
New Lidocaine-Based Pharmaceutical Cocrystals: Preparation, Characterization, and Influence of the Racemic vs. Enantiopure Coformer on the Physico-Chemical Properties

 ,
,  ,
, 
 , , ,
, , ,  ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cocrystal Preparation and Storage
2.3. Single Cocrystal Engineering
2.4. Physical Mixtures Preparation for the Establishment of the Stable Lidocaine/dl-Menthol Phase Diagram
2.5. Buffer Preparation
2.6. X-ray Diffraction
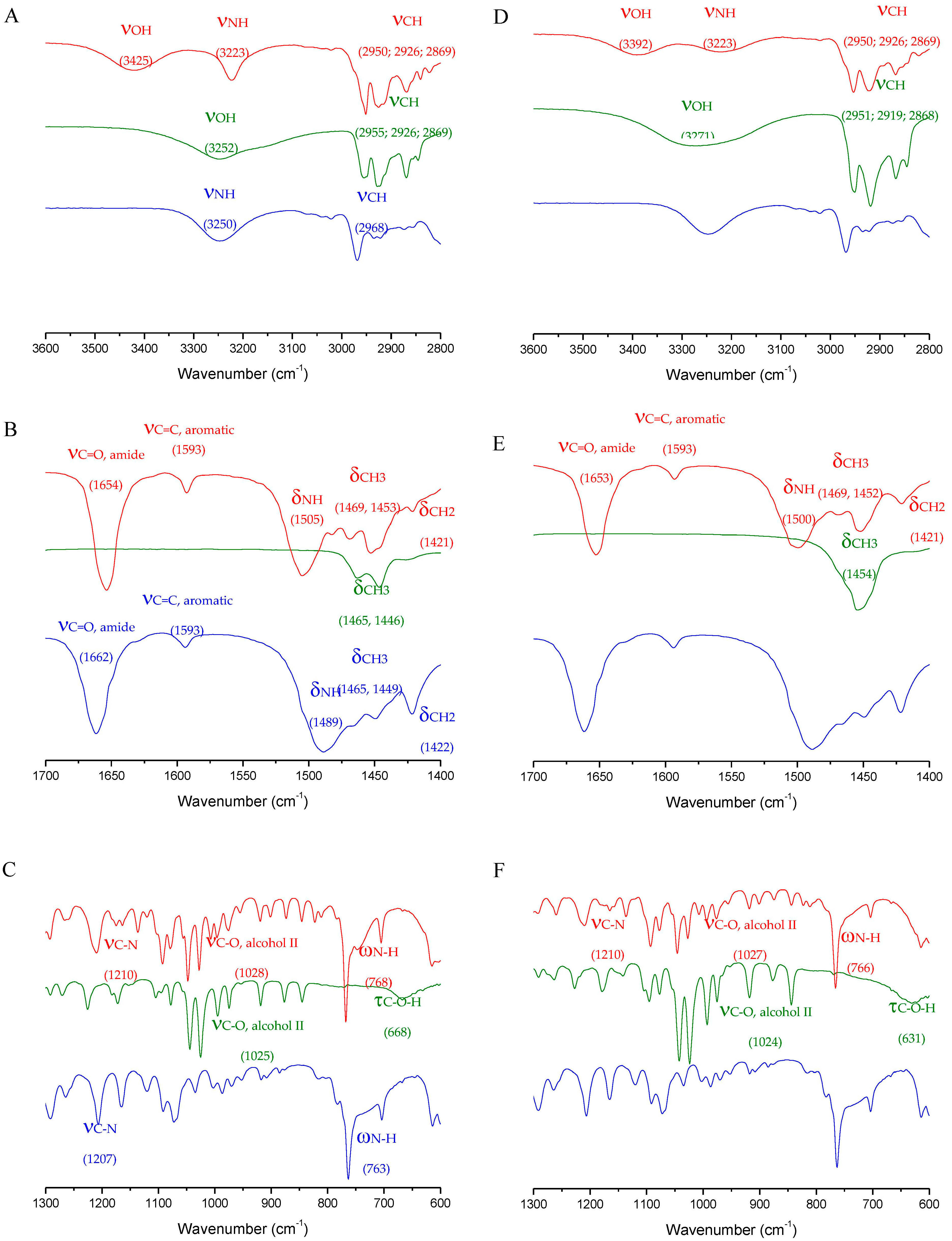
2.7. Spectroscopy Experiments
2.8. Thermal Analysis Experiments
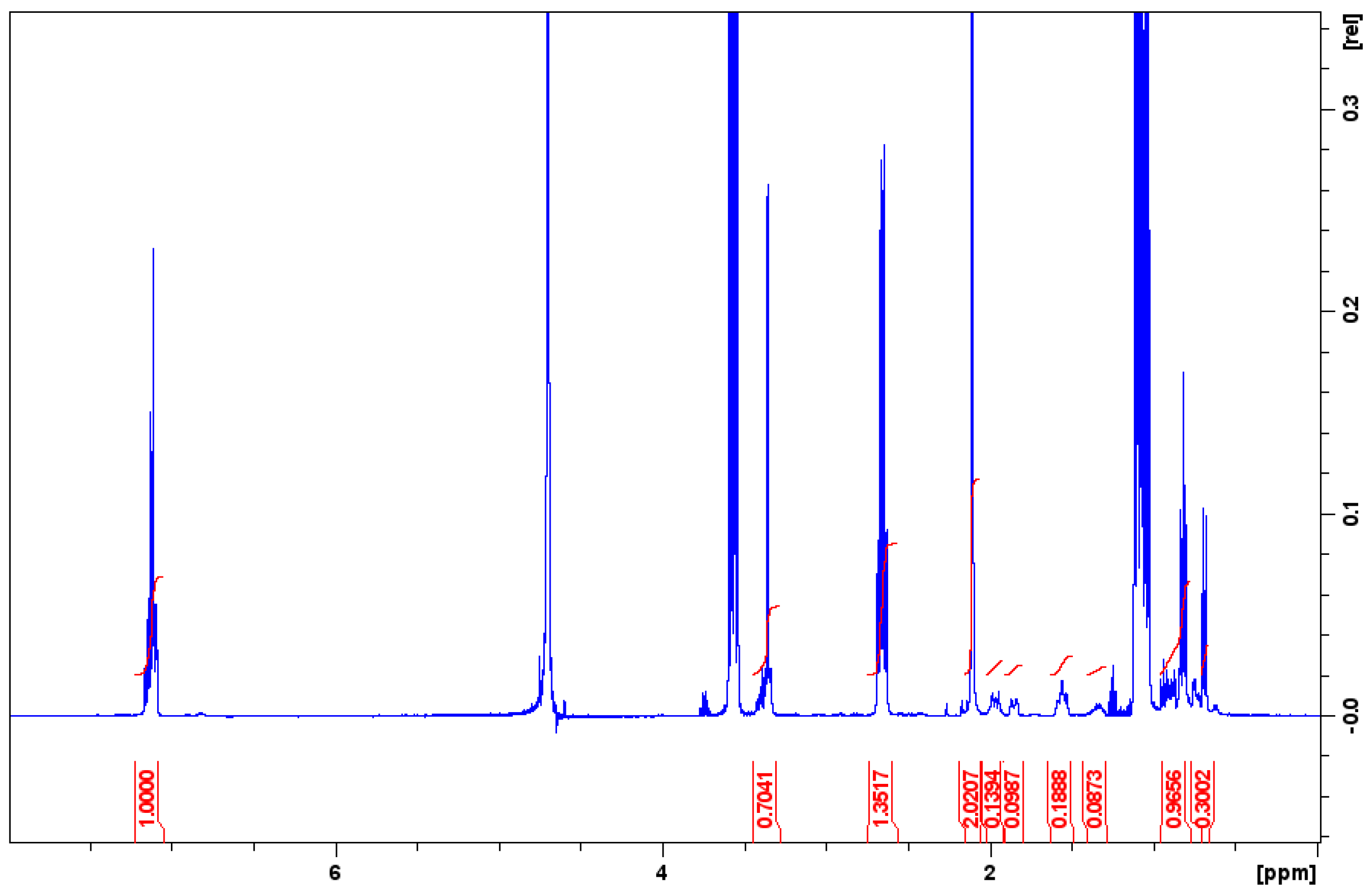
2.9. Nuclear Magnetic Resonance Experiments
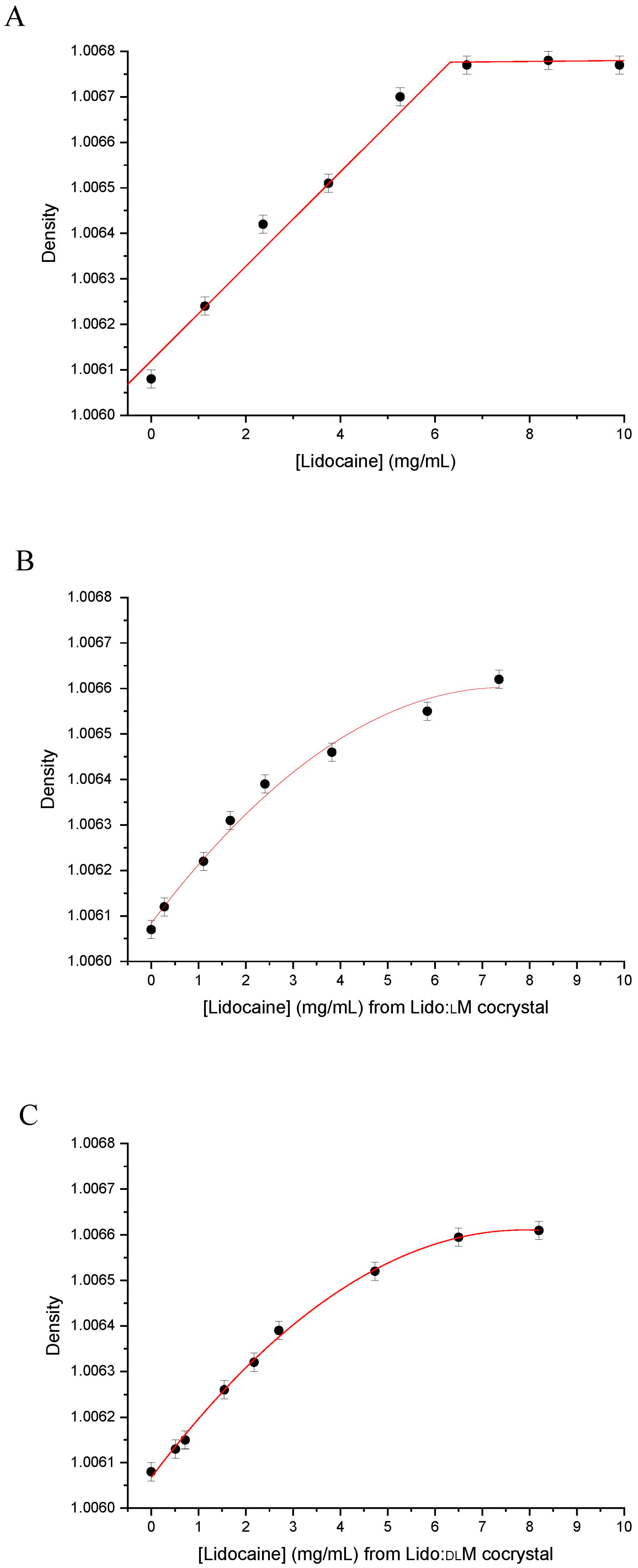
2.10. Density Measurement
2.11. Kinetics of Dissolution
3. Results and Discussion
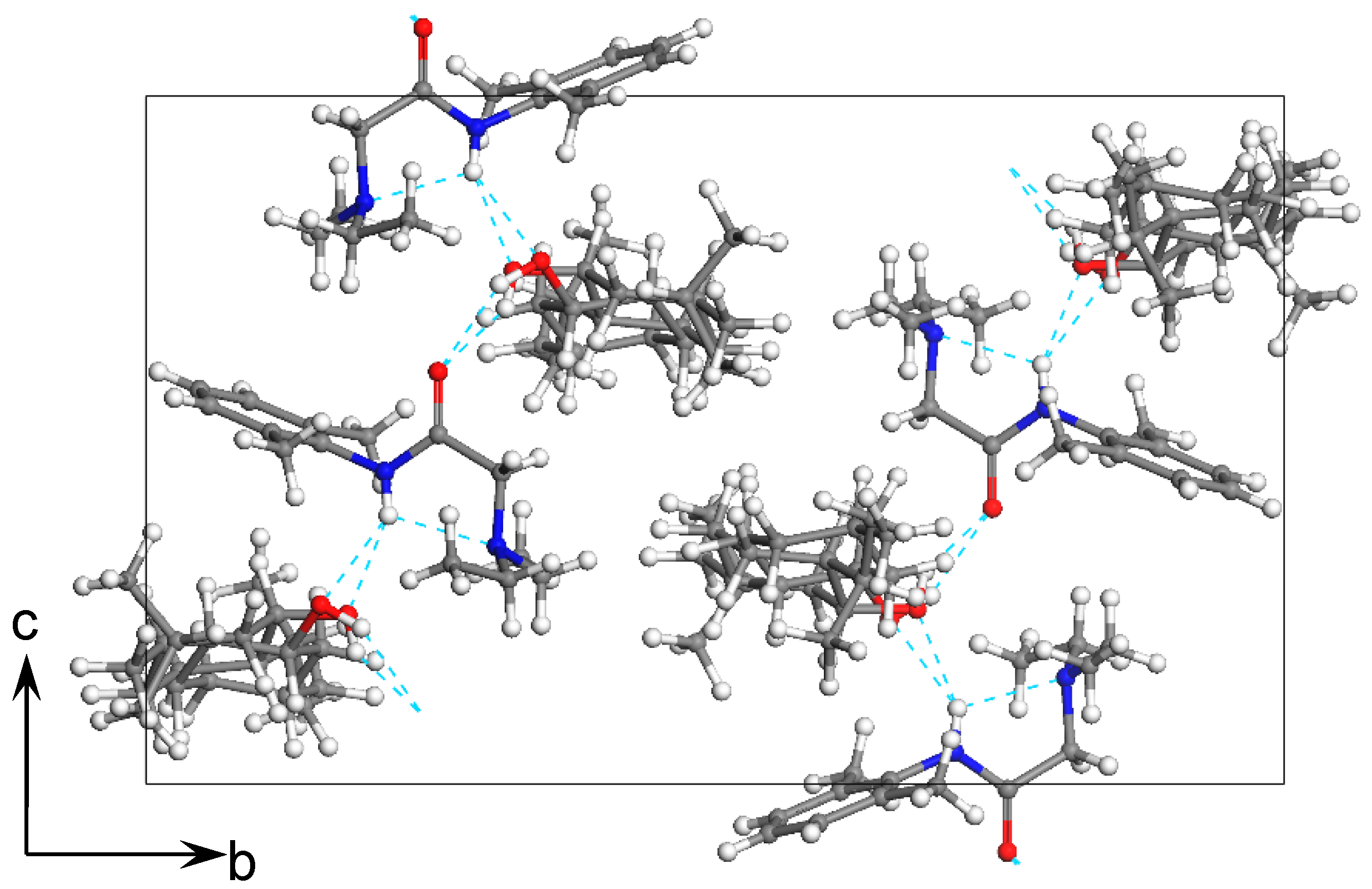
3.1. Design and Structure of Two New Cocrystals
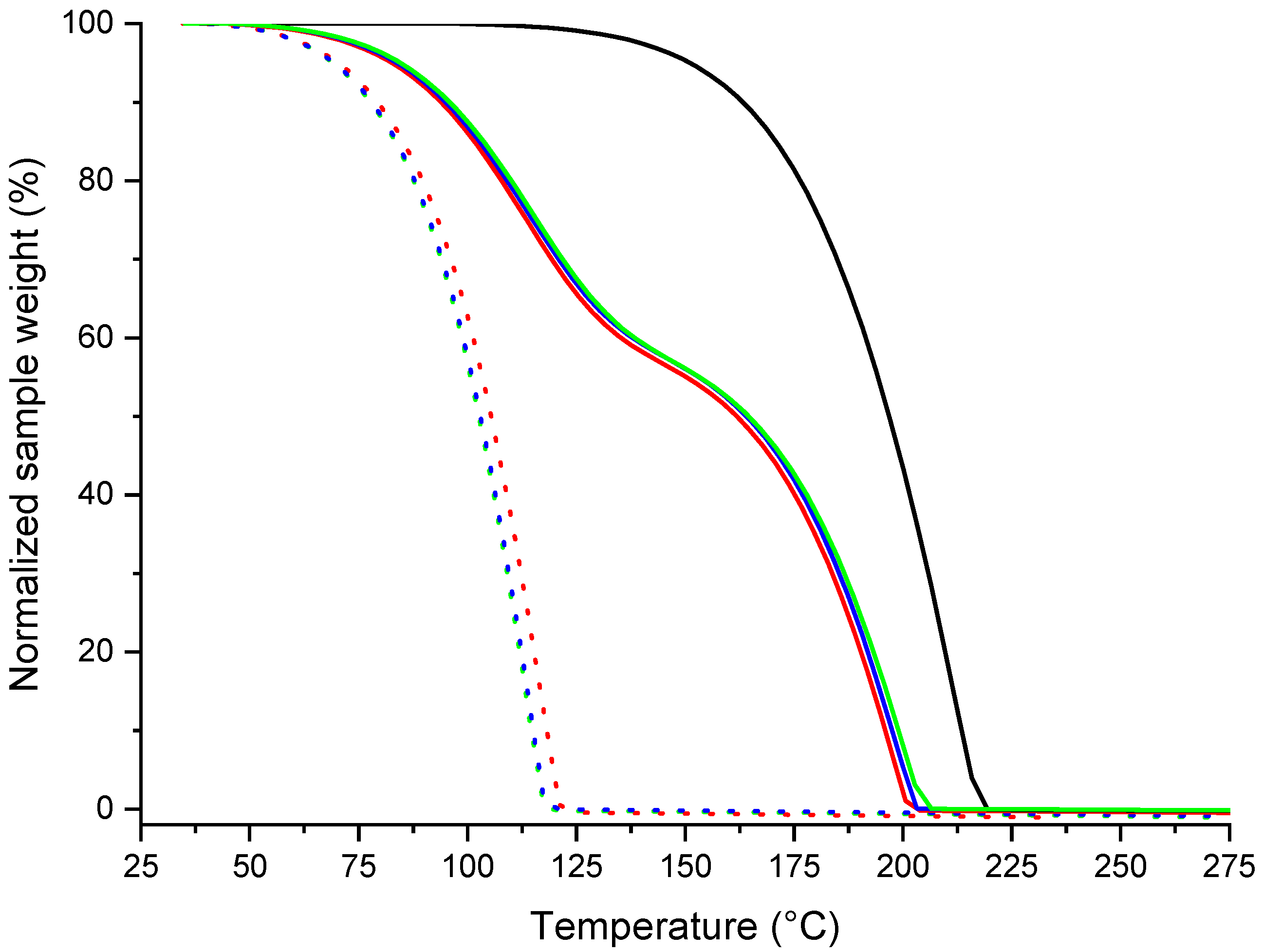
3.2. Physico-Chemical Properties of the l-, d- and dl-Menthol-Based Cocrystals
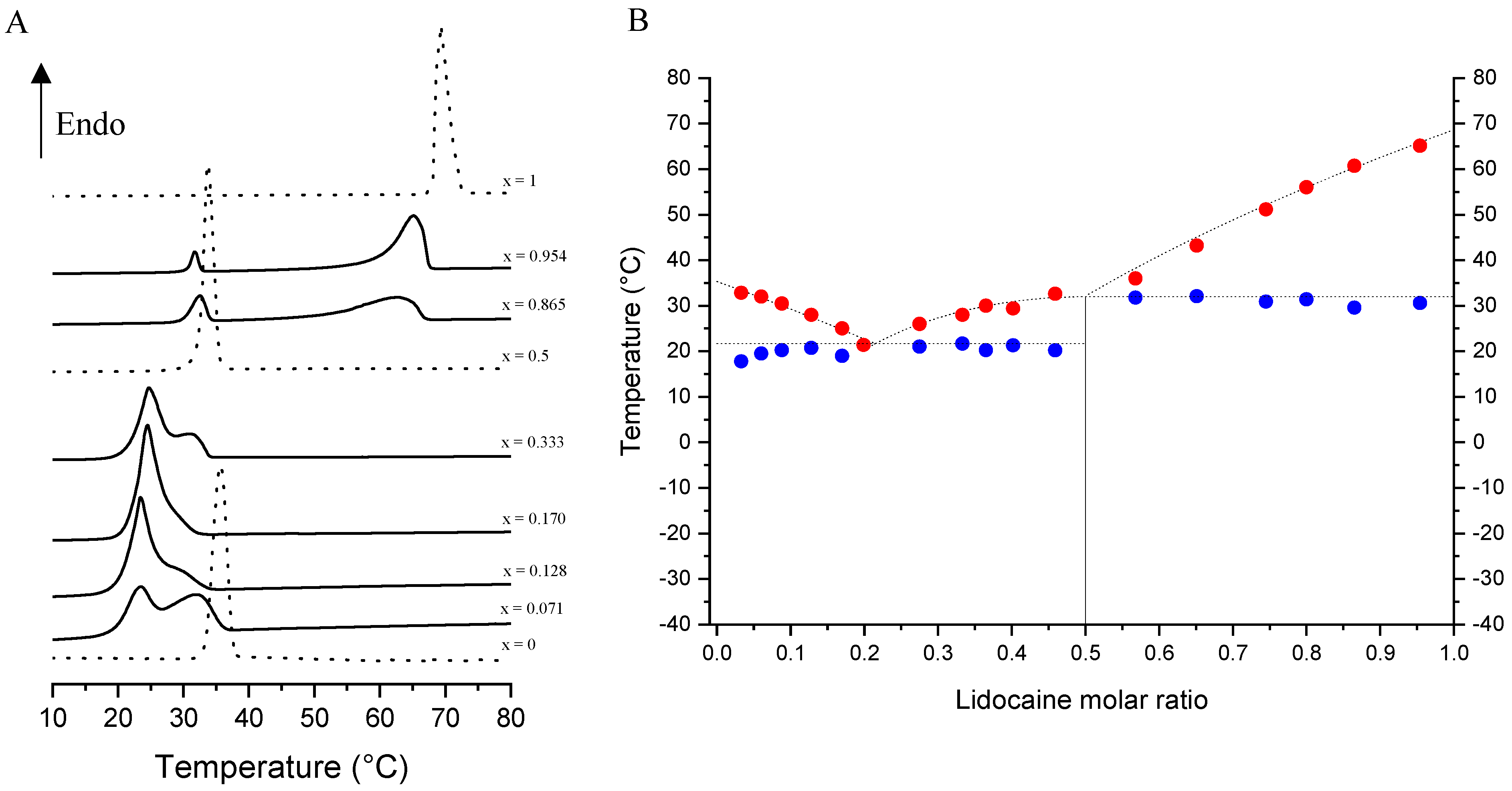
3.3. Screening of the Solid–Liquid Equilibria for the dl-Menthol-Based Cocrystal
3.4. Screening of the Solid–Liquid and Gas–Liquid Equilibria for the Racemic and Enantiopure Cocrystals
3.5. Dissolution Behavior Assessment and Comparison of the Racemic and Enantiopure Cocrystals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and In Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, P.; Seguin, J.; Ly, N.K.; Henríquez, L.C.; Plansart, E.; Hammad, K.; Gahoual, R.; Dhôtel, H.; Izabelle, C.; Saubamea, B.; et al. Designing fisetin nanocrystals for enhanced in cellulo anti-angiogenic and anticancer efficacy. Int. J. Pharm. X 2022, 4, 100138. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A. Cocrystal solubility product analysis—Dual concentration-pH mass action model not dependent on explicit solubility equations. Eur. J. Pharm. Sci. 2017, 110, 2–18. [Google Scholar] [CrossRef]
- Lu, J.; Rohani, S. Preparation and Characterization of Theophylline−Nicotinamide Cocrystal. Org. Process. Res. Dev. 2009, 13, 1269–1275. [Google Scholar] [CrossRef]
- Schultheiss, N.; Newman, A. Pharmaceutical Cocrystals and Their Physicochemical Properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef] [Green Version]
- Bandaru, R.K.; Rout, S.R.; Kenguva, G.; Gorain, B.; Alhakamy, N.A.; Kesharwani, P.; Dandela, R. Recent Advances in Pharmaceutical Cocrystals: From Bench to Market. Front. Pharmacol. 2021, 12, 780582. [Google Scholar] [CrossRef]
