009, 7, 874–881. [Google Scholar] [CrossRef] [Green Version]
- Nyboe Andersen, N.; Pasternak, B.; Friis-Møller, N.; Andersson, M.; Jess, T. Association between tumour necrosis factor-α inhibitors and risk of serious infections in people with inflammatory bowel disease: Nationwide Danish cohort study. BMJ 2015, 350, h2809. [Google Scholar] [CrossRef] [PubMed]
- Ben Musa, R.; Usha, L.; Hibbeln, J.; Mutlu, E.A. TNF inhibitors to treat ulcerative colitis in a metastatic breast cancer patient: A case report and literature review. World J. Gastroenterol. 2014, 20, 5912–5917. [Google Scholar] [CrossRef] [PubMed]
- Lindhaus, C.; Tittelbach, J.; Elsner, P. Cutaneous side effects of TNF-alpha inhibitors. J. Dtsch. Dermatol. Ges. 2017, 15, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Giat, E.; Ehrenfeld, M.; Shoenfeld, Y. Cancer and autoimmune diseases. Autoimmun. Rev. 2017, 16, 1049–1057. [Google Scholar] [CrossRef]
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef]
- Kennedy, L.C.; Bhatia, S.; Thompson, J.A.; Grivas, P. Preexisting Autoimmune Disease: Implications for Immune Checkpoint Inhibitor Therapy in Solid Tumors. J. Natl. Compr. Cancer Netw. 2019, 17, 750–757. [Google Scholar] [CrossRef] [Green Version]
- Tison, A.; Quéré, G.; Misery, L.; Funck-Brentano, E.; Danlos, F.-X.; Routier, E.; Robert, C.; Loriot, Y.; Lambotte, O.; Bonniaud, B.; et al. Safety and Efficacy of Immune Checkpoint Inhibitors in Patients with Cancer and Preexisting Autoimmune Disease: A Nationwide, Multicenter Cohort Study. Arthritis Rheumatol. 2019, 71, 2100–2111. [Google Scholar] [CrossRef]
- Cortellini, A.; Buti, S.; Santini, D.; Perrone, F.; Giusti, R.; Tiseo, M.; Bersanelli, M.; Michiara, M.; Grassadonia, A.; Brocco, D.; et al. Clinical Outcomes of Patients with Advanced Cancer and Pre-Existing Autoimmune Diseases Treated with Anti-Programmed Death-1 Immunotherapy: A Real-World Transverse Study. Oncologist 2019, 24, e327–e337. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, G.C.; Gainor, J.F.; Altan, M.; Kravets, S.; Dahlberg, S.E.; Gedmintas, L.; Azimi, R.; Rizvi, H.; Riess, J.W.; Hellmann, M.D.; et al. Safety of Programmed Death-1 Pathway Inhibitors Among Patients with Non-Small-Cell Lung Cancer and Preexisting Autoimmune Disorders. J. Clin. Oncol. 2018, 36, 1905–1912. [Google Scholar] [CrossRef]
- Martinez Chanza, N.; Xie, W.; Issa, M.; Dzimitrowicz, H.; Tripathi, A.; Beuselinck, B.; Lam, E.; Zakharia, Y.; Mckay, R.; Shah, S.; et al. Safety and efficacy of immune checkpoint inhibitors in advanced urological cancers with pre-existing autoimmune disorders: A retrospective international multicenter study. J. Immunother. Cancer 2020, 8, e000538. [Google Scholar] [CrossRef] [Green Version]
- Danlos, F.-X.; Voisin, A.-L.; Dyevre, V.; Michot, J.-M.; Routier, E.; Taillade, L.; Champiat, S.; Aspeslagh, S.; Haroche, J.; Albiges, L.; et al. Safety and efficacy of anti-programmed death 1 antibodies in patients with cancer and pre-existing autoimmune or inflammatory disease. Eur. J. Cancer 2018, 91, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.J.; Weppler, A.; Bhave, P.; Allayous, C.; Patrinely, J.R.; Ott, P.; Sandhu, S.; Haydon, A.; Lebbe, C.; Johnson, D.B.; et al. Combination anti-PD1 and ipilimumab therapy in patients with advanced melanoma and pre-existing autoimmune disorders. J. Immunother. Cancer 2021, 9, e002121. [Google Scholar] [CrossRef] [PubMed]
