


Molecular Hybridization as a Strategy for Developing Artemisinin-Derived Anticancer Candidates




Abstract
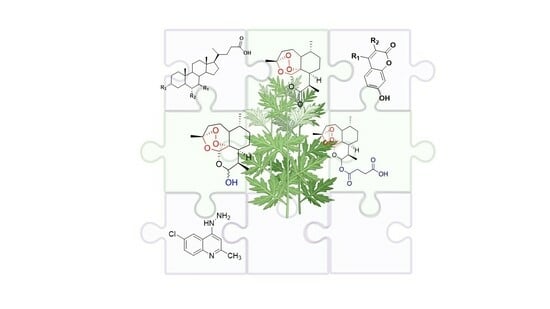
:
1. Introduction
2. Chemical Features of Artemisinin and Selected Artemisinin Derivatives
3. Artemisinin Hybrids Based on Natural and Synthetic Pharmacophores
3.1. Artemisinin–Bile Acid Hybrids
3.2. Artemisinin–Quinoline and Quinazoline Hybrids
3.3. Artemisinin–Nitrogen Mustard Hybrid
3.4. Artemisinin–Tyrosol Hybrids
3.5. Artemisinin–Camptothecin Hybrids
3.6. Artemisinin–Thymoquinone Hybrids
3.7. Artemisinin–Chalcone Hybrids
3.8. Artemisinin–Coumarin Hybrids
3.9. Artemisinin–Tamoxifen Hybrids
3.10. Artemisinin–Steroid Hybrids
3.11. Artemisinin–Cinnamic Acid Hybrids
3.12. Artemisinin–Acridine Hybrids
3.13. Artemisinin–Isatin Hybrids
3.14. Artemisinin–Sulfasalazine Hybrid
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Penny, L.K.; Wallace, H.M. The challenges for cancer chemoprevention. Chem. Soc. Rev. 2015, 44, 8836–8847. [Google Scholar] [CrossRef] [PubMed]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Rahman, T. The difficulties in cancer treatment. Ecancermedicalscience 2012, 6, ed16. [Google Scholar] [CrossRef]
- Zimmermann, G.R.; Lehár, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef]
- Gediya, L.K.; Njar, V.C. Promise and challenges in drug discovery and development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2009, 4, 1099–1111. [Google Scholar] [CrossRef]
- Meunier, B. Hybrid molecules with a dual mode of action: Dream or reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef]
- Viegas-Junior, C.; Danuello, A.; da Bolzani, V.S.; Barreiro, E.J.; Manssour, C.A. Molecular Hybridization: A Useful Tool in the Design of New Drug Prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef]
- Shalini; Kumar, V. Have molecular hybrids delivered effective anti-cancer treatments and what should future drug discovery focus on? Expert Opin. Drug Discov. 2021, 16, 335–363. [Google Scholar] [CrossRef]
- Singh, A.K.; Kumar, A.; Singh, H.; Sonawane, P.; Paliwal, H.; Thareja, S.; Pathak, P.; Grishina, M.; Jaremko, M.; Emwas, A.H.; et al. Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids. Pharmaceuticals 2022, 15, 1071. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J.; Snader, K.M. Natural Products in Drug Discovery and Development. J. Nat. Prod. 1997, 60, 52–60. [Google Scholar] [CrossRef]
- Cordell, G.A. Biodiversity and drug discovery—A symbiotic relationship. Phytochemistry 2000, 55, 463–480. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Strobl, J.S.; Bane, S.; Schilling, J.K.; McCracken, M.; Chatterjee, S.K.; Rahim-Bata, R.; Kingston, D.G.I. Design, Synthesis, and Bioactivities of Steroid-Linked Taxol Analogues as Potential Targeted Drugs for Prostate and Breast Cancer. J. Nat. Prod. 2004, 67, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Wittman, M.D.; Kadow, J.F.; Vyas, D.M.; Lee, F.L.; Rose, W.C.; Long, B.H.; Johnston, K. Synthesis and Antitumor Activity of Novel paclitaxel ± Chlorambucil Hybrids. Bioorg. Med. Chem. Lett. 2001, 11, 811–814. [Google Scholar] [CrossRef]
- Kingston, D.G.I.; Snyder, J.P. The quest for a simple bioactive analog of paclitaxel as a potential anticancer agent. Acc. Chem. Res. 2014, 47, 2682–2691. [Google Scholar] [CrossRef]