- Kavanagh, O.N.; Croker, D.M.; Walker, G.M.; Zaworotko, M.J. Pharmaceutical cocrystals: From serendipity to design to application. Drug Discov. Today 2019, 24, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.J.; Steed, J.W. Pharmaceutical cocrystals, salts and multicomponent systems; intermolecular interactions and property based design. Adv. Drug Deliv. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef] [Green Version]
- Healy, A.M.; Worku, Z.A.; Kumar, D.; Madi, A.M. Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals. Adv. Drug Deliv. Rev. 2017, 117, 25–46. [Google Scholar] [CrossRef] [Green Version]
- Kuminek, G.; Cao, F.; de Oliveira da Rocha, A.B.; Cardoso, S.G.; Rodríguez-Hornedo, N. Cocrystals to facilitate delivery of poorly soluble compounds beyond-rule-of-5. Adv. Drug Deliv. Rev. 2016, 101, 143–166. [Google Scholar] [CrossRef] [Green Version]
- Qiao, N.; Li, M.; Schlindwein, W.; Malek, N.; Davies, A.; Trappitt, G. Pharmaceutical cocrystals: An overview. Int. J. Pharm. 2011, 419, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Regulatory Classification of Pharmaceutical Co-Crystals Guidance for Industry, FDA. 2018. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM281764.pdf (accessed on 22 March 2023).
- Dalpiaz, A.; Ferretti, V.; Bertolasi, V.; Pavan, B.; Monari, A.; Pastore, M. From Physical Mixtures to Co-Crystals: How the Coformers Can Modify Solubility and Biological Activity of Carbamazepine. Mol. Pharm. 2017, 15, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Gagnière, E.; Mangin, D.; Puel, F.; Valour, J.-P.; Klein, J.-P.; Monnier, O. Cocrystal formation in solution: Inducing phase transition by manipulating the amount of cocrystallizing agent. J. Cryst. Growth 2011, 316, 118–125. [Google Scholar] [CrossRef]
- Rager, T.; Hilfiker, R. Cocrystal Formation from Solvent Mixtures. Cryst. Growth Des. 2010, 10, 3237–3241. [Google Scholar] [CrossRef]
- Lee, H.L.; Lee, T. Direct co-crystal assembly from synthesis to co-crystallization. CrystEngComm 2015, 17, 9002–9006. [Google Scholar] [CrossRef]
- Ancheria, R.K.; Jain, S.; Kumar, D.; Soni, S.L.; Sharma, M. An Overview of Pharmaceutical Co-Crystal. Asian J. Pharm. Res. Dev. 2019, 7, 39–46. [Google Scholar] [CrossRef]
- Sathisaran, I.; Dalvi, S.V. Engineering Cocrystals of Poorly Water-Soluble Drugs to Enhance Dissolution in Aqueous Medium. Pharmaceutics 2018, 10, 108. [Google Scholar] [CrossRef] [Green Version]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef] [Green Version]