- Van der Kooij, M.K.; Suijkerbuijk, K.P.M.; Aarts, M.J.B.; van den Berkmortel, F.W.P.J.; Blank, C.U.; Boers-Sonderen, M.J.; van Breeschoten, J.; van den Eertwegh, A.J.M.; de Groot, J.W.B.; Haanen, J.B.A.G.; et al. Safety and Efficacy of Checkpoint Inhibition in Patients with Melanoma and Preexisting Autoimmune Disease: A Cohort Study. Ann. Intern. Med. 2021, 174, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.B. Mechanism-based target identification and drug discovery in cancer research. Science 2000, 287, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Gashaw, I.; Ellinghaus, P.; Sommer, A.; Asadullah, K. What makes a good drug target? Drug Discov. Today 2012, 17, S24–S30. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Kumbhar, P.; Kole, K.; Yadav, T.; Bhavar, A.; Waghmare, P.; Bhokare, R.; Manjappa, A.; Jha, N.K.; Chellappan, D.K.; Shinde, S.; et al. Drug repurposing: An emerging strategy in alleviating skin cancer. Eur. J. Pharmacol. 2022, 926, 175031. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- To, K.K.W.; Cho, W.C.S. Drug Repurposing for Cancer Therapy in the Era of Precision Medicine. Curr. Mol. Pharmacol. 2022, 15, 895–903. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ding, H.; Quan, H.; Han, J. Anthelminthic niclosamide inhibits tumor growth and invasion in cisplatin-resistant human epidermal growth factor receptor 2-positive breast cancer. Oncol. Lett. 2021, 22, 666. [Google Scholar] [CrossRef] [PubMed]
- Chien, M.-H.; Ho, Y.-C.; Yang, S.-F.; Yang, Y.-C.; Lai, S.-Y.; Chen, W.-S.; Chen, M.-J.; Yeh, C.-B. Niclosamide, an oral antihelmintic drug, exhibits antimetastatic activity in hepatocellular carcinoma cells through downregulating twist-mediated CD10 expression. Environ. Toxicol. 2018, 33, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zuo, W.; Chen, L.; Bian, S.; Jing, J.; Gan, C.; Wu, X.; Liu, H.; Su, X.; Hu, W.; et al. Repurposing of the anti-helminthic drug niclosamide to treat melanoma and pulmonary metastasis via the STAT3 signaling pathway. Biochem. Pharmacol. 2019, 169, 113610. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Luo, M.; Rong, Q.-X.; Zhang, H.; Chen, Z.; Wang, F.; Zhao, H.-Y.; Fu, L.-W. Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 245. [Google Scholar] [CrossRef]
- Burock, S.; Daum, S.; Keilholz, U.; Neumann, K.; Walther, W.; Stein, U. Phase II trial to investigate the safety and efficacy of orally applied niclosamide in patients with metachronous or sychronous metastases of a colorectal cancer progressing after therapy: The NIKOLO trial. BMC Cancer 2018, 18, 297. [Google Scholar] [CrossRef] [Green Version]
- Riesenberg, B.P.; Ansa-Addo, E.A.; Gutierrez, J.; Timmers, C.D.; Liu, B.; Li, Z. Cutting Edge: Targeting Thrombocytes to Rewire Anticancer Immunity in the Tumor Microenvironment and Potentiate Efficacy of PD-1 Blockade. J. Immunol. 2019, 203, 1105–1110. [Google Scholar] [CrossRef]
- Johnson, K.E.; Ceglowski, J.R.; Roweth, H.G.; Forward, J.A.; Tippy, M.D.; El-Husayni, S.; Kulenthirarajan, R.; Malloy, M.W.; Machlus, K.R.; Chen, W.Y.; et al. Aspirin inhibits platelets from reprogramming breast tumor cells and promoting metastasis. Blood Adv. 2019, 3, 198–211. [Google Scholar] [CrossRef] [Green Version]
- Gilligan, M.M.; Gartung, A.; Sulciner, M.L.; Norris, P.C.; Sukhatme, V.P.; Bielenberg, D.R.; Huang, S.; Kieran, M.W.; Serhan, C.N.; Panigrahy, D. Aspirin-triggered proresolving mediators stimulate resolution in cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 6292–6297. [Google Scholar] [CrossRef] [Green Version]
- De Jaeghere, E.A.; Tuyaerts, S.; van Nuffel, A.M.T.; Belmans, A.; Bogaerts, K.; Baiden-Amissah, R.; Lippens, L.; Vuylsteke, P.; Henry, S.; Trinh, X.B.; et al. Pembrolizumab, radiotherapy, and an immunomodulatory five-drug cocktail in pretreated patients with persistent, recurrent, or metastatic cervical or endometrial carcinoma: Results of the phase II PRIMMO study. Cancer Immunol. Immunother. 2022, 1–7. [Google Scholar] [CrossRef]
