
Characterization of Potent ABCG2 Inhibitor Derived from Chromone: From the Mechanism of Inhibition to Human Extracellular Vesicles for Drug Delivery
 , , ,
, , ,  , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. ATPase Activity
2.2. Thermostabilization Assay
2.3. Cell Lines
2.4. Cell-Based Transport Assay by Flow Cytometry
2.5. Cell-Based Transport Assay by Confocal Microscopy
2.6. D3 Shift Assay
2.7. Molecular Modelling
2.8. Preparation of Liposomes
2.9. Extracellular Vesicles from Giardia Intestinalis
2.10. MTT Assay
2.11. Extracellular Vesicles from Human Blood
3. Results
3.1. Interaction of Chromone 4a (C4a) with ABCG2 and P-Glycoprotein
3.2. Cell-Based Studies of the Interaction of Chromone 4a (C4a) with ABCG2
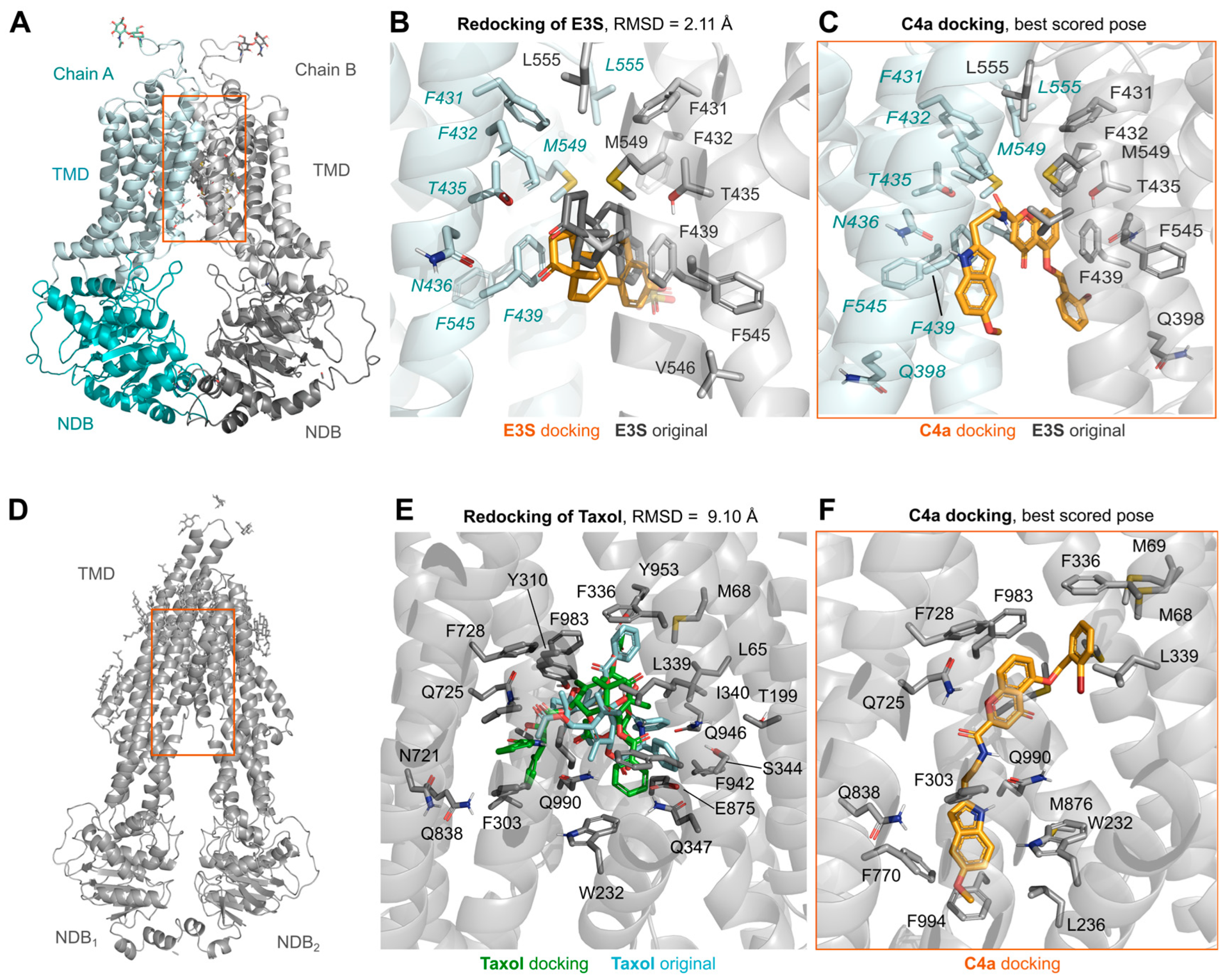
3.3. Molecular Dynamic Simulations of ABCG2 in Presence of Chromone (C4a)
3.4. Lipid-Nanoparticles-Based Delivery of Chromone (C4a)
3.5. G. intestinalis and Human Extracellular Vesicles (EVs) for Delivery of Chromone (C4a)
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gottesman, M.M. Mechanisms of Cancer Drug Resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The Human ATP-Binding Cassette (ABC) Transporter Superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Locher, K.P. Structure and mechanism of ABC transporters. Curr. Opin. Struct. Biol. 2004, 14, 426–431. [Google Scholar] [CrossRef]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Linton, K.J. Structure and Function of ABC Transporters. Physiology 2007, 22, 122–130. [Google Scholar] [CrossRef]
- Zattoni, I.F.; Delabio, L.C.; Dutra, J.D.P.; Kita, D.H.; Scheiffer, G.; Hembecker, M.; Pereira, G.D.S.; Moure, V.R.; Valdameri, G. Targeting breast cancer resistance protein (BCRP/ABCG2): Functional inhibitors and expression modulators. Eur. J. Med. Chem. 2022, 237. [Google Scholar] [CrossRef]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef]
- Miyake, K.; Mickley, L.; Litman, T.; Zhan, Z.; Robey, R.; Cristensen, B.; Brangi, M.; Greenberger, L.; Dean, M.; Fojo, T.; et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: Demonstration of homology to ABC transport genes. Cancer Res. 1999, 59, 8–13. [Google Scholar]
- Allikmets, R.; Schriml, L.; Hutchinson, A.; Romano-Spica, V.; Dean, M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998, 58, 5337–5339. [Google Scholar] [PubMed]
- Tamaki, A.; Ierano, C.; Szakacs, G.; Robey, R.; Bates, S.E. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011, 50, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Boumendjel, A.; Macalou, S.; Ahmed-Belkacem, A.; Blanc, M.; Di Pietro, A. Acridone derivatives: Design, synthesis, and inhibition of breast cancer resistance protein ABCG2. Bioorganic Med. Chem. 2007, 15, 2892–2897. [Google Scholar] [CrossRef]