- Delori, A.; Friščić, T.; Jones, W. The role of mechanochemistry and supramolecular design in the development of pharmaceutical materials. CrystEngComm 2012, 14, 2350–2362. [Google Scholar] [CrossRef]
- George, F.; Tumanov, N.; Norberg, B.; Robeyns, K.; Filinchuk, Y.; Wouters, J.; Leyssens, T. Does Chirality Influence the Tendency toward Cocrystal Formation? Cryst. Growth Des. 2014, 14, 2880–2892. [Google Scholar] [CrossRef]
- Trask, A.V.; Motherwell, W.D.S.; Jones, W. Solvent-drop grinding: Green polymorph control of cocrystallisation. Chem. Commun. 2004, 7, 890–891. [Google Scholar] [CrossRef] [PubMed]
- Fong, S.Y.K.; Ibisogly, A.; Bauer-Brandl, A. Solubility enhancement of BCS Class II drug by solid phospholipid dispersions: Spray drying versus freeze-drying. Int. J. Pharm. 2015, 496, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.; Chen, B.; Hou, M.; Liu, T. The Controllable Preparation, Properties and Structural Characteristics of Xylitol/Menthol Co-crystals. Int. J. Food Eng. 2017, 13, 20170060. [Google Scholar] [CrossRef]
- Kiyonga, E.M.; Kekani, L.N.; Chidziwa, T.V.; Kahwenga, K.D.; Bronkhorst, E.; Milne, M.; Poka, M.S.; Mokhele, S.; Demana, P.H.; Witika, B.A. Nano- and Crystal Engineering Approaches in the Development of Therapeutic Agents for Neoplastic Diseases. Crystals 2022, 12, 926. [Google Scholar] [CrossRef]
- Avdeef, A. Cocrystal Solubility Product Prediction Using an in combo Model and Simulations to Improve Design of Experiments. Pharm. Res. 2018, 35, 40. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhang, Q.; Zhu, B.; Zhang, Z.; Bao, J.; Ding, Q.; Ren, G.; Mei, X. Pharmaceutical Cocrystals of Nicorandil with Enhanced Chemical Stability and Sustained Release. Cryst. Growth Des. 2020, 20, 6995–7005. [Google Scholar] [CrossRef]
- Wöhler, F. Untersuchungen über das Chinon. Annalen Chem. Pharm. 1844, 51, 145–163. [Google Scholar] [CrossRef] [Green Version]
- Couillaud, B.M.; Espeau, P.; Mignet, N.; Corvis, Y. State of the Art of Pharmaceutical Solid Forms: From Crystal Property Issues to Nanocrystals Formulation. Chemmedchem 2018, 14, 8–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robieux, I.; Eliopoulos, C.; Hwang, P.; Greenberg, M.; Blanchette, V.; Olivieri, N.; Klein, N.; Koren, G. Pain Perception and Effectiveness of the Eutectic Mixture of Local Anesthetics in Children Undergoing Venipuncture. Pediatr. Res. 1992, 32, 520–523. [Google Scholar] [CrossRef] [Green Version]
- Buckley, M.M.; Benfield, P. Eutectic lidocaine/prilocaine cream. A review of the topical anaesthetic/analgesic efficacy of a eutectic mixture of local anaesthetics (EMLA). Drugs 1993, 46, 126–151. [Google Scholar] [CrossRef]