- Hanada, R.; Hanada, T.; Sigl, V.; Schramek, D.; Penninger, J.M. RANKL/RANK-beyond bones. J. Mol. Med. 2011, 89, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Clézardin, P.; Márquez-Rodas, I.; Niepel, D.; Gedye, C. The RANK-RANKL axis: An opportunity for drug repurposing in cancer? Clin. Transl. Oncol. 2019, 21, 977–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, L.A. Anti-tumor role of metformin. Nat. Immunol. 2018, 19, 1039. [Google Scholar] [CrossRef] [PubMed]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savic Prince, S.; Bubendorf, L. Predictive potential and need for standardization of PD-L1 immunohistochemistry. Virchows Arch. 2019, 474, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhao, Y.; Liu, G.; Zhou, H.-L.; Fan, J.; Zhang, L.; Li, Y.-L.; Wang, Y.; Liang, J.; Xu, Z.-X. Upregulation of programmed death ligand 1 by liver kinase B1 and its implication in programmed death 1 blockade therapy in non-small cell lung cancer. Life Sci. 2020, 256, 117923. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Wang, Y.; Luo, J.; Liu, M.; Luo, Z. Pleiotropic Effects of Metformin on the Antitumor Efficiency of Immune Checkpoint Inhibitors. Front. Immunol. 2020, 11, 586760. [Google Scholar] [CrossRef]
- Eleutherakis-Papaiakovou, V.; Bamias, A.; Dimopoulos, M.A. Thalidomide in cancer medicine. Ann. Oncol. 2004, 15, 1151–1160. [Google Scholar] [CrossRef]
- Rauf, A.; Abu-Izneid, T.; Khalil, A.A.; Imran, M.; Shah, Z.A.; Emran, T.B.; Mitra, S.; Khan, Z.; Alhumaydhi, F.A.; Aljohani, A.S.M.; et al. Berberine as a Potential Anticancer Agent: A Comprehensive Review. Molecules 2021, 26, 7368. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ji, Q.; Ye, N.; Sui, H.; Zhou, L.; Zhu, H.; Fan, Z.; Cai, J.; Li, Q. Berberine Inhibits Invasion and Metastasis of Colorectal Cancer Cells via COX-2/PGE2 Mediated JAK2/STAT3 Signaling Pathway. PLoS ONE 2015, 10, e01234782015. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Zhu, M.; Zhang, D.; Yang, L.; Yang, T.; Li, X.; Zhang, Y. Berberine inhibits the proliferation and migration of breast cancer ZR-75-30 cells by targeting Ephrin-B2. Phytomedicine 2017, 25, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Kim, M.-J.; Kim, E.J.; Yang, Y.; Lee, M.-S.; Lim, J.-S. Berberine-induced AMPK activation inhibits the metastatic potential of melanoma cells via reduction of ERK activity and COX-2 protein expression. Biochem. Pharmacol. 2012, 83, 385–394. [Google Scholar] [CrossRef]
- Liu, J.-F.; Lai, K.C.; Peng, S.-F.; Maraming, P.; Huang, Y.-P.; Huang, A.-C.; Chueh, F.-S.; Huang, W.-W.; Chung, J.-G. Berberine Inhibits Human Melanoma A375.S2 Cell Migration and Invasion via Affecting the FAK, uPA, and NF-κB Signaling Pathways and Inhibits PLX4032 Resistant A375.S2 Cell Migration In Vitro. Molecules 2018, 23, 2019. [Google Scholar] [CrossRef] [Green Version]
- Tsang, C.M.; Cheung, Y.C.; Lui, V.W.-Y.; Yip, Y.L.; Zhang, G.; Lin, V.W.; Cheung, K.C.-P.; Feng, Y.; Tsao, S.W. Berberine suppresses tumorigenicity and growth of nasopharyngeal carcinoma cells by inhibiting STAT3 activation induced by tumor associated fibroblasts. BMC Cancer 2013, 13, 619. [Google Scholar] [CrossRef] [Green Version]
- Petitdemange, A.; Blaess, J.; Sibilia, J.; Felten, R.; Arnaud, L. Shared development of targeted therapies among autoimmune and inflammatory diseases: A systematic repurposing analysis. Ther. Adv. Musculoskelet. Dis. 2020, 12, 1759720X20969261. [Google Scholar] [CrossRef]
- Kingsmore, K.M.; Grammer, A.C.; Lipsky, P.E. Drug repurposing to improve treatment of rheumatic autoimmune inflammatory diseases. Nat. Rev. Rheumatol. 2020, 16, 32–52. [Google Scholar] [CrossRef]
- Okada, Y.; Di, W.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.; Quattrochi, B.; Everett, C.; Hong, B.-Y.; Cervantes, J. Gut commensals, dysbiosis, and immune response imbalance in the pathogenesis of multiple sclerosis. Mult. Scler. 2021, 27, 807–811. [Google Scholar] [CrossRef]
