
Investigating the Targeting Power to Brain Tissues of Intranasal Rasagiline Mesylate-Loaded Transferosomal In Situ Gel for Efficient Treatment of Parkinson’s Disease

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methodology
2.2.1. Preparation of RSM-Loaded Transferosomes
2.2.2. Characterization of RSM-Loaded Transferosomes
Assessment of Vesicle Size (VS), Polydispersity Index (PDI), and Zeta Potential (ZP)
Assessment of Entrapment Efficiency (%EE)
2.2.3. Characterization of the Optimized Transferosomal Formulation
Fourier Transform Infrared Spectroscopy (FTIR)
Transmission Electron Microscopy (TEM)
2.2.4. Preparation of RSM-Loaded Transferosomal In Situ Gels
2.2.5. Characterization of the Prepared RSM-Loaded Transferosomal In Situ Gels
Determination of Sol-to-Gel Transition Temperature and Gelation Time
Assessment of Rheological Properties
In Vitro Drug Release
2.2.6. Effect of Storage
2.2.7. In Vivo Studies
Assessment of Formulation Biocompatibility
In Vivo Pharmacokinetics
Chromatographic Conditions
Sample Preparation
Brain and Systemic Kinetic Analysis
Statistical Analysis
3. Results and Discussion
3.1. Characterization of RSM-Loaded Transferosomes
3.1.1. Assessment of Vesicle Size (VS), Polydispersity Index (PDI), and Zeta Potential (ZP)
3.1.2. Assessment of Entrapment Efficiency (%EE)
3.2. Optimization of the Formulation Variables
3.3. Characterization of the Optimized Transferosomal Formulation (F12)
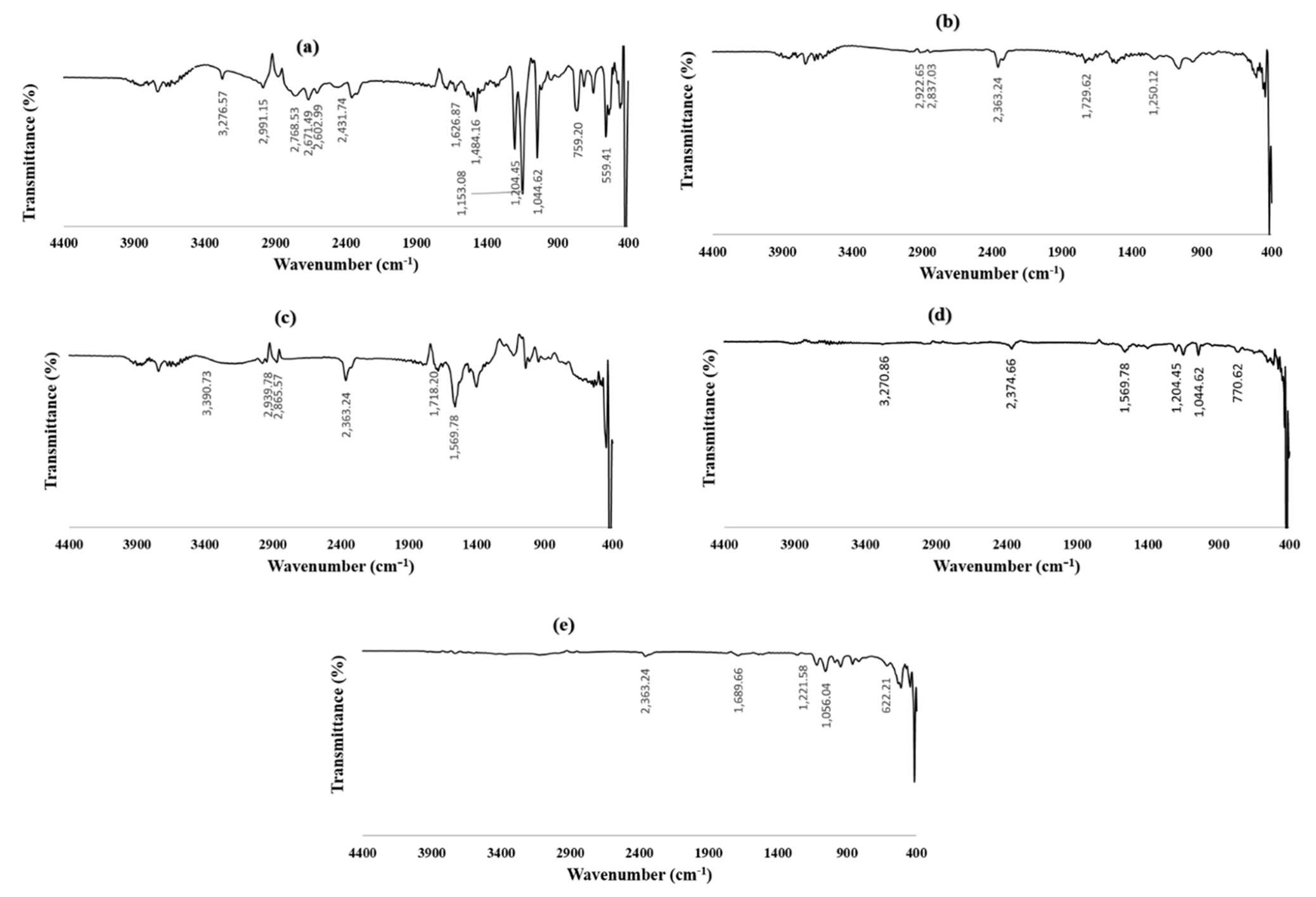
3.3.1. Fourier Transform Infrared Spectroscopy (FTIR)
3.3.2. Transmission Electron Microscopy (TEM)
3.4. Characterization of Mucoadhesive RSM-Loaded Transferosomal In Situ Gels
3.4.1. Sol-to-Gel Transition Temperature
3.4.2. Rheological Study
3.4.3. In Vitro Drug Release
3.5. Effect of Storage
3.6. In Vivo Studies
3.6.1. Assessment of Formulation Biocompatibility
3.6.2. In Vivo Pharmacokinetics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chougar, L.; Arsovic, E.; Gaurav, R.; Biondetti, E.; Faucher, A.; Valabrègue, R.; Pyatigorskaya, N.; Dupont, G.; Lejeune, F.X.; Cormier, F. Regional Selectivity of Neuromelanin Changes in the Substantia Nigra in Atypical Parkinsonism. Mov. Disord. 2022, 37, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Bartos, C.; Pallagi, E.; Szabó-Révész, P.; Ambrus, R.; Katona, G.; Kiss, T.; Rahimi, M.; Csóka, I. Formulation of levodopa containing dry powder for nasal delivery applying the quality-by-design approach. Eur. J. Pharm. Sci. 2018, 123, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Md, S.; Hasan, Q.; Fazil, M.; Ali, A.; Baboota, S.; Ali, J. Brain targeted nanoparticulate drug delivery system of rasagiline via intranasal route. Drug Deliv. 2016, 23, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.Q.; Wang, H.K.; Wang, Y.; Li, M.X.; Jiang, L.L.; Wang, Y. Rasagiline combined with levodopa therapy versus levodopa monotherapy for patients with Parkinson’s disease: A systematic review. Neurol. Sci. 2020, 41, 101–109. [Google Scholar] [CrossRef]
- Cereda, E.; Cilia, R.; Canesi, M.; Tesei, S.; Mariani, C.B.; Zecchinelli, A.L.; Pezzoli, G. Efficacy of rasagiline and selegiline in Parkinson’s disease: A head-to-head 3-year retrospective case—Control study. J. Neurol. 2017, 264, 1254–1263. [Google Scholar] [CrossRef]
- Müller, T. Pharmacokinetic/pharmacodynamic evaluation of rasagiline mesylate for Parkinson’s disease. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1423–1432. [Google Scholar] [CrossRef]
- Chen, J.J.; Swope, D.M.; Dashtipour, K. Comprehensive review of rasagiline, a second-generation monoamine oxidase inhibitor, for the treatment of Parkinson’s disease. Clin. Ther. 2007, 29, 1825–1849. [Google Scholar] [CrossRef]
- Robakis, D.; Fahn, S. Defining the role of the monoamine oxidase-B inhibitors for Parkinson’s disease. CNS Drugs 2015, 29, 433–441. [Google Scholar] [CrossRef]