- Leitch, D.G.; Wicks, J.; El Beshir, O.A.; Ali, S.A.; Chaudhury, B.K. Topical anesthesia with 50 mg of lidocaine spray facilitates upper gastrointestinal endoscopy. Gastrointest. Endosc. 1993, 39, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.-C.; Yew, M.; Lin, Y.-T.; Chen, J.-Y.; Wang, L.-K.; Chang, Y.-J.; Chang, Y.-P.; Lan, K.-M.; Ho, C.-N.; Sun, C.-K. Impact of intravenous and topical lidocaine on clinical outcomes in patients receiving propofol for gastrointestinal endoscopic procedures: A meta-analysis of randomised controlled trials. Br. J. Anaesth. 2021, 128, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, D.P.; McMeekin, T.O. A case of lidocaine absorption from topical administration of 40% lidocaine cream. J. Am. Acad. Dermatol. 1999, 41, 280–281. [Google Scholar] [CrossRef] [PubMed]
- Comba, C.; Demirayak, G.; Erdogan, S.V.; Karaca, I.; Demir, O.; Guler, O.; Ozdemir, I.A. Comparison of pain and proper sample status according to usage of tenaculum and analgesia: A randomized clinical trial. Obstet. Gynecol. Sci. 2020, 63, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Seangrung, R.; Pasutharnchat, K.; Injampa, S.; Kumdang, S.; Komonhirun, R. Comparison of the hemodynamic response of dexmedetomidine versus additional intravenous lidocaine with propofol during tracheal intubation: A randomized controlled study. BMC Anesthesiol. 2021, 21, 265. [Google Scholar] [CrossRef] [PubMed]
- Corvis, Y.; Négrier, P.; Massip, S.; Leger, J.-M.; Espeau, P. Insights into the crystal structure, polymorphism and thermal behavior of menthol optical isomers and racemates. CrystEngComm 2012, 14, 7055–7064. [Google Scholar] [CrossRef] [Green Version]
- Corvis, Y.; Wurm, A.; Schick, C.; Espeau, P. New menthol polymorphs identified by flash scanning calorimetry. CrystEngComm 2015, 17, 5357–5359. [Google Scholar] [CrossRef]
- Kamal, M.A.H.M.; Iimura, N.; Nabekura, T.; Kitagawa, S. Enhanced Skin Permeation of Salicylate by Ion-Pair Formation in Non-aqueous Vehicle and Further Enhancement by Ethanol and l-Menthol. Chem. Pharm. Bull. 2006, 54, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Tokuoka, Y.; Suzuki, M.; Ohsawa, Y.; Ochiai, A.; Ishizuka, M.; Kawashima, N. Enhancement in Skin Permeation of 5-Aminolevulinic Acid Using l-Menthol and its Derivatives. Drug Dev. Ind. Pharm. 2008, 34, 595–601. [Google Scholar] [CrossRef]