- Absinta, M.; Lassmann, H.; Trapp, B.D. Mechanisms underlying progression in multiple sclerosis. Curr. Opin. Neurol. 2020, 33, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Amadio, S.; Conte, F.; Esposito, G.; Fiscon, G.; Paci, P.; Volonté, C. Repurposing Histaminergic Drugs in Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 6347. [Google Scholar] [CrossRef] [PubMed]
- Garantziotis, P.; Nikolakis, D.; Doumas, S.; Frangou, E.; Sentis, G.; Filia, A.; Fanouriakis, A.; Bertsias, G.; Boumpas, D.T. Molecular Taxonomy of Systemic Lupus Erythematosus Through Data-Driven Patient Stratification: Molecular Endotypes and Cluster-Tailored Drugs. Front. Immunol. 2022, 13, 860726. [Google Scholar] [CrossRef] [PubMed]
- Psarras, A.; Emery, P.; Vital, E.M. Type I interferon-mediated autoimmune diseases: Pathogenesis, diagnosis and targeted therapy. Rheumatology 2017, 56, 1662–1675. [Google Scholar] [CrossRef] [Green Version]
- Ruiz de Morales, J.M.G.; Puig, L.; Daudén, E.; Cañete, J.D.; Pablos, J.L.; Martín, A.O.; Juanatey, C.G.; Adán, A.; Montalbán, X.; Borruel, N.; et al. Critical role of interleukin (IL)-17 in inflammatory and immune disorders: An updated review of the evidence focusing in controversies. Autoimmun. Rev. 2020, 19, 102429. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [Green Version]
- Sohraby, F.; Bagheri, M.; Aryapour, H. Performing an In Silico Repurposing of Existing Drugs by Combining Virtual Screening and Molecular Dynamics Simulation. Methods Mol. Biol. 2019, 1903, 23–43. [Google Scholar] [CrossRef]
- Nagaraj, A.B.; Wang, Q.Q.; Joseph, P.; Zheng, C.; Chen, Y.; Kovalenko, O.; Singh, S.; Armstrong, A.; Resnick, K.; Zanotti, K.; et al. Using a novel computational drug-repositioning approach (DrugPredict) to rapidly identify potent drug candidates for cancer treatment. Oncogene 2018, 37, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Tanoli, Z.-U.-R.; Ravikumar, B.; Alam, Z.; Rebane, A.; Vähä-Koskela, M.; Peddinti, G.; van Adrichem, A.J.; Wakkinen, J.; Jaiswal, A.; et al. Drug Target Commons: A Community Effort to Build a Consensus Knowledge Base for Drug-Target Interactions. Cell Chem. Biol. 2018, 25, 224–229.e2. [Google Scholar] [CrossRef] [PubMed]
- Khaladkar, M.; Koscielny, G.; Hasan, S.; Agarwal, P.; Dunham, I.; Rajpal, D.; Sanseau, P. Uncovering novel repositioning opportunities using the Open Targets platform. Drug Discov. Today 2017, 22, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Patel, C.J. A standard database for drug repositioning. Sci. Data 2017, 4, 170029. [Google Scholar] [CrossRef] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Shameer, K.; Glicksberg, B.S.; Hodos, R.; Johnson, K.W.; Badgeley, M.A.; Readhead, B.; Tomlinson, M.S.; O’Connor, T.; Miotto, R.; Kidd, B.A.; et al. Systematic analyses of drugs and disease indications in RepurposeDB reveal pharmacological, biological and epidemiological factors influencing drug repositioning. Brief. Bioinform. 2018, 19, 656–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, L.; Kehl, T.; Thedinga, K.; Grammes, N.L.; Backes, C.; Mohr, C.; Schubert, B.; Lenhof, K.; Gerstner, N.; Hartkopf, A.D.; et al. ClinOmicsTrailbc: A visual analytics tool for breast cancer treatment stratification. Bioinformatics 2019, 35, 5171–5181. [Google Scholar] [CrossRef] [Green Version]
- Zong, N.; Wen, A.; Moon, S.; Fu, S.; Wang, L.; Zhao, Y.; Yu, Y.; Huang, M.; Wang, Y.; Zheng, G.; et al. Computational drug repurposing based on electronic health records: A scoping review. NPJ Digit. Med. 2022, 5, 77. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, Q.; Ji, X.; Zhang, Y.; Li, W.; Peng, S.; Xue, F. Machine Learning Applications in Drug Repurposing. Interdiscip. Sci. 2022, 14, 15–21. [Google Scholar] [CrossRef]