- Güneş, M.; Karavana, S.Y. Non-Oral Drug Delivery in Parkinson’s Disease: Current Applications and Future. Turk. J. Pharm. Sci. 2022, 19, 343. [Google Scholar] [CrossRef]
- Tandarić, T.; Vianello, R. Computational insight into the mechanism of the irreversible inhibition of monoamine oxidase enzymes by the antiparkinsonian propargylamine inhibitors rasagiline and selegiline. ACS Chem. Neurosci. 2019, 10, 3532–3542. [Google Scholar] [CrossRef]
- Zubiaur, P.; Matas, M.; Martín-Vílchez, S.; Soria-Chacartegui, P.; Villapalos-García, G.; Figueiredo-Tor, L.; Calleja, S.; Navares-Gómez, M.; de Miguel, A.; Novalbos, J. Polymorphism of Drug Transporters, Rather Than Metabolizing Enzymes, Conditions the Pharmacokinetics of Rasagiline. Pharmaceutics 2022, 14, 2001. [Google Scholar] [CrossRef] [PubMed]
- Bali, N.R.; Salve, P.S. Impact of rasagiline nanoparticles on brain targeting efficiency via gellan gum based transdermal patch: A nanotheranostic perspective for Parkinsonism. Int. J. Biol. Macromol. 2020, 164, 1006–1024. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Das, S.; Sarma, K.N. Nose-to-brain drug delivery: An update to the alternative path to successful targeted antimigraine drugs. Int. J. Appl. Pharm. 2021, 13, 67–75. [Google Scholar] [CrossRef]
- Lombardo, R.; Musumeci, T.; Carbone, C.; Pignatello, R. Nanotechnologies for intranasal drug delivery: An update of literature. Pharm. Dev. Technol. 2021, 26, 824–845. [Google Scholar] [CrossRef]
- Dufes, C.; Olivier, J.C.; Gaillard, F.; Gaillard, A.; Couet, W.; Muller, J.M. Brain delivery of vasoactive intestinal peptide (VIP) following nasal administration to rats. Int. J. Pharm. 2003, 255, 87–97. [Google Scholar] [CrossRef]
- Fonseca, L.C.; Lopes, J.A.; Vieira, J.; Viegas, C.; Oliveira, C.S.; Hartmann, R.P.; Fonte, P. Intranasal drug delivery for treatment of Alzheimer’s disease. Drug Deliv. Transl. Res. 2021, 11, 411–425. [Google Scholar] [CrossRef]
- Salama, A.H.; Salama, A.A.; Elhabak, M. Single step nanospray drying preparation technique of gabapentin-loaded nanoparticles-mediated brain delivery for effective treatment of PTZ-induced seizures. Int. J. Pharm. 2021, 602, 120604. [Google Scholar] [CrossRef]
- Gartziandia, O.; Egusquiaguirre, S.P.; Bianco, J.; Pedraz, J.L.; Igartua, M.; Hernandez, R.M.; Préat, V.; Beloqui, A. Nanoparticle transport across in vitro olfactory cell monolayers. Int. J. Pharm. 2016, 499, 81–89. [Google Scholar] [CrossRef]
- Alberto, M.; Paiva-Santos, A.C.; Veiga, F.; Pires, P.C. Lipid and Polymeric Nanoparticles: Successful Strategies for Nose-to-Brain Drug Delivery in the Treatment of Depression and Anxiety Disorders. Pharmaceutics 2022, 14, 2742. [Google Scholar] [CrossRef]
- Erdő, F.; Bors, L.A.; Farkas, D.; Bajza, Á.; Gizurarson, S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef]
- Dholakia, J.; Prabhakar, B.; Shende, P. Strategies for the delivery of antidiabetic drugs via intranasal route. Int. J. Pharm. 2021, 608, 121068. [Google Scholar] [CrossRef] [PubMed]
- Ozsoy, Y.; Gungor, S.; Cevher, E. Nasal delivery of high molecular weight drugs. Molecules 2009, 14, 3754–3779. [Google Scholar] [CrossRef] [PubMed]
- Yurtdaş-Kırımlıoğlu, G. A promising approach to design thermosensitive in situ gel based on solid dispersions of desloratadine with Kolliphor® 188 and Pluronic® F127. J. Therm. Anal. Calorim. 2022, 147, 1307–1327. [Google Scholar] [CrossRef]
- Vigani, B.; Rossi, S.; Sandri, G.; Bonferoni, M.C.; Caramella, C.M.; Ferrari, F. Recent advances in the development of in situ gelling drug delivery systems for non-parenteral administration routes. Pharmaceutics 2020, 12, 859. [Google Scholar] [CrossRef]
- Tengamnuay, P.; Sahamethapat, A.; Sailasuta, A.; Mitra, A.K. Chitosans as nasal absorption enhancers of peptides: Comparison between free amine chitosans and soluble salts. Int. J. Pharm. 2000, 197, 53–67. [Google Scholar] [CrossRef]