- Valdameri, G.; Rangel, L.P.; Spatafora, C.; Guitton, J.; Gauthier, C.; Arnaud, O.; Ferreira-Pereira, A.; Falson, P.; Winnischofer, S.M.B.; Rocha, M.E.M.; et al. Methoxy Stilbenes as Potent, Specific, Untransported, and Noncytotoxic Inhibitors of Breast Cancer Resistance Protein. ACS Chem. Biol. 2011, 7, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Valdameri, G.; Gauthier, C.; Terreux, R.; Kachadourian, R.; Day, B.J.; Winnischofer, S.M.B.; Rocha, M.E.M.; Frachet, V.; Ronot, X.; Di Pietro, A.; et al. Investigation of Chalcones as Selective Inhibitors of the Breast Cancer Resistance Protein: Critical Role of Methoxylation in both Inhibition Potency and Cytotoxicity. J. Med. Chem. 2012, 55, 3193–3200. [Google Scholar] [CrossRef]
- Vesga, L.C.; Kronenberger, T.; Tonduru, A.K.; Kita, D.H.; Zattoni, I.F.; Bernal, C.C.; Bohórquez, A.R.R.; Mendez-Sánchez, S.C.; Ambudkar, S.V.; Valdameri, G.; et al. Tetrahydroquinoline/4,5-Dihydroisoxazole Molecular Hybrids as Inhibitors of Breast Cancer Resistance Protein (BCRP/ABCG2). Chemmedchem 2021, 16, 2686–2694. [Google Scholar] [CrossRef]
- Kita, D.H.; Guragossian, N.; Zattoni, I.F.; Moure, V.R.; Rego, F.G.D.M.; Lusvarghi, S.; Moulenat, T.; Belhani, B.; Picheth, G.; Bouacida, S.; et al. Mechanistic basis of breast cancer resistance protein inhibition by new indeno[1,2-b]indoles. Sci. Rep. 2021, 11, 1788. [Google Scholar] [CrossRef]
- Zattoni, I.F.; Kronenberger, T.; Kita, D.H.; Guanaes, L.D.; Guimarães, M.M.; Prado, L.D.O.; Ziasch, M.; Vesga, L.C.; Rego, F.G.D.M.; Picheth, G.; et al. A new porphyrin as selective substrate-based inhibitor of breast cancer resistance protein (BCRP/ABCG2). Chem. Interact. 2021, 351, 109718. [Google Scholar] [CrossRef]
- Valdameri, G.; Genoux-Bastide, E.; Peres, B.; Gauthier, C.; Guitton, J.; Terreux, R.; Winnischofer, S.M.B.; Rocha, M.E.M.; Boumendjel, A.; Di Pietro, A. Substituted Chromones as Highly Potent Nontoxic Inhibitors, Specific for the Breast Cancer Resistance Protein. J. Med. Chem. 2012, 55, 966–970. [Google Scholar] [CrossRef]
- Arnaud, O.; Boumendjel, A.; Gèze, A.; Honorat, M.; Matera, E.; Guitton, J.; Stein, W.; Bates, S.; Falson, P.; Dumontet, C.; et al. The acridone derivative MBLI-87 sensitizes breast cancer resistance protein-expressing xenografts to irinotecan. Eur. J. Cancer 2011, 47, 640–648. [Google Scholar] [CrossRef]