- Daniel, J.; Montaleytang, M.; Nagarajan, S.; Picard, S.; Clermont, G.; Lazar, A.N.; Dumas, N.; Correard, F.; Braguer, D.; Blanchard-Desce, M.; et al. Hydrophilic Fluorescent Nanoprodrug of paclitaxel for Glioblastoma Chemotherapy. ACS Omega 2019, 4, 18342–18354. [Google Scholar] [CrossRef]
- Roussi, F.; Quoc, A.N.; Thoret, S.; Guéritte, F.; Guénard, D. The design and synthesis of new steroidal compounds as potential mimics of taxoids. Eur. J. Org. Chem. 2005, 2005, 3952–3961. [Google Scholar] [CrossRef]
- Melloni, E.; Marchesi, E.; Preti, L.; Casciano, F.; Rimondi, E.; Romani, A.; Secchiero, P.; Navacchia, M.L.; Perrone, D. Synthesis and Biological Investigation of Bile Acid-Paclitaxel Hybrids. Molecules 2022, 27, 471. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew. Chem.-Int. Ed. 2003, 42, 3996–4028. [Google Scholar] [CrossRef]
- Tu, Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat. Med. 2011, 17, 1217–1220. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, D.; Goswami, A.; Saikia, P.P.; Barua, N.C.; Rao, P.G. Artemisinin and its derivatives: A novel class of anti-malarial and anti-cancer agents. Chem. Soc. Rev. 2010, 39, 435–454. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Woon, C.Y.-N.; Liu, C.-G.; Cheng, J.-T.; You, M.; Sethi, G.; Wong, A.L.-A.; Ho, P.C.-L.; Zhang, D.; Ong, P.; et al. Repurposing Artemisinin and its Derivatives as Anticancer Drugs: A Chance or Challenge? Front. Pharmacol. 2021, 12, 828856. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.C.; Singh, N.P.; Sasaki, T. Development of artemisinin compounds for cancer treatment. Investig. New Drugs 2013, 31, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.K.; Xu, C.; Kalesh, K.A.; He, Y.; Lin, Q.; Wong, W.S.F.; Shen, H.M.; Wang, J. Artemisinin as an anticancer drug: Recent advances in target profiling and mechanisms of action. Med. Res. Rev. 2017, 37, 1492–1517. [Google Scholar] [CrossRef]
- Dai, X.; Zhang, X.; Chen, W.; Chen, Y.; Zhang, Q.; Mo, S.; Lu, J. Dihydroartemisinin: A potential natural anticancer drug. Int. J. Biol. Sci. 2021, 17, 603–622. [Google Scholar] [CrossRef]
- Wen, L.; Liu, L.; Wen, L.; Yu, T.; Wei, F. Artesunate promotes G2/M cell cycle arrest in MCF7 breast cancer cells through ATM activation. Breast Cancer 2018, 25, 681–686. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Zhan, X.; Wang, L.; Ho, R.J.Y.; Sasaki, T. pH-responsive artemisinin dimer in lipid nanoparticles are effective against human breast cancer in a xenograft model. J. Pharm. Sci. 2015, 104, 1815–1824. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, X.; Chen, K.; Ba, Q.; Zhang, X.; Li, J.; Wang, J.; Wang, H.; Liu, H. Structural optimization and biological evaluation for novel artemisinin derivatives against liver and ovarian cancers. Eur. J. Med. Chem. 2021, 211, 113000. [Google Scholar] [CrossRef]
- Zhou, C.; Pan, W.; Wang, X.P.; Chen, T.S. Artesunate induces apoptosis via a Bak-mediated caspase-independent intrinsic pathway in human lung adenocarcinoma cells. J. Cell. Physiol. 2012, 227, 3778–3786. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, X.; Zhang, J.; He, A.; Wang, Y.L.; Han, K.; Su, Y.; Yin, J.; Lv, X.; Hu, H. Artesunate suppresses the viability and mobility of prostate cancer cells through UCA1, the sponge of miR-184. Oncotarget 2017, 8, 18260–18270. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zou, W.Q.; Du, S.J.; Wu, M.J.; Xiang, T.X.; Luo, Z.G. Mechanism of dihydroartemisinin-induced apoptosis in prostate cancer PC3 cells: An iTRAQ-based proteomic analysis. Life Sci. 2016, 157, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, B. Biological Actions of Artemisinin: Insights from Medicinal Chemistry Studies. Molecules 2010, 15, 1378–1397. [Google Scholar] [CrossRef] [PubMed]