- Cirri, M.; Maestrelli, F.; Nerli, G.; Mennini, N.; D’Ambrosio, M.; Luceri, C.; Mura, P.A. Development of a cyclodextrin-based mucoadhesive-thermosensitive in situ gel for clonazepam intranasal delivery. Pharmaceutics 2021, 13, 969. [Google Scholar] [CrossRef]
- Sideek, S.A.; El-Nassan, H.B.; Fares, A.R.; ElMeshad, A.N.; Elkasabgy, N.A. Different Curcumin-Loaded Delivery Systems for Wound Healing Applications: A Comprehensive Review. Pharmaceutics 2023, 15, 38. [Google Scholar] [CrossRef]
- Salama, A.H.; AbouSamra, M.M.; Awad, G.E.A.; Mansy, S.S. Promising bioadhesive ofloxacin-loaded polymeric nanoparticles for the treatment of ocular inflammation: Formulation and in vivo evaluation. Drug Deliv. Transl. Res. 2021, 11, 1943–1957. [Google Scholar] [CrossRef]
- AbouSamra, M.M.; Salama, A.H.; Awad, G.E.A.; Mansy, S.S. Formulation and evaluation of novel hybridized nanovesicles for enhancing buccal delivery of ciclopirox olamine. AAPS PharmSciTech 2020, 21, 283. [Google Scholar] [CrossRef]
- Baloglu, E.; Karavana, S.Y.; Senyigit, Z.A.; Guneri, T. Rheological and mechanical properties of poloxamer mixtures as a mucoadhesive gel base. Pharm. Dev. Technol. 2011, 16, 627–636. [Google Scholar] [CrossRef]
- Sherje, A.P.; Londhe, V. Development and evaluation of pH-responsive cyclodextrin-based in situ gel of paliperidone for intranasal delivery. AAPS PharmSciTech 2018, 19, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, F.; Feng, L.; Yang, L.; Chen, L.; Wei, G.; Lu, W. In vivo retention of poloxamer-based in situ hydrogels for vaginal application in mouse and rat models. Acta Pharm. Sin. B 2017, 7, 502–509. [Google Scholar] [CrossRef] [PubMed]
- De Jong, W.H.; Borm, P.J. Drug delivery and nanoparticles: Applications and hazards. Int. J. Nanomed. 2008, 3, 133. [Google Scholar] [CrossRef] [PubMed]
- Kamel, R.; Afifi, S.M.; Kassem, I.A.A.; Elkasabgy, N.A.; Farag, M.A. Arabinoxylan and rhamnogalacturonan mucilage: Outgoing and potential trends of pharmaceutical, environmental, and medicinal merits. Int. J. Biol. Macromol. 2020, 165, 2550–2564. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Pandey, V.K.; Tewari, R.P.; Agarwal, V. Nanoparticle based drug delivery system: Advantages and applications. Indian J. Sci. Technol. 2011, 4, 177–180. [Google Scholar] [CrossRef]
- Natsheh, H.; Touitou, E. Phospholipid vesicles for dermal/transdermal and nasal administration of active molecules: The effect of surfactants and alcohols on the fluidity of their lipid bilayers and penetration enhancement properties. Molecules 2020, 25, 2959. [Google Scholar] [CrossRef]
- Aboud, H.M.; Ali, A.A.; El-Menshawe, S.F.; Elbary, A.A. Nanotransfersomes of carvedilol for intranasal delivery: Formulation, characterization and in vivo evaluation. Drug Deliv. 2016, 23, 2471–2481. [Google Scholar] [CrossRef]
- Ibrahim, S.S.; Elseoud, O.G.A.; Mohamedy, M.H.; Amer, M.M.; Mohamed, Y.Y.; Elmansy, S.A.; Kadry, M.M.; Attia, A.A.; Fanous, R.A.; Kamel, M.S.; et al. Nose-to-brain delivery of chrysin transfersomal and composite vesicles in doxorubicin-induced cognitive impairment in rats: Insights on formulation, oxidative stress and TLR4/NF-kB/NLRP3 pathways. Neuropharmacology 2021, 197, 108738. [Google Scholar] [CrossRef]
- Toksoy, M.O.; Tirnaksiz, F.F. Development of rasagiline mesylate loaded solid lipid nanoparticles in a thermosensitive mucoadhesive gel: Formulation design using DoE, in-vitro and ex-vivo characterization. J. Res. Pharm. 2021, 25, 695–707. [Google Scholar]
- Mishra, N.; Tiwari, D.K.; Mishra, K. Development of intranasal deformable ethosomes of rasagiline mesylate for the effective management of parkinsonism. Int. J. Pharm. Biol. Sci. 2020, 10, 25–33. [Google Scholar]
- Raj, N.D.; Rao, A.R.; Bhimanadhuni, C.N. Development of RP-HPLC method for the estimation of Rasagiline mesylate in bulk and tablet dosage forms. Int. Curr. Pharm. J. 2012, 1, 285–287. [Google Scholar] [CrossRef]