- Eccles, R. What is the Role of Over 100 Excipients in Over the Counter (OTC) Cough Medicines? Lung 2020, 198, 727–734. [Google Scholar] [CrossRef]
- Kamatou, G.P.; Vermaak, I.; Viljoen, A.M.; Lawrence, B.M. Menthol: A simple monoterpene with remarkable biological properties. Phytochemistry 2013, 96, 15–25. [Google Scholar] [CrossRef]
- Oz, M.; El Nebrisi, E.G.; Yang, K.-H.S.; Howarth, F.C.; Al Kury, L.T. Cellular and Molecular Targets of Menthol Actions. Front. Pharmacol. 2017, 8, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvis, Y.; Négrier, P.; Lazerges, M.; Massip, S.; Léger, J.-M.; Espeau, P. Lidocaine/l-Menthol Binary System: Cocrystallization versus Solid-State Immiscibility. J. Phys. Chem. B 2010, 114, 5420–5426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, D.; Bhatia, D.; Dave, V.; Sutariya, V.; Gupta, S.V. Salts of Therapeutic Agents: Chemical, Physicochemical, and Biological Considerations. Molecules 2018, 23, 1719. [Google Scholar] [CrossRef] [Green Version]
- Fayed, N.D.; Arafa, M.F.; Essa, E.A.; El Maghraby, G.M. Lopinavir-menthol co-crystals for enhanced dissolution rate and intestinal absorption. J. Drug Deliv. Sci. Technol. 2022, 74, 103587. [Google Scholar] [CrossRef]
- Castro, E.; Dent, D. A comparison of transdermal over-the-counter lidocaine 3.6% menthol 1.25%, Rx lidocaine 5% and placebo for back pain and arthritis. Pain Manag. 2017, 7, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Alhadid, A.; Jandl, C.; Mokrushina, L.; Minceva, M. Experimental Investigation and Modeling of Cocrystal Formation in L-Menthol/Thymol Eutectic System. Cryst. Growth Des. 2021, 21, 6083–6091. [Google Scholar] [CrossRef]
- Alhadid, A.; Jandl, C.; Mokrushina, L.; Minceva, M. Cocrystal Formation in l-Menthol/Phenol Eutectic System: Experimental Study and Thermodynamic Modeling. Cryst. Growth Des. 2022, 22, 3973–3980. [Google Scholar] [CrossRef]
- Keating, L.; Harris, H.H.; Chickos, J.S. Vapor pressures and vaporization enthalpy of (−) α-bisabolol and (dl) menthol by correlation gas chromatography. J. Chem. Thermodyn. 2017, 107, 18–25. [Google Scholar] [CrossRef]
- Štejfa, V.; Bazyleva, A.; Fulem, M.; Rohlíček, J.; Skořepová, E.; Růžička, K.; Blokhin, A.V. Polymorphism and thermophysical properties of l- and dl-menthol. J. Chem. Thermodyn. 2018, 131, 524–543. [Google Scholar] [CrossRef]
- Wagner, Z.; Bendová, M.; Rotrekl, J.; Parmar, N.; Kočí, S.; Vrbka, P. Thermochemical Properties of Menthol and Terpineol. J. Solut. Chem. 2020, 49, 1267–1278. [Google Scholar] [CrossRef]