- Posadino, A.M.; Giordo, R.; Pintus, G.; Mohammed, S.A.; Orhan, I.E.; Fokou, P.V.T.; Sharopov, F.; Adetunji, C.O.; Gulsunoglu-Konuskan, Z.; Ydyrys, A.; et al. Medicinal and mechanistic overview of artemisinin in the treatment of human diseases. Biomed. Pharmacother. 2023, 163, 114866. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, H.; Mu, L.; Yang, X. Artemisinins as Anticancer Drugs: Novel Therapeutic Approaches, Molecular Mechanisms, and Clinical Trials. Front. Pharmacol. 2020, 11, 529881. [Google Scholar] [CrossRef]
- O’Neill, P.M.; Barton, V.E.; Ward, S.A. The molecular mechanism of action of artemisinin-The debate continues. Molecules 2010, 15, 1705–1721. [Google Scholar] [CrossRef]
- World Health Organization. WHO Calls for an Immediate Halt to Provision of Single-Drug Artemisinin Malaria Pills; World Health Organization: Washington, DC, USA, 2006; p. 2006. [Google Scholar]
- World Health Organization. WHO Resolution WHA6018 Malaria, including proposal for establishment of World Malaria Day In Sixtieth World Health Assembly, Geneva, Resolutions and Decisions, Annexes; World Health Organization: Geneva, Switzerland, 2007; (WHA60/2007/REC/1). [Google Scholar]
- Nosten, F.; White, N.J. Artemisinin-based combination treatment of falciparum malaria. Am. J. Trop. Med. Hyg. 2007, 77, 181–192. [Google Scholar] [CrossRef]
- Tsamesidis, I.; Reybier, K.; Marchetti, G.; Pau, M.C.; Virdis, P.; Fozza, C.; Nepveu, F.; Low, P.S.; Turrini, F.M.; Pantaleo, A. Syk Kinase Inhibitors Synergize with Artemisinins by Enhancing Oxidative Stress in Plasmodium falciparum-Parasitized Erythrocytes. Antioxidants 2020, 9, 753–774. [Google Scholar] [CrossRef]
- Chien, H.D.; Pantaleo, A.; Kesely, K.R.; Noomuna, P.; Putt, K.S.; Tuan, T.A.; Low, P.S.; Turrini, F.M. Imatinib augments standard malaria combination therapy without added toxicity. J. Exp. Med. 2021, 218, e20210724. [Google Scholar] [CrossRef]
- Li, Q.; Ma, Q.; Cheng, J.; Zhou, X.; Pu, W.; Zhong, X.; Guo, X. Dihydroartemisinin as a sensitizing agent in cancer therapies. OncoTargets Ther. 2021, 14, 2563–2573. [Google Scholar] [CrossRef]
- Navacchia, M.L.; Marchesi, E.; Mari, L.; Chinaglia, N.; Gallerani, E.; Gavioli, R.; Capobianco, M.L.; Perrone, D. Rational design of nucleoside-bile acid conjugates incorporating a triazole moiety for anticancer evaluation and SAR exploration. Molecules 2017, 22, 1710. [Google Scholar] [CrossRef]
- Marchesi, E.; Chinaglia, N.; Capobianco, M.L.; Marchetti, P.; Huang, T.E.; Weng, H.C.; Guh, J.H.; Hsu, L.C.; Perrone, D.; Navacchia, M.L. Dihydroartemisinin–Bile Acid Hybridization as an Effective Approach to Enhance Dihydroartemisinin Anticancer Activity. ChemMedChem 2019, 14, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.E.; Deng, Y.N.; Hsu, J.L.; Leu, W.J.; Marchesi, E.; Capobianco, M.L.; Marchetti, P.; Navacchia, M.L.; Guh, J.H.; Perrone, D.; et al. Evaluation of the Anticancer Activity of a Bile Acid-Dihydroartemisinin Hybrid Ursodeoxycholic-Dihydroartemisinin in Hepatocellular Carcinoma Cells. Front. Pharmacol. 