
Stabilized Astaxanthin Nanoparticles Developed Using Flash Nanoprecipitation to Improve Oral Bioavailability and Hepatoprotective Effects

 , , , , and
, , , , and 
Abstract
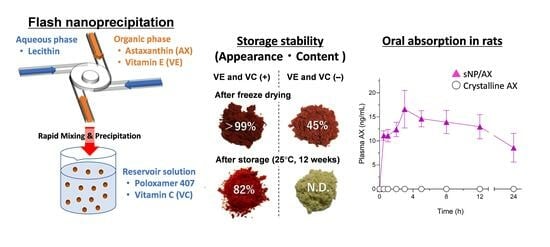
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of sNP/AX
2.3. Ultraperformance LC (UPLC)/Electrospray Ionization (ESI)–Mass Spectrometry (MS) System
2.4. Particle Size Distribution
2.5. Morphology of sNP/AX
2.6. In Vitro Release Study
2.7. Crystallinity
2.8. Stability Studies
2.9. Pharmacokinetic Study
2.9.1. Animals
2.9.2. Plasma Concentration of AX
2.10. Evaluation of Hepatoprotective Effects
2.10.1. Rat Model of Acute Hepatic Injury
2.10.2. Histopathological Observation
2.10.3. Plasma Biomarkers Level
2.10.4. Hepatic Tissue Distribution
2.11. Statistical Analysis
3. Results and Discussion
3.1. Development of sNP/AX
3.1.1. Selection of Amphiphilic Block Co-Polymer as Stabilizer
3.1.2. Stabilizing Effects of Antioxidative Additives during Preparation and Storage
3.2. Physicochemical Characterization of sNP/AX
3.3. Release Properties of sNP/AX
3.4. Pharmacokinetic Studies in Rats
3.5. Hepatoprotective Effects of sNP/AX
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miki, W. Biological Functions and Activities of Animal Carotenoids. Pure Appl. Chem. 1991, 63, 141–146. [Google Scholar] [CrossRef]
- Da Silva, F.O.; Tramonte, V.L.; Parisenti, J.; Lima-Garcia, J.F.; Maraschin, M.; da Silva, E.L. Litopenaeus vannamei Muscle Carotenoids versus Astaxanthin: A Comparison of Antioxidant Activity and in Vitro Protective Effects against Lipid Peroxidation. Food Biosci. 2015, 9, 12–19. [Google Scholar] [CrossRef]
- Goswami, G.; Chaudhuri, S.; Dutta, D. The Present Perspective of Astaxanthin with Reference to Biosynthesis and Pharmacological Importance. World J. Microbiol. Biotechnol. 2010, 26, 1925–1939. [Google Scholar] [CrossRef]
- Fakhri, S.; Abbaszadeh, F.; Dargahi, L.; Jorjani, M. Astaxanthin: A Mechanistic Review on Its Biological Activities and Health Benefits. Pharmacol. Res. 2018, 136, 1–20. [Google Scholar] [CrossRef]
- Hussein, G.; Sankawa, U.; Goto, H.; Matsumoto, K.; Watanabe, H. Astaxanthin, a Carotenoid with Potential in Human Health and Nutrition. J. Nat. Prod. 2006, 69, 443–449. [Google Scholar] [CrossRef]
- Preuss, H.G.; Echard, B.; Bagchi, D.; Perricone, N.V.; Yamashita, E. Astaxanthin Lowers Blood Pressure and Lessens the Activity of the Renin-Angiotensin System in Zucker Fatty Rats. J. Funct. Foods 2009, 1, 13–22. [Google Scholar] [CrossRef]
- Shakeri, M.; Razavi, S.H.; Shakeri, S. Carvacrol and Astaxanthin Co-Entrapment in Beeswax Solid Lipid Nanoparticles as an Efficient Nano-System with Dual Antioxidant and Anti-Biofilm Activities. LWT-Food Sci. Technol. 2019, 107, 280–290. [Google Scholar] [CrossRef]
- Liu, X.; McClements, D.J.; Cao, Y.; Xiao, H. Chemical and Physical Stability of Astaxanthin-Enriched Emulsion-Based Delivery Systems. Food Biophys. 2016, 11, 302–310. [Google Scholar] [CrossRef]