- Ballon, J.; Comparat, V.; Pouxe, J. The blade chamber: A solution for curved gaseous detectors. Nucl. Instruments Methods Phys. Res. 1983, 217, 213–216. [Google Scholar] [CrossRef]
- Evain, M.; Deniard, P.; Jouanneaux, A.; Brec, R. Potential of the INEL X-ray position-sensitive detector: A general study of the Debye–Scherrer setting. J. Appl. Crystallogr. 1993, 26, 563–569. [Google Scholar] [CrossRef] [Green Version]
- MS Modeling (Materials Studio), Version 5.5. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-materials-studio/ (accessed on 22 March 2023).
- CrysAlis PRO Software, Version 1.171.42.63a; Rigaku Oxford Diffraction/Agilent Technologies UK Ltd.: Yarnton, England, 2022.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, L.J. WinGXsuite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Veesler, S.; Lafferrere, L.; Garcia, E.; Hoff, C. Phase transitions in supersaturated drug solution. Org. Process Res. Dev. 2003, 7, 983–989. [Google Scholar] [CrossRef]
- Svärd, M.; Ahuja, D.; Rasmuson, C. Calorimetric Determination of Cocrystal Thermodynamic Stability: Sulfamethazine–Salicylic Acid Case Study. Cryst. Growth Des. 2020, 20, 4243–4251. [Google Scholar] [CrossRef]
- Taylor, C.R.; Day, G.M. Evaluating the Energetic Driving Force for Cocrystal Formation. Cryst. Growth Des. 2017, 18, 892–904. [Google Scholar] [CrossRef] [Green Version]
- Bakonyi, M.; Gácsi, A.; Kovács, A.; Szűcs, M.-B.; Berkó, S.; Csányi, E. Following-up skin penetration of lidocaine from different vehicles by Raman spectroscopic mapping. J. Pharm. Biomed. Anal. 2018, 154, 1–6. [Google Scholar] [CrossRef]
- Corvis, Y.; Espeau, P. Incidence of chirality on the properties of mixtures containing an amide type anesthetic compound. Thermochim. Acta 2012, 539, 39–43. [Google Scholar] [CrossRef]
- Schröder, I.Z. Über die Abhängigkeit der Löslichkeit eines festen Körpers von seiner Schmelztemperatur. Z. Phys. Chem. 1893, 11, 449. [Google Scholar]
- Le Châtelier, H.C.R. Acad. Sci. 1894, 118, 638.
- van Laar, J.J. Process of the fusion curves of firm alloys and amalgams. [machine translation]. Arch. Neerl. II 1903, 8, 264–284. [Google Scholar]
- Haneef, J.; Amir, M.; Sheikh, N.A.; Chadha, R. Mitigating Drug Stability Challenges Through Cocrystallization. AAPS PharmSciTech 2023, 24, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Devogelaer, J.-J.; Charpentier, M.D.; Tijink, A.; Dupray, V.; Coquerel, G.; Johnston, K.; Meekes, H.; Tinnemans, P.; Vlieg, E.; ter Horst, J.H.; et al. Cocrystals of Praziquantel: Discovery by Network-Based Link Prediction. Cryst. Growth Des. 2021, 21, 3428–3437. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | C24H42N2O2 | |