2020, 11, 599067. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.F.; Kung, F.L.; Huang, T.E.; Deng, Y.N.; Guh, J.H.; Marchetti, P.; Marchesi, E.; Perrone, D.; Navacchia, M.L.; Hsu, L.C. Anticancer Activity and Molecular Mechanisms of an Ursodeoxycholic Acid Methyl Ester-Dihydroartemisinin Hybrid via a Triazole Linkage in Hepatocellular Carcinoma Cells. Molecules 2023, 28, 2358. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Ortiz, M.P.; Wei, M.Q. Antitumor activity of artemisinin and its derivatives: From a well-known antimalarial agent to a potential anticancer drug. J. Biomed. Biotechnol. 2012, 2012, 247597. [Google Scholar] [CrossRef] [PubMed]
- Galal, A.M.; Ross, S.A.; ElSohly, M.A.; Hala, N.; ElSohly, H.N.; . El-Feraly, F.S.; Ahmed, M.S.; McPhail, E.T. Deoxyartemisinin derivatives from photooxygenation of anhydrodeoxydihydroartemisinin and their cytotoxic evaluation. J. Nat. Prod. 2002, 65, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, P.; Chen, Y.-X.; Li, L.-Q.; Gai, D.-S.; Wang, Y.-P.Z. Synthesis of some derivatives of artemisinine. Chin. Sci. Bull. (Repr. Kexue Tongbao) 1979, 24, 667–669. [Google Scholar]
- Posner, G.H.; Paik, I.H.; Chang, W.; Borstnik, K.; Sinishtaj, S.; Rosenthal, A.S.; Shapiro, T.A. Malaria-infected mice are cured by a single dose of novel artemisinin derivatives. J. Med. Chem. 2007, 50, 2516–2519. [Google Scholar] [CrossRef]
- Presser, A.; Feichtinger, A.; Buzzi, S. A simplified and scalable synthesis of artesunate. Monatshefte Fur Chem. 2017, 148, 63–68. [Google Scholar] [CrossRef]
- Li, Q.G.; Peggins, J.O.; Fleckenstein, L.L.; Masonic, K.; Heiffer, M.H.; Brewer, T.G. The pharmacokinetics and bioavailability of dihydroartemisinin, arteether, artemether, artesunic acid and artelinic acid in rats. J. Pharm. Pharmacol. 1998, 50, 173–182. [Google Scholar] [CrossRef]
- Navacchia, M.L.; Marchesi, E.; Perrone, D. Bile Acid Conjugates with Anticancer Activity: Most Recent Research. Molecules 2021, 26, 25. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Liu, C.; Li, C.; Fu, R.; Xu, W.; Bian, H.; Dong, X.; Zhao, X.; Xu, Z.; Zhang, J.; et al. Study on the structure-activity relationship of dihydroartemisinin derivatives: Discovery, synthesis, and biological evaluation of dihydroartemisinin-bile acid conjugates as potential anticancer agents. Eur. J. Med. Chem. 2021, 225, 113754. [Google Scholar] [CrossRef] [PubMed]
- Letis, A.S.; Seo, E.J.; Nikolaropoulos, S.S.; Efferth, T.; Giannis, A.; Fousteris, M.A. Synthesis and cytotoxic activity of new artemisinin hybrid molecules against human leukemia cells. Bioorg. Med. Chem. 2017, 25, 3357–3367. [Google Scholar] [CrossRef]
- Herrmann, L.; Yaremenko, I.A.; Çapcı, A.; Struwe, J.; Tailor, D.; Dheeraj, A.; Hodek, J.; Belyakova, Y.Y.; Radulov, P.S.; Weber, J.; et al. Synthesis and in vitro Study of Artemisinin/Synthetic Peroxide-Based Hybrid Compounds against SARS-CoV-2 and Cancer. ChemMedChem 2022, 17, e202200005. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.; Kim, M.; Chung, W.Y.; Park, K.K.; Jung, M. Discovery of artemisinin-glycolipid hybrids as anti-oral cancer agents. Chem. Pharm. Bull. 2011, 59, 1471–1475. [Google Scholar] [CrossRef]