- Tanoli, Z.; Vähä-Koskela, M.; Aittokallio, T. Artificial intelligence, machine learning, and drug repurposing in cancer. Expert Opin. Drug Discov. 2021, 16, 977–989. [Google Scholar] [CrossRef]
- Issa, N.T.; Stathias, V.; Schürer, S.; Dakshanamurthy, S. Machine and deep learning approaches for cancer drug repurposing. Semin. Cancer Biol. 2021, 68, 132–142. [Google Scholar] [CrossRef]
- Koromina, M.; Pandi, M.-T.; Patrinos, G.P. Rethinking Drug Repositioning and Development with Artificial Intelligence, Machine Learning, and Omics. OMICS 2019, 23, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A. Drug repositioning: Current approaches and their implications in the precision medicine era. Expert Rev. Precis. Med. Drug Dev. 2018, 3, 49–61. [Google Scholar] [CrossRef]
- Napolitano, F.; Zhao, Y.; Moreira, V.M.; Tagliaferri, R.; Kere, J.; D’Amato, M.; Greco, D. Drug repositioning: A machine-learning approach through data integration. J. Cheminform. 2013, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Bambrah, R.K.; Pham, D.C.; Rana, F. Argatroban in heparin-induced thrombocytopenia: Rationale for use and place in therapy. Ther. Adv. Chronic Dis. 2013, 4, 302–304. [Google Scholar] [CrossRef]
- Koster, A.; Fischer, K.-G.; Harder, S.; Mertzlufft, F. The direct thrombin inhibitor argatroban: A review of its use in patients with and without HIT. Biologics 2007, 1, 105–112. [Google Scholar]
- Mercader, A.G.; Duchowicz, P.R.; Sivakumar, P.M. (Eds.) Chemometrics Applications and Research: QSAR in Medicinal Chemistry; Apple Academic Press: Palm Bay, FL, USA, 2021; ISBN 9781774633878. [Google Scholar]
- Akhoon, B.A.; Singh, K.P.; Karmakar, M.; Smita, S.; Pandey, R.; Gupta, S.K. Virtual screening and prediction of the molecular mechanism of bioactive compounds in silico. In Biotechnology of Bioactive Compounds; Gupta, V.K., Tuohy, M.G., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2015; pp. 371–394. ISBN 9781118733103. [Google Scholar]
- Dror, O.; Shulman-Peleg, A.; Nussinov, R.; Wolfson, H.J. Predicting molecular interactions in silico: I. A guide to pharmacophore identification and its applications to drug design. Curr. Med. Chem. 2004, 11, 71–90. [Google Scholar] [CrossRef] [Green Version]
- Meslamani, J.; Li, J.; Sutter, J.; Stevens, A.; Bertrand, H.-O.; Rognan, D. Protein-ligand-based pharmacophores: Generation and utility assessment in computational ligand profiling. J. Chem. Inf. Model. 2012, 52, 943–955. [Google Scholar] [CrossRef]
- Falchi, F.; Caporuscio, F.; Recanatini, M. Structure-based design of small-molecule protein-protein interaction modulators: The story so far. Future Med. Chem. 2014, 6, 343–357. [Google Scholar] [CrossRef]
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins 2007, 69 (Suppl. S8), 108–117. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Spassov, V.Z.; Flook, P.K.; Yan, L. LOOPER: A molecular mechanics-based algorithm for protein loop prediction. Protein Eng. Des. Sel. 2008, 21, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spassov, V.Z.; Yan, L.; Flook, P.K. The dominant role of side-chain backbone interactions in structural realization of amino acid code. ChiRotor: A side-chain prediction algorithm based on side-chain backbone interactions. Protein Sci. 2007, 16, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Li, L.; Weng, Z. ZDOCK: An initial-stage protein-docking algorithm. Proteins 2003, 52, 80–87. [Google Scholar] [CrossRef]
- Li, L.; Chen, R.; Weng, Z. RDOCK: Refinement of rigid-body protein docking predictions. Proteins 2003, 53, 693–707. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Database | Description | Website |
|---|---|---|