- Pires, A.D.R.A.; Lecerf-Schmidt, F.; Guragossian, N.; Pazinato, J.; Gozzi, G.J.; Winter, E.; Valdameri, G.; Veale, A.; Boumendjel, A.; Di Pietro, A.; et al. New, highly potent and non-toxic, chromone inhibitors of the human breast cancer resistance protein ABCG2. Eur. J. Med. Chem. 2016, 122, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Honorat, M.; Guitton, J.; Gauthier, C.; Bouard, C.; Lecerf-Schmidt, F.; Peres, B.; Terreux, R.; Gervot, H.; Rioufol, C.; Boumendjel, A.; et al. MBL-II-141, a chromone derivative, enhances irinotecan (CPT-11) anticancer efficiency in ABCG2-positive xenografts. Oncotarget 2014, 5, 11957–11970. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.; Wu, W.; Tony, T.; Zhao, H.-F.; Wang, J. Advances in lipid-based drug delivery: Enhancing efficiency for hydrophobic drugs. Expert Opin. Drug Deliv. 2015, 12, 1475–1499. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- N’diaye, E.-R.; Orefice, N.S.; Ghezzi, C.; Boumendjel, A. Chemically Modified Extracellular Vesicles and Applications in Radiolabeling and Drug Delivery. Pharmaceutics 2022, 14, 653. [Google Scholar] [CrossRef] [PubMed]
- EL Andaloussi, S.; Lakhal, S.; Mäger, I.; Wood, M.J. Exosomes for targeted siRNA delivery across biological barriers. Adv. Drug Deliv. Rev. 2013, 65, 391–397. [Google Scholar] [CrossRef]
- Rufino-Ramos, D.; Albuquerque, P.R.; Carmona, V.; Perfeito, R.; Nobre, R.J.; de Almeida, L.P. Extracellular vesicles: Novel promising delivery systems for therapy of brain diseases. J. Control. Release 2017, 262, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V. Drug-Stimulatable ATPase Activity in Crude Membranes of Human MDR1-Transfected Mammalian Cells. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1998; Volume 292, pp. 504–514. [Google Scholar] [CrossRef]
- Lusvarghi, S.; Ambudkar, S.V. ATP-dependent thermostabilization of human P-glycoprotein (ABCB1) is blocked by modulators. Biochem. J. 2019, 476, 3737–3750. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Liao, M. ABCG2 transports anticancer drugs via a closed-to-open switch. Nat. Commun. 2020, 11, 2264. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand / Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC’06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Inglut, C.T.; Sorrin, A.J.; Kuruppu, T.; Vig, S.; Cicalo, J.; Ahmad, H.; Huang, H.-C. Immunological and Toxicological Considerations for the Design of Liposomes. Nanomaterials 2020, 10, 190. [Google Scholar] [CrossRef]
- Hadjeri, M.; Barbier, M.; Ronot, X.; Mariotte, A.-M.; Boumendjel, A.; Boutonnat, J. Modulation of P-Glycoprotein-Mediated Multidrug Resistance by Flavonoid Derivatives and Analogues. J. Med. Chem. 2003, 46, 2125–2131. [Google Scholar] [CrossRef]