- Wang, L.; Switalska, M.; Wang, N.; Du, Z.J.; Fukumoto, Y.; Diep, N.K.; Kiguchi, R.; Nokami, J.; Wietrzyk, J.; Inokuchi, T. Design, synthesis, and biological evaluation of artemisinin-indoloquinoline hybrids as potent antiproliferative agents. Molecules 2014, 19, 19021–19035. [Google Scholar] [CrossRef]
- Yao, G.; Chen, H.; Chen, L.; Ge, M.; Yang, J.; Liu, W.; Xia, M.; Hayashi, T.; Guo, C.; Ikejima, T. Autophagy promotes apoptosis induction through repressed nitric oxide generation in the treatment of human breast cancer MCF-7 cells with L-A03, a dihydroartemisinin derivative. Med. Chem. Res. 2017, 26, 1427–1436. [Google Scholar] [CrossRef]
- Yao, G.D.; Ge, M.Y.; Li, D.Q.; Chen, L.; Hayashi, T.; Tashiro, S.I.; Onodera, S.; Guo, C.; Song, S.J.; Ikejima, T. L-A03, a dihydroartemisinin derivative, promotes apoptotic cell death of human breast cancer MCF-7 cells by targeting c-Jun N-terminal kinase. Biomed. Pharmacother. 2018, 105, 320–325. [Google Scholar] [CrossRef]
- Lombard, M.C.; N’Da, D.D.; Breytenbach, J.C.; Smith, P.J.; Lategan, C.A. Artemisinin-quinoline hybrid-dimers: Synthesis and in vitro antiplasmodial activity. Bioorg. Med. Chem. Lett. 2010, 20, 6975–6977. [Google Scholar] [CrossRef]
- Lombard, M.C.; N’Da, D.D.; Breytenbach, J.C.; Kolesnikova, N.I.; Tran Van Ba, C.; Wein, S.; Norman, J.; Denti, P.; Vial, H.; Wiesner, L. Antimalarial and anticancer activities of artemisinin-quinoline hybrid-dimers and pharmacokinetic properties in mice. Eur. J. Pharm. Sci. 2012, 47, 834–841. [Google Scholar] [CrossRef]
- Fröhlich, T.; Reiter, C.; Ibrahim, M.M.; Beutel, J.; Hutterer, C.; Zeitträger, I.; Bahsi, H.; Leidenberger, M.; Friedrich, O.; Kappes, B.; et al. Synthesis of Novel Hybrids of Quinazoline and Artemisinin with High Activities against Plasmodium falciparum, Human Cytomegalovirus, and Leukemia Cells. ACS Omega 2017, 2, 2422–2431. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Kong, L.; Liu, H.; Zhang, Y.; Zhang, L.; Liu, X.; Yuan, F.; Li, Y.; Zuo, Z. Design and synthesis of novel artemisinin derivatives with potent activities against colorectal cancer in vitro and in vivo. Eur. J. Med. Chem. 2019, 182, 111665. [Google Scholar] [CrossRef]
- Dai, T.; Lin, L.; Chen, H.; Lu, W.; Yang, X.; Yang, L.; Liu, Y.; Cui, J.; Sun, D. Novel nitrogen mustard-artemisinin hybrids with potent anti-leukemia action through DNA damage and activation of GPx. Eur. J. Med. Chem. 2022, 244, 114783. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, Y.; Liu, Y.; Zhang, X.; Chen, T.; Chen, K.; Ba, Q.; Li, J.; Liu, H.; Wang, H. Preclinical Efficacy and Safety Assessment of Artemisinin-Chemotherapeutic Agent Conjugates for Ovarian Cancer. EBioMedicine 2016, 14, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Botta, L.; Filippi, S.; Bizzarri, B.M.; Zippilli, C.; Meschini, R.; Pogni, R.; Baratto, M.C.; Villanova, L.; Saladino, R. Synthesis and Evaluation of Artemisinin-Based Hybrid and Dimer Derivatives as Antimelanoma Agents. ACS Omega 2020, 5, 243–251. [Google Scholar] [CrossRef]
- Botta, L.; Cesarini, S.; Zippilli, C.; Filippi, S.; Bizzarri, B.M.; Baratto, M.C.; Pogni, R.; Saladino, R. Stereoselective Access to Antimelanoma Agents by Hybridization and Dimerization of Dihydroartemisinin and Artesunic acid. ChemMedChem 2021, 16, 2270–2277. [Google Scholar] [CrossRef]