| Drug library databases | ||
| PubChem | Larger molecules such as nucleotides, polysaccharides, lipids, peptides, and chemically altered macromolecules are also present in the National Institutes of Health’s (NIH) open chemistry database alongside smaller molecules. | http://pubchem.ncbi.nlm.nih.gov |
| Drugbank | Contains information on drugs and drug targets. Database is a comprehensive, freely accessible, online. Created and maintained by the University of Alberta and The Metabolomics Innovation Centre. | http://www.drugbank.ca/ |
| Chemspider | Chemical structures are included in databases, giving users quick text and structure search access to more than 100 million structures from countless data sources. | http://www.chemspider.com |
| ChemDB | Databases have all the commercially available important as well as small molecules which play important role for building useful blocks for drug discovery. | http://www.chemdb.com |
| ZINC | ZINC database contains over 230 million compounds for virtual screening. | https://zinc.docking.org |
| COCONUT | The collection of open natural products (COCONUT) database provides over 400 thousand natural compounds for virtual screening and drug repurposing. | https://coconut.naturalproducts.net |
| STITCH | STITCH databases provide known and predicted interactions between chemicals and proteins which can be used for drug repurposing. | http://stitch.embl.de |
| Target 3D structure databases | ||
| RCSB Protein Data Bank (PDB) | Information of three-dimensional structural of large biological molecules, such as proteins and nucleic acids. | http://www.rcsb.org |
| OCA | Rapidly search through the contents of the entire PDB Archive. | http://oca.weizmann.ac.il/oca-bin/ocamain |
| Proteopedia | Structural and functional knowledge about biomacromolecules, their assemblies and interactions with small molecules. | http://proteopedia.org |
| Drug-target databases | ||
| Drugbank | Data related to drug interactions, pharmacology, chemical structures, targets, metabolism and identify repurposing opportunities, or build predictive machine learning models. | http://www.drugbank.ca/ |
| Pharmacogenetics Knowledge Base (PharmGKB) | Information on pharmacogenomics that is used to collect, organize, evaluate, and scientific information about how human genetic variation influences drug response. | http://www.pharmgkb.org/ |
| Therapeutic Target Database (TTD) | TTD contains the information on the targeted disease, pathways, and known and under-researched therapeutic protein and nucleic acid targets. | https://bidd.group/group/cjttd/ |
| Drug Target Commons | Improved consensus and utilization of drug–target interactions thanks to a crowdsourcing platform. | https://drugtargetcommons.fimm.fi/ |
| Open Targets | Integrates public domain data to enable target identification and prioritization. | https://www.opentargets.org/ |
| Protein interaction databases | ||
| Human Protein Atlas | Central platform for all human proteins in cells, tissues, and organs using combination of various omics technologies, including antibody-based imaging, mass spectrometry-based proteomics, transcriptomics, and systems biology. | https://www.proteinatlas.org/ |
| Biological General Repository for Interaction (BIOGRID) | Theme of the database is to curate on specific biological processes with disease relevance and curated for biological interactions. | http://thebiogrid.org/ |
| Database of Interacting Proteins (DIP) | DIP include experimentally determined interactions be-tween proteins and collected a single, unified set of protein–protein interactions by combining data from many sources. | http://dip.doe-mbi.ucla.edu/dip/Main.cgi |
| STRING | Biological database and web resource of known and predicted protein–protein interactions. | http://string-db.org/ |