- Boumendjel, A.; Nicolle, E.; Moraux, T.; Gerby, B.; Blanc, M.; Ronot, X.; Boutonnat, J. Piperazinobenzopyranones and Phenalkylaminobenzopyranones: Potent Inhibitors of Breast Cancer Resistance Protein (ABCG2). J. Med. Chem. 2005, 48, 7275–7281. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ohnuma, S.; Fukuda, M.; Chufan, E.E.; Kudoh, K.; Kanehara, K.; Sugisawa, N.; Ishida, M.; Naitoh, T.; Shibata, H.; et al. Synthetic Analogs of Curcumin Modulate the Function of Multidrug Resistance–Linked ATP-Binding Cassette Transporter ABCG2. Drug Metab. Dispos. 2017, 45, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Tiwari, A.K.; Shukla, S.; Robey, R.W.; Singh, S.; Kim, I.-W.; Bates, S.E.; Peng, X.; Abraham, I.; Ambudkar, S.V.; et al. Sildenafil Reverses ABCB1- and ABCG2-Mediated Chemotherapeutic Drug Resistance. Cancer Res. 2011, 71, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Valdameri, G.; Genoux-Bastide, E.; Gauthier, C.; Peres, B.; Terreux, R.; Winnischofer, S.M.B.; Rocha, M.E.M.; Di Pietro, A.; Boumendjel, A. 6-Halogenochromones Bearing Tryptamine: One-Step Access to Potent and Highly Selective Inhibitors of Breast Cancer Resistance Protein. Chemmedchem 2012, 7, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Telbisz, Á.; Hegedüs, C.; Özvegy-Laczka, C.; Goda, K.; Várady, G.; Takáts, Z.; Szabó, E.; Sorrentino, B.P.; Váradi, A.; Sarkadi, B. Antibody binding shift assay for rapid screening of drug interactions with the human ABCG2 multidrug transporter. Eur. J. Pharm. Sci. 2012, 45, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Zylberberg, C.; Matosevic, S. Pharmaceutical liposomal drug delivery: A review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016, 23, 3319–3329. [Google Scholar] [CrossRef]
- Alberro, A.; Iparraguirre, L.; Fernandes, A.; Otaegui, D. Extracellular Vesicles in Blood: Sources, Effects, and Applications. Int. J. Mol. Sci. 2021, 22, 8163. [Google Scholar] [CrossRef]
- Kuo, W.P.; Tigges, J.C.; Toxavidis, V.; Ghiran, I. Red Blood Cells: A Source of Extracellular Vesicles. Methods Mol. Biol. 2017, 1660, 15–22. [Google Scholar] [CrossRef]
- Wang, H.; Li, X.; Chen, T.; Wang, W.; Liu, Q.; Li, H.; Yi, J.; Wang, J. Mechanisms of verapamil-enhanced chemosensitivity of gallbladder cancer cells to platinum drugs: Glutathione reduction and MRP1 downregulation. Oncol. Rep. 2012, 29, 676–684. [Google Scholar] [CrossRef]