| FW (g·mol−1) | 390.59 | |
| Temperature | 233 K | |
| Wavelength | 1.54184 Å | |
| Cryst. Syst. | Monoclinic | |
| Space group | P 21/c | |
| Unit cell Dimensions | a (Å) b (Å) c (Å) | 8.4947(3) 22.2025(6) 13.4443(3) |
| α (°) β (°) γ (°) | 90 93.162(3) 90 | |
| Volume (Å3) | 2531.78(13) | |
| Z | 4 | |
| Dx (g·cm−3) | 1.025 | |
| µ (mm−1) | 0.496 | |
| Final R1 [I > 2σ(I)] | 0.1053 | |
| wR2 (all data) | 0.1971 | |
| S | 1.038 | |
| Lido:dlM | dl-Menthol | Lidocaine | Lido:lM | l-Menthol | |
|---|---|---|---|---|---|
| Tfus (°C) | 32.0 ± 0.3 | 33.8 ± 0.3 | 68.6 ± 0.5 | 39.1 ± 0.2 | 42.9 ± 0.3 |
| ΔfusH (kJ per mol of pure component) | 28.6 ± 0.3 | 14.2 ± 0.2 | 16.9 ± 0.2 | 38.3 ± 0.3 | 14.1 ± 0.2 |
| Ref. | This work | [37] | [44] | [44] | [44] |
| Signal Attribution | Wavenumber (cm−1) | ||||
|---|---|---|---|---|---|
| Lidocaine | l-Menthol | dl-Menthol | Lido:lM | Lido:dlM | |
| υNH | 3226 | 3232 | |||
| υCH | 3043 | 3045 | 3043 | ||
| υCH | 2967 | 2964 | 2958 | 2953 | 2955 |
| υCH | 2921 | 2931 | 2926 | 2932 | 2923 |
| υCH | 2874 | 2859 | 2867 | 2856 | 2872 |
| υCH | 2722 | 2722 | 2725 | ||
| υC=O | 1660 | 1660 | 1661 | ||
| δHNC | 1652 | 1651 | |||
| υCN, δHNC | 1591 | 1590 | 1592 | ||
| δCH | 1450 | 1455/1445 | 1455/1443 | 1447 | 1450 |
| δCH | 1377/1371 | 1377/1361 | 1374/1361 | ||
| δOH | 1345 | 1345 | 1343 | 1343 | |
| δCH | 1304 | 1307 | 1303 | 1307 | |
| υring | 1261 | 1264 | 1261 | ||
| υCO | 1240 | 1240 | 1239 | 1240 | |
| υCN | 1208 | 1208 | 1210 | ||
| υCN | 1161 | 1161 | 1163 | ||
| δCH | 1092 | 1090 | 1093 | ||
| νCC, δNCO | 989 | 990 | 989 | ||
| υCC | 966/954 | 965/952 | 966/956 | 967/955 | |
| υCC | 919 | 919 | 918 | 920 | |
| δring, υCC | 875 | 876 | 877 | 873 | 875 |
| γCH | 809 | 809 | 803 | 808 | |
| γCH | 768 | 767 | 767 | 766 | |
| ωHNC. δring | 752 | 752 | 752 | ||
| δring | 703 | 704 | 702 | ||
| ωNCO | 616 | 616 | 614 | ||
| τNCO, δring | 546/540 | 553 | 543 | 546 | |
| τring | 499 | 500 | 508/501 | 509/501 | |
| δring, δCH2 | 487 | 470 | 488 | ||
| ωNC2 | 402 | 404 | 408 | 404 | 405 |
| δCC | 324 | ||||
| Lattice vib. | 291 | 289 | 293 | 287 | |
| Lattice vib. | 264 | 258 | 258 | ||
| ωCC | 227 | 230 | 229 | ||
| Lidocaine/dl-Menthol | Lidocaine/l-Menthol | ||
|---|---|---|---|
| xe | Te (°C) | xe | Te (°C) |
| 0.215 ± 0.003 | 21.7 ± 0.3 | 0.20 | 28.6 |
| ∈ [0.50, 0.57] | 32.0 ± 0.3 | 0.61 | 37.6 |
| Lidocaine | Lido:lM | Lido:dlM | |
|---|---|---|---|