- Taksima, T.; Limpawattana, M.; Klaypradit, W. Astaxanthin Encapsulated in Beads Using Ultrasonic Atomizer and Application in Yogurt as Evaluated by Consumer Sensory Profile. LWT-Food Sci. Technol. 2015, 62, 431–437. [Google Scholar] [CrossRef]
- Pan, L.; Zhang, S.; Gu, K.; Zhang, N. Preparation of Astaxanthin-Loaded Liposomes: Characterization, Storage Stability and Antioxidant Activity. CyTA-J. Food 2018, 16, 607–618. [Google Scholar] [CrossRef]
- Jinno, J.; Kamada, N.; Miyake, M.; Yamada, K.; Mukai, T.; Odomi, M.; Toguchi, H.; Liversidge, G.G.; Higaki, K.; Kimura, T. Effect of Particle Size Reduction on Dissolution and Oral Absorption of a Poorly Water-Soluble Drug, Cilostazol, in Beagle Dogs. J. Control. Release 2006, 111, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca Antunes, A.B.; De Geest, B.G.; Vervaet, C.; Remon, J.P. Solvent-Free Drug Crystal Engineering for Drug Nano- and Micro Suspensions. Eur. J. Pharm. Sci. 2013, 48, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Galli, C. Experimental Determination of the Diffusion Boundary Layer Width of Micron and Submicron Particles. Int. J. Pharm. 2006, 313, 114–122. [Google Scholar] [CrossRef]
- Matteucci, M.E.; Brettmann, B.K.; Rogers, T.L.; Elder, E.J.; Williams, R.O., III; Johnston, K.P. Design of Potent Amorphous Drug Nanoparticles for Rapid Generation of Highly Supersaturated Media. Mol. Pharm. 2007, 4, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Auweter, H.; Haberkorn, H.; Heckmann, W.; Horn, D.; Lüddecke, E.; Rieger, J.; Weiss, H. Supramolecular Structure of Precipitated Nanosize β-Carotene Particles. Angew Chem. Int. Ed. Engl. 1999, 38, 2188–2191. [Google Scholar] [CrossRef]
- Markwalter, C.E.; Prud’homme, R.K. Design of a Small-Scale Multi-Inlet Vortex Mixer for Scalable Nanoparticle Production and Application to the Encapsulation of Biologics by Inverse Flash Nanoprecipitation. J. Pharm. Sci. 2018, 107, 2465–2471. [Google Scholar] [CrossRef]
- Johnson, B.K.; Prud’homme, R.K. Chemical Processing and Micromixing in Confined Impinging Jets. AIChE J. 2003, 49, 2264–2282. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, C.; Prud’homme, R.K.; Fox, R.O. Mixing in a Multi-Inlet Vortex Mixer (MIVM) for Flash Nano-Precipitation. Chem. Eng. Sci. 2008, 63, 2829–2842. [Google Scholar] [CrossRef]
- Chow, S.F.; Wan, K.Y.; Cheng, K.K.; Wong, K.W.; Sun, C.C.; Baum, L.; Chow, A.H.L. Development of Highly Stabilized Curcumin Nanoparticles by Flash Nanoprecipitation and Lyophilization. Eur. J. Pharm. Biopharm. 2015, 94, 436–449. [Google Scholar] [CrossRef]
- Zhu, Z.; Anacker, J.L.; Ji, S.; Hoye, T.R.; Macosko, C.W.; Prud’homme, R.K. Formation of Block Copolymer-Protected Nanoparticles via Reactive Impingement Mixing. Langmuir 2007, 23, 10499–10504. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, R.; Guo, Z.; Li, C.; Li, P. The Preparation and Stability of the Inclusion Complex of Astaxanthin with β-Cyclodextrin. Food Chem. 2007, 101, 1580–1584. [Google Scholar] [CrossRef]