- Yaghoobian, M.; Haeri, A.; Bolourchian, N.; Shahhosseni, S.; Dadashzadeh, S. The impact of surfactant composition and surface charge of niosomes on the oral absorption of repaglinide as a BCS II model drug. Int. J. Nanomed. 2020, 15, 8767. [Google Scholar] [CrossRef] [PubMed]
- Balata, G.F.; Faisal, M.M.; Elghamry, H.A.; Sabry, S.A. Preparation and characterization of ivabradine HCl transfersomes for enhanced transdermal delivery. J. Drug Deliv. Sci. Technol. 2020, 60, 101921. [Google Scholar] [CrossRef]
- Tavano, L.; Vivacqua, M.; Carito, V.; Muzzalupo, R.; Caroleo, M.C.; Nicoletta, F. Doxorubicin loaded magneto-niosomes for targeted drug delivery. Colloids Surf. B Biointerfaces 2013, 102, 803–807. [Google Scholar] [CrossRef]
- ElKasabgy, N.A.; Elsayed, I.; Elshafeey, A.H. Design of lipotomes as a novel dual functioning nanocarrier for bioavailability enhancement of lacidipine: In-vitro and in-vivo characterization. Int. J. Pharm. 2014, 472, 369–379. [Google Scholar] [CrossRef]
- Zaki, N.M.; Awad, G.A.; Mortada, N.D.; Abd ElHady, S.S. Enhanced bioavailability of metoclopramide HCl by intranasal administration of a mucoadhesive in situ gel with modulated rheological and mucociliary transport properties. Eur. J. Pharm. Sci. 2007, 32, 296–307. [Google Scholar] [CrossRef]
- Jeong, B.; Choi, Y.K.; Bae, Y.H.; Zentner, G.; Kim, S.W. New biodegradable polymers for injectable drug delivery systems. J. Control. Release 1999, 62, 109–114. [Google Scholar] [CrossRef]
- Vadnere, M.; Amidon, G.; Lindenbaum, S.; Haslam, J.L. Thermodynamic studies on the gel-sol transition of some pluronic polyols. Int. J. Pharm. 1984, 22, 207–218. [Google Scholar] [CrossRef]
- Gilbert, J.C.; Richardson, J.L.; Davies, M.C.; Palin, K.J.; Hadgraft, J. The effect of solutes and polymers on the gelation properties of pluronic F-127 solutions for controlled drug delivery. J. Control. Release 1987, 5, 113–118. [Google Scholar] [CrossRef]
- Liu, L.; Tang, X.; Wang, Y.; Guo, S. Smart gelation of chitosan solution in the presence of NaHCO3 for injectable drug delivery system. Int. J. Pharm. 2011, 414, 6–15. [Google Scholar] [CrossRef]
- Krtalić, I.; Radošević, S.; Hafner, A.; Grassi, M.; Nenadić, M.; Cetina-Čižmek, B.; Filipović-Grčić, J.; Pepić, I.; Lovrić, J. D-optimal design in the development of rheologically improved in situ forming ophthalmic gel. J. Pharm. Sci. 2018, 107, 1562–1571. [Google Scholar] [CrossRef]
- Hirun, N.; Kraisit, P.; Tantishaiyakul, V. Thermosensitive Polymer Blend Composed of Poloxamer 407, Poloxamer 188 and Polycarbophil for the Use as Mucoadhesive In Situ Gel. Polymers 2022, 14, 1836. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, N.; Bhandari, R.; Kuhad, A.; Sinha, V.R. Polycaprolactone-based neurotherapeutic delivery of rasagiline targeting behavioral and biochemical deficits in Parkinson’s disease. Drug Deliv. Transl. Res. 2019, 9, 891–905. [Google Scholar] [CrossRef] [PubMed]
- Hammad, R.W.; Sanad, R.A.; Abdelmalak, N.S.; Torad, F.A.; Latif, R. New intranasal cross-linked mosapride xyloglucan pluronics micelles (MOS-XPMs) for reflux esophagitis disease: In-vitro optimization and improved therapeutic efficacy. J. Adv. Res. 2020, 23, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Hammad, R.W.; Sanad, R.A.; Abdelmalk, N.S.; Aziz, R.L.; Torad, F.A. Intranasal surface-modified mosapride citrate-loaded nanostructured lipid carriers (MOS-SMNLCs) for treatment of reflux diseases: In vitro optimization, pharmacodynamics, and pharmacokinetic studies. AAPS PharmSciTech 2018, 19, 3791–3808. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Z.; Zhang, Y.Q.; Wang, Z.Z.; Wu, K.; Lou, J.N.; Qi, X.R. Enhanced brain distribution and pharmacodynamics of rivastigmine by liposomes following intranasal administration. Int. J. Pharm. 