| Lidocaine solubility (mg·mL−1) | 6.3 ± 0.1 | – | – |
| Cocrystal estimated solubility (mg·mL−1) | – | 11.5 | 15.2 |
| Lidocaine solubility from the cocrystal (mg·mL−1) | – | 6.9 ± 0.1 | 9.1 ± 0.1 |
| Slope of the d = f(C) curve (×104 mL·mg−1) | 1.04 ± 0.02 | 1.05 ± 0.02 | 1.16 ± 0.01 |
| Lido:dlM | Lido:lM | Lidocaine | dl-Menthol | ||
|---|---|---|---|---|---|
| Anhydrous medium |  |  |  |  | |
| Time after hydration (min) | |||||
| 0 |  |  |  |  | |
| 10 |  |  |  |  | |
| 50 |  |  |  |  | |
| 380 |  |  | – | – | |
| 560 |  |  | – | – | |
| Lido:dlM | Lidocaine | dl-menthol | ||
|---|---|---|---|---|
| Anhydrous medium |  |  |  | |
| Time after hydration (min) | ||||
| 0 |  |  |  | |
| 4 |  |  |  | |
| 60 |  |  |  | |
| 268 |  |  |  | |
| 569 |  |  |  | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, P.; Toussaint, B.; Roberti, E.A.; Scornet, N.; Santos Silva, A.; Castillo Henríquez, L.; Cadasse, M.; Négrier, P.; Massip, S.; Dufat, H.; et al. New Lidocaine-Based Pharmaceutical Cocrystals: Preparation, Characterization, and Influence of the Racemic vs. Enantiopure Coformer on the Physico-Chemical Properties. Pharmaceutics 2023, 15, 1102. https://doi.org/10.3390/pharmaceutics15041102
Ma P, Toussaint B, Roberti EA, Scornet N, Santos Silva A, Castillo Henríquez L, Cadasse M, Négrier P, Massip S, Dufat H, et al. New Lidocaine-Based Pharmaceutical Cocrystals: Preparation, Characterization, and Influence of the Racemic vs. Enantiopure Coformer on the Physico-Chemical Properties. Pharmaceutics. 2023; 15(4):1102. https://doi.org/10.3390/pharmaceutics15041102
Chicago/Turabian StyleMa, Panpan, Balthazar Toussaint, Enrica Angela Roberti, Noémie Scornet, Axel Santos Silva, Luis Castillo Henríquez, Monique Cadasse, Philippe Négrier, Stéphane Massip, Hanh Dufat, and et al. 2023. "New Lidocaine-Based Pharmaceutical Cocrystals: Preparation, Characterization, and Influence of the Racemic vs. Enantiopure Coformer on the Physico-Chemical Properties" Pharmaceutics 15, no. 4: 1102. https://doi.org/10.3390/pharmaceutics15041102
APA StyleMa, P., Toussaint, B., Roberti, E. A., Scornet, N., Santos Silva, A., Castillo Henríquez, L., Cadasse, M., Négrier, P., Massip, S., Dufat, H., Hammad, K., Baraldi, C., Gamberini, M. C., Richard, C., Veesler, S., Espeau, P., Lee, T., & Corvis, Y. (2023). New Lidocaine-Based Pharmaceutical Cocrystals: Preparation, Characterization, and Influence of the Racemic vs. Enantiopure Coformer on the Physico-Chemical Properties. Pharmaceutics, 15(4), 1102. https://doi.org/10.3390/pharmaceutics15041102