- Honda, M.; Kageyama, H.; Hibino, T.; Osawa, Y.; Kawashima, Y.; Hirasawa, K.; Kuroda, I. Evaluation and Improvement of Storage Stability of Astaxanthin Isomers in Oils and Fats. Food Chem. 2021, 352, 129371. [Google Scholar] [CrossRef] [PubMed]
- Meléndez-Martínez, A.J.; Vicario, I.M.; Heredia, F.J. Estabilidad de los pigmentos carotenoides en los alimentos. Arch. Latinoam. Nutr. 2004, 54, 209–215. [Google Scholar]
- Tamjidi, F.; Shahedi, M.; Varshosaz, J.; Nasirpour, A. EDTA and α-Tocopherol Improve the Chemical Stability of Astaxanthin Loaded into Nanostructured Lipid Carriers. Eur. J. Lipid Sci. Technol. 2014, 116, 968–977. [Google Scholar] [CrossRef]
- Caggiano, N.J.; Wilson, B.K.; Priestley, R.D.; Prud’homme, R.K. Development of an In Vitro Release Assay for Low-Density Cannabidiol Nanoparticles Prepared by Flash NanoPrecipitation. Mol. Pharm. 2022, 19, 1515–1525. [Google Scholar] [CrossRef]
- Freag, M.S.; Elnaggar, Y.S.; Abdelmonsif, D.A.; Abdallah, O.Y. Stealth, Biocompatible Monoolein-Based Lyotropic Liquid Crystalline Nanoparticles for Enhanced Aloe-Emodin Delivery to Breast Cancer Cells: In Vitro and in Vivo Studies. Int. J. Nanomed. 2016, 11, 4799. [Google Scholar] [CrossRef]
- Directive 2010/63/eu of the European Parliament and of the Council of 22 September 2010 on the Protection of Animals Used for Scientific Purposes. Off. J. Eur. Union 2010, L276, 33–79. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:276:0033:0079:en:PDF (accessed on 24 October 2023).
- Choi, H.D.; Kang, H.E.; Yang, S.H.; Lee, M.G.; Shin, W.G. Pharmacokinetics and First-Pass Metabolism of Astaxanthin in Rats. Br. J. Nutr. 2011, 105, 220–227. [Google Scholar] [CrossRef]
- Telange, D.R.; Patil, A.T.; Pethe, A.M.; Fegade, H.; Anand, S.; Dave, V.S. Formulation and Characterization of an Apigenin-Phospholipid Phytosome (APLC) for Improved Solubility, in Vivo Bioavailability, and Antioxidant Potential. Eur. J. Pharm. Sci. 2017, 108, 36–49. [Google Scholar] [CrossRef]
- Dumortier, G.; Grossiord, J.L.; Agnely, F.; Chaumeil, J.C. A Review of Poloxamer 407 Pharmaceutical and Pharmacological Characteristics. Pharm. Res. 2006, 23, 2709–2728. [Google Scholar] [CrossRef] [PubMed]
- Caris-Veyrat, C.; Schmid, A.; Carail, M.; Böhm, V. Cleavage Products of Lycopene Produced by in Vitro Oxidations: Characterization and Mechanisms of Formation. J. Agric. Food Chem. 2003, 51, 7318–7325. [Google Scholar] [CrossRef] [PubMed]
- Lutz-Röder, A.; Jezussek, M.; Winterhalter, P. Nickel Peroxide Induced Oxidation of Canthaxanthin. J. Agric. Food Chem. 1999, 47, 1887–1891. [Google Scholar] [CrossRef] [PubMed]
- Waché, Y.; Bosser-DeRatuld, A.; Lhuguenot, J.-C.; Belin, J.-M. Effect of Cis/Trans Isomerism of β-Carotene on the Ratios of Volatile Compounds Produced during Oxidative Degradation. J. Agric. Food Chem. 2003, 51, 1984–1987. [Google Scholar] [CrossRef]
- Etoh, H.; Suhara, M.; Tokuyama, S.; Kato, H.; Nakahigashi, R.; Maejima, Y.; Ishikura, M.; Terada, Y.; Maoka, T. Auto-Oxidation Products of Astaxanthin. J. Oleo Sci. 2012, 61, 17–21. [Google Scholar] [CrossRef]