2013, 452, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Albash, R.; Yousry, C.; Al-Mahallawi, A.M.; Alaa-Eldin, A.A. Utilization of PEGylated cerosomes for effective topical delivery of fenticonazole nitrate: In-vitro characterization, statistical optimization, and in-vivo assessment. Drug Deliv. 2021, 28, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Barakat, S.S.; Nasr, M.; Ahmed, R.F.; Badawy, S.S.; Mansour, S. Intranasally administered in situ gelling nanocomposite system of dimenhydrinate: Preparation, characterization and pharmacodynamic applicability in chemotherapy induced emesis model. Sci. Rep. 2017, 7, 9910. [Google Scholar] [CrossRef]
- Moore, J.; Flanner, H. Mathematical comparison of dissolution profiles. Pharm. Technol. 1996, 20, 64–74. [Google Scholar]
- Samaha, D.; Shehayeb, R.; Kyriacos, S. Modeling and comparison of dissolution profiles of diltiazem modified-release formulations. Dissolution Technol. 2009, 16, 41–46. [Google Scholar] [CrossRef]
- ElShagea, H.N.; ElKasabgy, N.A.; Fahmy, R.H.; Basalious, E.B. Freeze-dried self-nanoemulsifying self-nanosuspension (snesns): A new approach for the preparation of a highly drug-loaded dosage form. AAPS PharmSciTech 2019, 20, 258. [Google Scholar] [CrossRef] [PubMed]
- Ravi, P.R.; Aditya, N.; Patil, S.; Cherian, L. Nasal in-situ gels for delivery of rasagiline mesylate: Improvement in bioavailability and brain localization. Drug Deliv. 2015, 22, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Bancroft, J.D.; Gamble, M. Theory and Practice of Histological Techniques; Elsevier Health Sciences, Churchill Livingstone: London, UK, 2008. [Google Scholar]
- El-Mahrouk, G.; Aboul-Einien, M.H.; Elkasabgy, N.A. Formulation and evaluation of meloxicam orally dispersible capsules. Asian J. Pharm. Sci. 2009, 4, 8–22. [Google Scholar]
- Fatouh, A.M.; Elshafeey, A.H.; Abdelbary, A. Agomelatine-based in situ gels for brain targeting via the nasal route: Statistical optimization, in vitro, and in vivo evaluation. Drug Deliv. 2017, 24, 1077–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Chen, X.; Duan, X.; Deng, P.; Wang, H.; Zhong, D. Validated LC–MS/MS method for quantitative determination of rasagiline in human plasma and its application to a pharmacokinetic study. J. Chromatogr. B 2008, 873, 203–208. [Google Scholar] [CrossRef]
- Vyas, T.K.; Babbar, A.K.; Sharma, R.K.; Misra, A. Intranasal mucoadhesive microemulsions of zolmitriptan: Preliminary studies on brain-targeting. J. Drug Target. 2005, 13, 317–324. [Google Scholar] [CrossRef]
- Kassem, M.A.; Aboul-Einien, M.H.; El Taweel, M.M. Dry gel containing optimized felodipine-loaded transferosomes: A promising transdermal delivery system to enhance drug bioavailability. AAPS PharmSciTech 2018, 19, 2155–2173. [Google Scholar] [CrossRef]
- Mu, X.; Zhong, Z. Preparation and properties of poly (vinyl alcohol)-stabilized liposomes. Int. J. Pharm. 2006, 318, 55–61. [Google Scholar] [CrossRef]
- Al-Mahallawi, A.M.; Khowessah, O.M.; Shoukri, R.A. Nano-transfersomal ciprofloxacin loaded vesicles for non-invasive trans-tympanic ototopical delivery: In-vitro optimization, ex-vivo permeation studies, and in-vivo assessment. Int. J. Pharm. 2014, 472, 304–314. [Google Scholar] [CrossRef]
- Duangjit, S.; Pamornpathomkul, B.; Opanasopit, P.; Rojanarata, T.; Obata, Y.; Takayama, K.; Ngawhirunpat, T. Role of the charge, carbon chain length, and content of surfactant on the skin penetration of meloxicam-loaded liposomes. Int. J. Nanomed. 2014, 9, 2005. [Google Scholar] [CrossRef]
- Adel, I.M.; ElMeligy, M.F.; Abdelrahim, M.E.; Maged, A.; Abdelkhalek, A.A.; Abdelmoteleb, A.M.; Elkasabgy, N.A. Design and characterization of spray-dried proliposomes for the pulmonary delivery of curcumin. Int. J. Nanomed. 2021, 16, 2667–2687. [Google Scholar] [CrossRef]
- Kreuter, J. Colloidal Drug Delivery Systems; CRC Press: Boca Raton, FL, USA, 1994; Volume 66. [Google Scholar]