- Botta, L.; Filippi, S.; Zippilli, C.; Cesarini, S.; Bizzarri, B.M.; Cirigliano, A.; Rinaldi, T.; Paiardini, A.; Fiorucci, D.; Saladino, R.; et al. Artemisinin Derivatives with Antimelanoma Activity Show Inhibitory Effect against Human DNA Topoisomerase 1. ACS Med. Chem. Lett. 2020, 11, 1035–1040. [Google Scholar] [CrossRef]
- Fröhlich, T.; Ndreshkjana, B.; Muenzner, J.K.; Reiter, C.; Hofmeister, E.; Mederer, S.; Fatfat, M.; El-Baba, C.; Gali-Muhtasib, H.; Schneider-Stock, R.; et al. Synthesis of Novel Hybrids of Thymoquinone and Artemisinin with High Activity and Selectivity Against Colon Cancer. ChemMedChem 2017, 12, 226–234. [Google Scholar] [CrossRef]
- Fröhlich, T.; Reiter, C.; Saeed, M.E.M.; Hutterer, C.; Hahn, F.; Leidenberger, M.; Friedrich, O.; Kappes, B.; Marschall, M.; Efferth, T.; et al. Synthesis of Thymoquinone-Artemisinin Hybrids: New Potent Antileukemia, Antiviral, and Antimalarial Agents. ACS Med. Chem. Lett. 2018, 9, 534–539. [Google Scholar] [CrossRef]
- Çapcı Karagöz, A.; Reiter, C.; Seo, E.J.; Gruber, L.; Hahn, F.; Leidenberger, M.; Klein, V.; Hampel, F.; Friedrich, O.; Marschall, M.; et al. Access to new highly potent antileukemia, antiviral and antimalarial agents via hybridization of natural products (homo)egonol, thymoquinone and artemisinin. Bioorg. Med. Chem. 2018, 26, 3610–3618. [Google Scholar] [CrossRef]
- Xie, L.; Zhai, X.; Ren, L.; Meng, H.; Liu, C.; Zhu, W.; Zhao, Y. Design, synthesis and antitumor activity of novel artemisinin derivatives using hybrid approach. Chem. Pharm. Bull. 2011, 59, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhai, X.; Liu, C.; Li, P.; Li, Y.; Guo, G.; Gong, P. Anti-tumor activity of new artemisinin-chalcone hybrids. Arch. Pharm. 2011, 344, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Smit, F.J.; Van Biljon, R.A.; Birkholtz, L.M.; N’da, D.D. Synthesis and in vitro biological evaluation of dihydroartemisinyl-chalcone esters. Eur. J. Med. Chem. 2015, 90, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liang, Z.; Xu, H.; Mou, Y.; Guo, C. Design, synthesis and cytotoxicity of novel dihydroartemisinin-coumarin hybrids via click chemistry. Molecules 2016, 21, 758. [Google Scholar] [CrossRef]
- Yu, H.; Hou, Z.; Yang, X.; Mou, Y.; Guo, C. Design, Synthesis, and Mechanism of Dihydroartemisinin–Coumarin Hybrids as Potential Anti-Neuroinflammatory AgentsNo Title. Molecules 2019, 24, 1672. [Google Scholar] [CrossRef]
- Yu, H.; Hou, Z.; Tian, Y.; Mou, Y.; Guo, C. Design, synthesis, cytotoxicity and mechanism of novel dihydroartemisinin-coumarin hybrids as potential anti-cancer agents. Eur. J. Med. Chem. 2018, 151, 434–449. [Google Scholar] [CrossRef]
- Fröhlich, T.; Mai, C.; Bogautdinov, R.P.; Morozkina, S.N.; Shavva, A.G.; Friedrich, O.; Gilbert, D.F.; Tsogoeva, S.B. Synthesis of Tamoxifen-Artemisinin and Estrogen-Artemisinin Hybrids Highly Potent Against Breast and Prostate Cancer. ChemMedChem 2020, 15, 1473–1479. [Google Scholar] [CrossRef]
- Fröhlich, T.; Kiss, A.; Wölfling, J.; Mernyák, E.; Kulmány, Á.E.; Minorics, R.; Zupkó, I.; Leidenberger, M.; Friedrich, O.; Kappes, B.; et al. Synthesis of Artemisinin-Estrogen Hybrids Highly Active against HCMV, P. falciparum, and Cervical and Breast Cancer. ACS Med. Chem. Lett. 2018, 9, 1128–1133. [Google Scholar] [CrossRef]
- Xu, C.C.; Deng, T.; Fan, M.L.; Lv, W.B.; Liu, J.H.; Yu, B.Y. Synthesis and in vitro antitumor evaluation of dihydroartemisinin-cinnamic acid ester derivatives. Eur. J. Med. Chem. 2016, 107, 192–203. [Google Scholar] [CrossRef]