- Niamnuy, C.; Devahastin, S.; Soponronnarit, S.; Raghavan, G.V. Kinetics of Astaxanthin Degradation and Color Changes of Dried Shrimp during Storage. J. Food Eng. 2008, 87, 591–600. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, T.; Yu, J.; Liu, Y.; Wang, Y.; He, Z. In Vitro/Vivo Assessment of Praziquantel Nanocrystals: Formulation, Characterization, and Pharmacokinetics in Beagle Dogs. Asian J. Pharm. Sci. 2019, 14, 321–328. [Google Scholar] [CrossRef]
- Hariharan, S.; Bhardwaj, V.; Bala, I.; Sitterberg, J.; Bakowsky, U.; Ravi Kumar, M.N.V. Design of Estradiol Loaded PLGA Nanoparticulate Formulations: A Potential Oral Delivery System for Hormone Therapy. Pharm. Res. 2006, 23, 184–195. [Google Scholar] [CrossRef]
- Furr, H.C.; Clark, R.M. Intestinal Absorption and Tissue Distribution of Carotenoids. J. Nutr. Biochem. 1997, 8, 364–377. [Google Scholar] [CrossRef]
- He, C.; Yin, L.; Tang, C.; Yin, C. Size-Dependent Absorption Mechanism of Polymeric Nanoparticles for Oral Delivery of Protein Drugs. Biomaterials 2012, 33, 8569–8578. [Google Scholar] [CrossRef]
- De Stefani, C.; Lodovichi, J.; Albonetti, L.; Salvatici, M.C.; Quintela, J.C.; Bilia, A.R.; Bergonzi, M.C. Solubility and Permeability Enhancement of Oleanolic Acid by Solid Dispersion in Poloxamers and γ-CD. Molecules 2022, 27, 3042. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Qiao, X.; Nan, H.; Cao, Y.; Xu, J.; Xue, C. MPEG-Carboxymethyl Astaxanthin Monoester: A Novel Hydrophilic Astaxanthin with Increased Water Solubility and Bioavailability. LWT-Food Sci. Technol. 2021, 143, 111134. [Google Scholar] [CrossRef]
- Zhu, Y.; Gu, Z.; Liao, Y.; Li, S.; Xue, Y.; Firempong, M.A.; Xu, Y.; Yu, J.; Smyth, H.D.; Xu, X. Improved Intestinal Absorption and Oral Bioavailability of Astaxanthin Using Poly (Ethylene Glycol)-Graft-Chitosan Nanoparticles: Preparation, In Vitro Evaluation, and Pharmacokinetics in Rats. J. Sci. Food Agric. 2022, 102, 1002–1011. [Google Scholar] [CrossRef]
- Yuan, J.-P.; Peng, J.; Yin, K.; Wang, J.-H. Potential Health-Promoting Effects of Astaxanthin: A High-Value Carotenoid Mostly from Microalgae. Mol. Nutr. Food Res. 2011, 55, 150–165. [Google Scholar]
- Lee, D.-H.; Kim, C.-S.; Lee, Y.J. Astaxanthin Protects against MPTP/MPP+-Induced Mitochondrial Dysfunction and ROS Production In Vivo and In Vitro. Food Chem. Toxicol. 2011, 49, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Itoh, A.; Isoda, K.; Kondoh, M.; Kawase, M.; Yagi, K. Fucoidan Partly Prevents CCl4-Induced Liver Fibrosis. Eur. J. Pharmacol. 2008, 580, 380–384. [Google Scholar] [CrossRef]
- Kalegari, M.; Gemin, C.A.B.; Araújo-Silva, G.; De Brito, N.J.N.; López, J.A.; de Oliveira Tozetto, S.; das Graças Almeida, M.; Miguel, M.D.; Stien, D.; Miguel, O.G. Chemical Composition, Antioxidant Activity and Hepatoprotective Potential of Rourea induta Planch. (Connaraceae) against CCl4-Induced Liver Injury in Female Rats. Nutrition 2014, 30, 713–718. [Google Scholar] [CrossRef]
- Banik, S.; Yamada, K.; Sato, H.; Onoue, S. Development of Poly (Lipoic Acid) Nanoparticles with Improved Oral Bioavailability and Hepatoprotective Effects of Quercetin. Mol. Pharm. 2022, 19, 1468–1476. [Google Scholar] [CrossRef]