- Salama, A.H.; Abdelkhalek, A.A.; Elkasabgy, N.A. Etoricoxib-loaded bio-adhesive hybridized polylactic acid-based nanoparticles as an intra-articular injection for the treatment of osteoarthritis. Int. J. Pharm. 2020, 578, 119081. [Google Scholar] [CrossRef]
- Joseph, E.; Singhvi, G. Multifunctional nanocrystals for cancer therapy: A potential nanocarrier. In Nanomaterials for Drug Delivery and Therapy; Elsevier: Amsterdam, The Netherlands, 2019; pp. 91–116. [Google Scholar]
- Müller, R.H.; Jacobs, C.; Kayser, O. Nanosuspensions as particulate drug formulations in therapy: Rationale for development and what we can expect for the future. Adv. Drug Deliv. Rev. 2001, 47, 3–19. [Google Scholar] [CrossRef]
- Kosmulski, M.; Mączka, E. Zeta potential in dispersions of titania nanoparticles in moderately polar solvents stabilized with anionic surfactants. J. Mol. Liq. 2022, 355, 118972. [Google Scholar] [CrossRef]
- Asasutjarit, R.; Lorenzen, S.I.; Sirivichayakul, S.; Ruxrungtham, K.; Ruktanonchai, U.; Ritthidej, G.C. Effect of solid lipid nanoparticles formulation compositions on their size, zeta potential and potential for in vitro pHIS-HIV-hugag transfection. Pharm. Res. 2007, 24, 1098–1107. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W.; Shamoto-Nagai, M. Neuroprotective Function of Rasagiline and Selegiline, Inhibitors of Type B Monoamine Oxidase, and Role of Monoamine Oxidases in Synucleinopathies. Int. J. Mol. Sci. 2022, 23, 11059. [Google Scholar] [CrossRef]
- Sodium Deoxycholate. Available online: https://www.sigmaaldrich.com/EG/en/search/sodium-deoxycholate?focus=products&gclid=CjwKCAiAkrWdBhBkEiwAZ9cdcMalphdZ3qU7FBfsxyfSrsQs9w2u0ToGV1kDGBhBIVXLPXcj7abd1RoCMIYQAvD_BwE&page=1&perpage=30&sort=relevance&term=sodium%20deoxycholate&type=product_name (accessed on 29 December 2022).
- Sodium Cholate. Available online: https://www.sigmaaldrich.com/EG/en/search/sodium-cholate?focus=products&page=1&perpage=30&sort=relevance&term=sodium%20cholate&type=product_name (accessed on 29 December 2022).
- Shatalebi, M.; Mostafavi, S.; Moghaddas, A. Niosome as a drug carrier for topical delivery of N-acetyl glucosamine. Res. Pharm. Sci. 2010, 5, 107. [Google Scholar]
- Torres-Flores, G.; Gonzalez-Horta, A.; Vega-Cantu, Y.I.; Rodriguez, C.; Rodriguez-Garcia, A. Preparation and characterization of liposomal everolimus by thin-film hydration technique. Adv. Polym. Technol. 2020, 2020, 5462949. [Google Scholar] [CrossRef]
- Abdelbary, G. Ocular ciprofloxacin hydrochloride mucoadhesive chitosan-coated liposomes. Pharm. Dev. Technol. 2011, 16, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Basalious, E.B.; Shawky, N.; Badr-Eldin, S.M. SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I: Development and optimization. Int. J. Pharm. 2010, 391, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Dong, W.; Ye, J.; Hao, H.; Zhou, J.; Wang, R.; Liu, Y. A novel matrix dispersion based on phospholipid complex for improving oral bioavailability of baicalein: Preparation, in vitro and in vivo evaluations. Drug Deliv. 2017, 24, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Lasch, P.; Pacifico, A.; Diem, M. Spatially resolved IR microspectroscopy of single cells. Biopolym. Orig. Res. Biomol. 2002, 67, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Depciuch, J.; Sowa-Kućma, M.; Nowak, G.; Dudek, D.; Siwek, M.; Styczeń, K.; Parlińska-Wojtan, M. Phospholipid-protein balance in affective disorders: Analysis of human blood serum using Raman and FTIR spectroscopy. A pilot study. J. Pharm. Biomed. Anal. 2016, 131, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, A.E.; Salah, S.; Amer, M.S.; Elkasabgy, N.A. 3D nanocomposite alginate hydrogel loaded with pitavastatin nanovesicles as a functional wound dressing with controlled drug release; preparation, in-vitro and in-vivo evaluation. J. Drug Deliv. Sci. Technol. 2022, 71, 103292. [Google Scholar] [CrossRef]