- Usman, W.M.; Pham, T.C.; Kwok, Y.Y.; Vu, L.T.; Ma, V.; Peng, B.; Chan, Y.S.; Wei, L.; Chin, S.M.; Azad, A.; et al. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 2359. [Google Scholar] [CrossRef]
- Malhotra, S.; Dumoga, S.; Sirohi, P.; Singh, N. Red Blood Cells-Derived Vesicles for Delivery of Lipophilic Drug Camptothecin. ACS Appl. Mater. Interfaces 2019, 11, 22141–22151. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Lin, Q.; Huang, L.; Fu, Y.; Wang, L.; He, S.; Fu, Y.; Yang, S.; Zhang, Z.; Zhang, L.; et al. Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson’s disease. J. Control. Release 2018, 287, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.; Du, D.; Pu, L.; Song, H.; Deng, M.; Long, Q.; Yin, X.; Wang, Y.; Rao, L. SPION-Decorated Exosome Delivered BAY55-9837 Targeting the Pancreas through Magnetism to Improve the Blood GLC Response. Small 2019, 15, e1903135. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdameri, G.; Kita, D.H.; Dutra, J.d.P.; Gomes, D.L.; Tonduru, A.K.; Kronenberger, T.; Gavinho, B.; Rossi, I.V.; Carvalho, M.M.d.; Pérès, B.; et al. Characterization of Potent ABCG2 Inhibitor Derived from Chromone: From the Mechanism of Inhibition to Human Extracellular Vesicles for Drug Delivery. Pharmaceutics 2023, 15, 1259. https://doi.org/10.3390/pharmaceutics15041259
Valdameri G, Kita DH, Dutra JdP, Gomes DL, Tonduru AK, Kronenberger T, Gavinho B, Rossi IV, Carvalho MMd, Pérès B, et al. Characterization of Potent ABCG2 Inhibitor Derived from Chromone: From the Mechanism of Inhibition to Human Extracellular Vesicles for Drug Delivery. Pharmaceutics. 2023; 15(4):1259. https://doi.org/10.3390/pharmaceutics15041259
Chicago/Turabian StyleValdameri, Glaucio, Diogo Henrique Kita, Julia de Paula Dutra, Diego Lima Gomes, Arun Kumar Tonduru, Thales Kronenberger, Bruno Gavinho, Izadora Volpato Rossi, Mariana Mazetto de Carvalho, Basile Pérès, and et al. 2023. "Characterization of Potent ABCG2 Inhibitor Derived from Chromone: From the Mechanism of Inhibition to Human Extracellular Vesicles for Drug Delivery" Pharmaceutics 15, no. 4: 1259. https://doi.org/10.3390/pharmaceutics15041259
APA StyleValdameri, G., Kita, D. H., Dutra, J. d. P., Gomes, D. L., Tonduru, A. K., Kronenberger, T., Gavinho, B., Rossi, I. V., Carvalho, M. M. d., Pérès, B., Zattoni, I. F., Rego, F. G. d. M., Picheth, G., Freitas, R. A. d., Poso, A., Ambudkar, S. V., Ramirez, M. I., Boumendjel, A., & Moure, V. R. (2023). Characterization of Potent ABCG2 Inhibitor Derived from Chromone: From the Mechanism of Inhibition to Human Extracellular Vesicles for Drug Delivery. Pharmaceutics, 15(4), 1259. https://doi.org/10.3390/pharmaceutics15041259