- Shen, M.; Chen, K.; Lu, J.; Cheng, P.; Xu, L.; Dai, W.; Wang, F.; He, L.; Zhang, Y.; Chengfen, W. Protective Effect of Astaxanthin on Liver Fibrosis through Modulation of TGF-1 Expression and Autophagy. Mediat. Inflamm. 2014, 2014, 954502. [Google Scholar] [CrossRef]
- Li, J.; Xia, Y.; Liu, T.; Wang, J.; Dai, W.; Wang, F.; Zheng, Y.; Chen, K.; Li, S.; Abudumijiti, H. Protective Effects of Astaxanthin on ConA-Induced Autoimmune Hepatitis by the JNK/p-JNK Pathway-Mediated Inhibition of Autophagy and Apoptosis. PLoS ONE 2015, 10, e0120440. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stabilizer | Particle Size (nm) | PDI | AX Recovery (%) |
|---|---|---|---|
| P188 | 186 ± 2.6 | 0.19 ± 0.01 | 73 ± 0.6 |
| P407 | 187 ± 1.5 | 0.14 ± 0.03 | 89 ± 7.3 |
| TPGS | 170 ± 6.5 | 0.38 ± 0.03 | 82 ± 6.2 |
| Soluplus® | 155 ± 1.5 | 0.19 ± 0.01 | 68 ± 3.5 |
| Stabilizers | Initial Samples | Stored Samples (4 Weeks, 4 °C) | Stored Samples (4 Weeks, 25 °C) | |||
|---|---|---|---|---|---|---|
| Particle Size (nm) | AX Recovery (%) | Particle Size (nm) | Remaining AX (%) | Particle Size (nm) | Remaining AX (%) | |
| P407 | 236 ± 3.6 | 45 | 278 ± 12 | 11 | 289 ± 15 | N.D. |
| P407 + VC (2.5%) | 289 ± 7.8 | 54 | - | - | - | - |
| P407 + VC (5.0%) | 266 ± 5.5 | 54 | 270 ± 50 | 7 | 299 ± 45 | N.D. |
| P407 + VE (2.5%) | 260 ± 5.0 | 46 | - | - | - | - |
| P407 + VE (5.0%) | 255 ± 5.5 | 40 | 270 ± 45 | 34 | 266 ± 49 | 34 |
| P407 + VE (2.5%) + VC (2.5%) | 215 ± 6.8 | >99 | 212 ± 12 | 94 | 242 ± 13 | 82 |
| Parameters | Crystalline AX | sNP/AX |
|---|---|---|
| Cmax (ng/mL) | <LOD | 17 ± 3.5 |
| Tmax (h) | <LOD | 3.0 ± 0 |
| AUC0–24 h (ng·h/mL) | – | 298 ± 12 |
| BA (%) | – | 2.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, A.; Banik, S.; Yamada, K.; Misaka, S.; Prud’homme, R.K.; Sato, H.; Onoue, S. Stabilized Astaxanthin Nanoparticles Developed Using Flash Nanoprecipitation to Improve Oral Bioavailability and Hepatoprotective Effects. Pharmaceutics 2023, 15, 2562. https://doi.org/10.3390/pharmaceutics15112562
Ghosh A, Banik S, Yamada K, Misaka S, Prud’homme RK, Sato H, Onoue S. Stabilized Astaxanthin Nanoparticles Developed Using Flash Nanoprecipitation to Improve Oral Bioavailability and Hepatoprotective Effects. Pharmaceutics. 2023; 15(11):2562. https://doi.org/10.3390/pharmaceutics15112562
Chicago/Turabian StyleGhosh, Antara, Sujan Banik, Kohei Yamada, Shingen Misaka, Robert K. Prud’homme, Hideyuki Sato, and Satomi Onoue. 2023. "Stabilized Astaxanthin Nanoparticles Developed Using Flash Nanoprecipitation to Improve Oral Bioavailability and Hepatoprotective Effects" Pharmaceutics 15, no. 11: 2562. https://doi.org/10.3390/pharmaceutics15112562
APA StyleGhosh, A., Banik, S., Yamada, K., Misaka, S., Prud’homme, R. K., Sato, H., & Onoue, S. (2023). Stabilized Astaxanthin Nanoparticles Developed Using Flash Nanoprecipitation to Improve Oral Bioavailability and Hepatoprotective Effects. Pharmaceutics, 15(11), 2562. https://doi.org/10.3390/pharmaceutics15112562