- Kaur, M.; Sharma, S.; Sinha, V. Polymer based microspheres of aceclofenac as sustained release parenterals for prolonged anti-inflammatory effect. Mater. Sci. Eng. C 2017, 72, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Adel, S.; ElKasabgy, N.A. Design of innovated lipid-based floating beads loaded with an antispasmodic drug: In-vitro and in-vivo evaluation. J. Liposome Res. 2014, 24, 136–149. [Google Scholar] [CrossRef]
- Karavasili, C.; Fatouros, D.G. Smart materials: In situ gel-forming systems for nasal delivery. Drug Discov. Today 2016, 21, 157–166. [Google Scholar] [CrossRef]
- Yuan, Y.; Cui, Y.; Zhang, L.; Zhu, H.P.; Guo, Y.S.; Zhong, B.; Hu, X.; Zhang, L.; Wang, X.H.; Chen, L. Thermosensitive and mucoadhesive in situ gel based on poloxamer as new carrier for rectal administration of nimesulide. Int. J. Pharm. 2012, 430, 114–119. [Google Scholar] [CrossRef]
- Hasan, M.; Elkhoury, K.; Kahn, C.J.; Arab-Tehrany, E.; Linder, M. Preparation, characterization, and release kinetics of chitosan-coated nanoliposomes encapsulating curcumin in simulated environments. Molecules 2019, 24, 2023. [Google Scholar] [CrossRef]
- Abo Elela, M.M.; ElKasabgy, N.A.; Basalious, E.B. Bio-shielding in situ forming gels (BSIFG) loaded with lipospheres for depot injection of quetiapine fumarate: In vitro and in vivo evaluation. AAPS PharmSciTech 2017, 18, 2999–3010. [Google Scholar] [CrossRef]
- Din, F.U.; Mustapha, O.; Kim, D.W.; Rashid, R.; Park, J.H.; Choi, J.Y.; Ku, S.K.; Yong, C.S.; Kim, J.O.; Choi, H.G. Novel dual-reverse thermosensitive solid lipid nanoparticle-loaded hydrogel for rectal administration of flurbiprofen with improved bioavailability and reduced initial burst effect. Eur. J. Pharm. Biopharm. 2015, 94, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Hawthorne, G.H.; Bernuci, M.P.; Bortolanza, M.; Tumas, V.; Issy, A.C.; Del-Bel, E. Nanomedicine to overcome current Parkinson’s treatment liabilities: A systematic review. Neurotox. Res. 2016, 30, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Salem, H.F.; Kharshoum, R.M.; Abou-Taleb, H.A.; Naguib, D.M. Nanosized transferosome-based intranasal in situ gel for brain targeting of resveratrol: Formulation, optimization, in vitro evaluation, and in vivo pharmacokinetic study. AAPS PharmSciTech 2019, 20, 181. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Saraf, S.; Saraf, S.; Dubey, S.K.; Puri, A.; Gupta, U.; Kesharwani, P.; Ravichandiran, V.; Kumar, P.; Naidu, V.G. Stimuli-responsive In situ gelling system for nose-to-brain drug delivery. J. Control. Release 2020, 327, 235–265. [Google Scholar] [CrossRef] [PubMed]
- Bulloch, M.N.; Hutchison, A.M. Fentanyl pectin nasal spray: A novel intranasal delivery method for the treatment of breakthrough cancer pain. Expert Rev. Clin. Pharmacol. 2013, 6, 9–22. [Google Scholar] [CrossRef] [PubMed]
- El Taweel, M.M.; Aboul-Einien, M.H.; Kassem, M.A.; Elkasabgy, N.A. Intranasal zolmitriptan-loaded bilosomes with extended nasal mucociliary transit time for direct nose to brain delivery. Pharmaceutics 2021, 13, 1828. [Google Scholar] [CrossRef] [PubMed]
- Muntimadugu, E.; Dhommati, R.; Jain, A.; Challa, V.G.; Shaheen, M.; Khan, W. Intranasal delivery of nanoparticle encapsulated tarenflurbil: A potential brain targeting strategy for Alzheimer’s disease. Eur. J. Pharm. Sci. 2016, 92, 224–234. [Google Scholar] [CrossRef]
- Seju, U.; Kumar, A.; Sawant, K. Development and evaluation of olanzapine-loaded PLGA nanoparticles for nose-to-brain delivery: In vitro and in vivo studies. Acta Biomater. 2011, 7, 4169–4176. [Google Scholar] [CrossRef]
- Chatterjee, B.; Amalina, N.; Sengupta, P.; Mandal, U.K. Mucoadhesive polymers and their mode of action: A recent update. J. Appl. Pharm. Sci. 2017, 7, 195–203. [Google Scholar]