| Pathway databases | ||
| NCI Pathway Interaction Database (NCI-PID) | Relevant information related to pathway interaction of human cellular signaling and contained the molecular interactions and processes that occur in cells, with a special emphasis on actions that may be important for the study and therapy of cancer. | https://www.ndexbio.org |
| Kyoto Encyclopedia of Genes and Genomes (KEGG) | Collection of genomes, biological pathways, diseases, drugs, and chemical substances. | http://www.genome.jp/kegg/ |
| PathwayCommons | Collection of the different biological pathways which includes proteins, DNA, RNA, and tiny molecules are involved in biochemical reactions, the building of bio-molecular complexes, transport and catalytic events, and physical interactions. | http://www.pathwaycommons.org/about/ |
| REACTOME | Peer-reviewed pathway database of disease, signaling cascades, metabolic networks. | https://reactome.org |
| Clinical trial information databases | ||
| Clinicaltrial.gov | The database includes international clinical trials that have been financed by both governmental and commercial sources. | http://clinicaltrials.gov |
| SIDER | Medicines that are marketed and their documented negative drug effects. | http://sideeffects.embl.de/ |
| Drug Repurposing Hub | Collection of curated and annotated FDA-approved medications and pharmaceuticals used in clinical trials, and preclinical studies. | https://clue.io/repurposing |
| repoDB | This database contains successful and failed drug repositioning studies. | https://unmtid-shinyapps.net/shiny/repodb/ |
| FDA label information | ||
| FDALabel (US FDA) | Over 140,000 human pharmaceutical, biological, over-the-counter (OTC), and animal medicine labelling documents can be searched in a variety of ways using this application. | https://nctr-crs.fda.gov/fdalabel/ui/search |
| DailyMed (US FDA) | Contains 143,950 labeling submitted to the Food and Drug Administration (FDA) by companies. | http://dailymed.nlm.nih.gov/dailymed/about.cfm |
| Omics data (Target/Drug) | ||
| NCBI-GEO | Store the gene expression profiling and RNA methylation profiling managed by the National Center for Biotechnology Information. | http://www.ncbi.nlm.nih.gov/geo/ |
| Sequence Read Archive (SRA) | Repository of high throughput sequencing data. | http://www.ncbi.nlm.nih.gov/Traces/sra/ |
| ArrayExpress | Contained functional genomics data which extracted from high-throughput functional genomics experiments. | http://www.ebi.ac.uk/arrayexpress/ |
| Cancer Cell Line Encyclopedia (CCLE) | Detailed genetic and chemical characterization of over 1100 cancer cell lines. | http://www.broadinstitute.org/ccle/home |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spitschak, A.; Gupta, S.; Singh, K.P.; Logotheti, S.; Pützer, B.M. Drug Repurposing at the Interface of Melanoma Immunotherapy and Autoimmune Disease. Pharmaceutics 2023, 15, 83. https://doi.org/10.3390/pharmaceutics15010083
Spitschak A, Gupta S, Singh KP, Logotheti S, Pützer BM. Drug Repurposing at the Interface of Melanoma Immunotherapy and Autoimmune Disease. Pharmaceutics. 2023; 15(1):83. https://doi.org/10.3390/pharmaceutics15010083
Chicago/Turabian StyleSpitschak, Alf, Shailendra Gupta, Krishna P. Singh, Stella Logotheti, and Brigitte M. Pützer. 2023. "Drug Repurposing at the Interface of Melanoma Immunotherapy and Autoimmune Disease" Pharmaceutics 15, no. 1: 83. https://doi.org/10.3390/pharmaceutics15010083
APA StyleSpitschak, A., Gupta, S., Singh, K. P., Logotheti, S., & Pützer, B. M. (2023). Drug Repurposing at the Interface of Melanoma Immunotherapy and Autoimmune Disease. Pharmaceutics, 15(1), 83. https://doi.org/10.3390/pharmaceutics15010083