- Al Khateb, K.; Ozhmukhametova, E.K.; Mussin, M.N.; Seilkhanov, S.K.; Rakhypbekov, T.K.; Lau, W.M.; Khutoryanskiy, V.V. In situ gelling systems based on Pluronic F127/Pluronic F68 formulations for ocular drug delivery. Int. J. Pharm. 2016, 502, 70–79. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Levels |
|---|---|
| X1:Type of EA | Sodium cholate, sodium deoxycholate, Pluronic® F-68, Pluronic® L-35, Pluronic® L-31, and Span® 60 |
| X2:Cholesterol | Absent Present |
| Dependent variables | Desirability Constraint |
| Y1:VS | Minimize |
| Y2:ZP | Maximize |
| Y3:%EE | Maximize |
| Formulation Code | Factor 1 X1:EA Type | Factor 2 X2:Cholesterol | Response 1 Y1:VS (nm) | Response 2 Y2:ZP (mV) | Response 3 Y3:EE% |
|---|---|---|---|---|---|
| F1 | Span® 60 | Present | 491.40 ± 48.79 | −32.60 ± 4.94 | 43.25 ± 5.02 |
| F2 | Pluronic® F-68 | Present | 347.26 ± 56.66 | −29.65 ± 0.49 | 52.33 ± 3.53 |
| F3 | Sodium cholate | Present | 283.30 ± 8.62 | −36.95 ± 1.06 | 54.80 ± 5.19 |
| F4 | Pluronic® L-35 | Present | 391.95 ± 56.17 | −39.60 ± 6.36 | 51.73 ± 5.46 |
| F5 | Pluronic® L-31 | Present | 387.95 ± 52.82 | −30.50 ± 1.27 | 48.51 ± 1.85 |
| F6 | Sodium deoxycholate | Present | 248.56 ± 32.47 | −35.45 ± 3.18 | 54.98 ± 6.52 |
| F7 | Span® 60 | Absent | 448.70 ± 14.84 | −24.90 ± 1.13 | 50.68 ± 1.24 |
| F8 | Pluronic® F-68 | Absent | 227.51 ± 13.69 | −26.25 ± 4.31 | 61.37 ± 0.53 |
| F9 | Sodium cholate | Absent | 208.42 ± 11.95 | −33.20 ± 0.28 | 69.40 ± 0.98 |
| F10 | Pluronic® L-35 | Absent | 276.00 ± 27.05 | −29.20 ± 2.26 | 57.47 ± 2.65 |
| F11 | Pluronic® L-31 | Absent | 352.90 ± 15.41 | −30.00 ± 0.42 | 55.45 ± 5.65 |
| F12 | Sodium deoxycholate | Absent | 198.63 ± 34.98 | −33.45 ± 4.73 | 95.73 ± 0.09 |
| Gel Formulation Code | Composition | |||
|---|---|---|---|---|
| Pectin (% w/v) | Pluronic® F-127 (% w/v) | Pluronic® F-68 (% w/v) | Sol to Gel Temperature (°C) | |
| G1 | 0.50 | 15 | 0 | 32.4 ± 0.56 |
| G2 | 0.50 | 15 | 5 | 36.2 ± 0.28 |
| G3 | 0.50 | 15 | 10 | 40.1 ± 1.69 |
| In Plasma | |||
|---|---|---|---|
| Parameter | IV Aqueous Solution | IN In Situ Gel | p-Value |
| Cmax (ng/mL) | 103.30 ± 6.95 | 14.01 ± 5.30 | 0.0012 |
| Tmax (h) | 0.25 | 0.25 | - |
| AUC0-12h (ng.h/mL) | 63.40 ± 3.00 | 27.31 ± 5.60 | 0.001 |
| Absolute bioavailability (%) | -- | 43.07% | |
| In Brain | |||
| Parameter | IV Aqueous Solution | IN In Situ Gel | p-Value |
| Cmax (ng/mL) | 241.93 ± 11.41 | 381.58 ± 10.15 | 0.0014 |
| Tmax (h) | 0.25 | 0.25 | - |
| AUC0-12h (ng.h/mL) | 447.08 ± 31.49 | 586.47 ± 31.50 | 0.002 |
| Brain bioavailability (%) | -- | 131.17% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
ElShagea, H.N.; Makar, R.R.; Salama, A.H.; Elkasabgy, N.A.; Basalious, E.B. Investigating the Targeting Power to Brain Tissues of Intranasal Rasagiline Mesylate-Loaded Transferosomal In Situ Gel for Efficient Treatment of Parkinson’s Disease. Pharmaceutics 2023, 15, 533. https://doi.org/10.3390/pharmaceutics15020533
ElShagea HN, Makar RR, Salama AH, Elkasabgy NA, Basalious EB. Investigating the Targeting Power to Brain Tissues of Intranasal Rasagiline Mesylate-Loaded Transferosomal In Situ Gel for Efficient Treatment of Parkinson’s Disease. Pharmaceutics. 2023; 15(2):533. https://doi.org/10.3390/pharmaceutics15020533
Chicago/Turabian StyleElShagea, Hala N., Rana R. Makar, Alaa H. Salama, Nermeen A. Elkasabgy, and Emad B. Basalious. 2023. "Investigating the Targeting Power to Brain Tissues of Intranasal Rasagiline Mesylate-Loaded Transferosomal In Situ Gel for Efficient Treatment of Parkinson’s Disease" Pharmaceutics 15, no. 2: 533. https://doi.org/10.3390/pharmaceutics15020533
APA StyleElShagea, H. N., Makar, R. R., Salama, A. H., Elkasabgy, N. A., & Basalious, E. B. (2023). Investigating the Targeting Power to Brain Tissues of Intranasal Rasagiline Mesylate-Loaded Transferosomal In Situ Gel for Efficient Treatment of Parkinson’s Disease. Pharmaceutics, 15(2), 533. https://doi.org/10.3390/pharmaceutics15020533