- Jones, M.; Mercer, A.E.; Stocks, P.A.; La Pensée, L.J.I.; Cosstick, R.; Kevin Park, B.K.; Kennedy, M.E.; Piantanida, I.; Ward, S.A.; Davies, J.; et al. Antitumour and antimalarial activity of artemisinin–acridine hybrids. Bioorg. Med. Chem. Lett. 2009, 19, 2033–2037. [Google Scholar] [CrossRef]
- Joubert, J.P.; Smit, F.J.; du Plessis, L.; Smith, P.J.; David, D. Synthesis and in vitro biological evaluation of aminoacridines and artemisinin–acridine hybrids. Eur. J. Pharm. Sci. 2014, 56, 16–27. [Google Scholar] [CrossRef]
- Hou, H.; Qu, B.; Su, C.; Hou, G.; Gao, F. Design, Synthesis and Anti-Lung Cancer Evaluation of 1, 2, 3-Triazole Tethered Dihydroartemisinin-Isatin Hybrids. Front. Pharmacol. 2021, 12, 801580. [Google Scholar] [CrossRef]
- Dong, M.; Zheng, G.; Gao, F.; Li, M.; Zhong, C. Three-Carbon Linked Dihydroartemisinin-Isatin Hybrids: Design, Synthesis and Their Antiproliferative Anticancer Activity. Front. Pharmacol. 2022, 13, 834317. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, H.; Xu, J.; Gao, F.; Cao, W. The Anti-Breast Cancer Activity of Dihydroartemisinin-5-methylisatin Hybrids Tethered via Different Carbon Spacers. Molecules 2022, 27, 7994. [Google Scholar] [CrossRef]
- Ding, F.; Chen, X.; Cao, W.; Dong, T.; Wang, P. The anti-breast cancer potential of dihydroartemisinin-isatin hybrids with hydrogen bond donors at C-3 position of isatin moiety. Fitoterapia 2023, 165, 105426. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, X.; Tang, M.; Liu, X.; Deng, J.; Zhou, W.; Xu, Z. Design, synthesis and anti-breast cancer properties of butyric ester tethered dihydroartemisinin-isatin hybrids. Med. Chem. Res. 2023, 32, 705–712. [Google Scholar] [CrossRef]
- Liu, S.; Wang, S.; Xu, D.; Pan, B.; Chen, L.; Zhao, S.; Xu, Z.; Zhou, W. Novel ester tethered dihydroartemisinin-3-(oxime/thiosemicarbazide)isatin hybrids as potential anti-breast cancer agents: Synthesis, in vitro cytotoxicity and structure–activity relationship. Drug Dev. Res. 2023, 2040, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, A.; Çapcı, A.; Buchfelder, M.; Tsogoeva, S.B.; Savaskan, N. Chemical hybridization of sulfasalazine and dihydroartemisinin promotes brain tumor cell death. Sci. Rep. 2021, 11, 20766. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Wang, Q.-H.; Molina-Molina, E.; Baccetto, R.L.; Calamita, G.; Palmieri, V.O.; Portincasa, P. Bile Acid and Cancer: Direct and environmental-dependent effects. Ann. Hepatol. 2017, 16, s87–s105. [Google Scholar] [CrossRef]
- Goossens, J.-F.; Bailly, C. Ursodeoxycholic acid and cancer: From chemoprevention to chemotherapy. Pharmacol. Ther. 2019, 203, 107396. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Paganetto, G.; Pavan, B.; Fogagnolo, M.; Medici, A.; Beggiato, S.; Perrone, D. Zidovudine and Ursodeoxycholic Acid Conjugation: Design of a New Prodrug Potentially Able To Bypass the Active Efflux Transport Systems of the Central Nervous System. Mol. Pharm. 2012, 9, 957–968. [Google Scholar] [CrossRef]
- Faustino, C.; Serafim, C.; Rijo, P.; Reis, C.P. Bile acids and bile acid derivatives: Use in drug delivery systems and as therapeutic agents. Expert Opin. Drug Deliv. 2016, 13, 1133–1148. [Google Scholar] [CrossRef]
- Pavlovic, N.; Golocorbin-Kon, S.; Danic, M.; Stanimirov, B.; Al-Salami, H.; Stankov, K.; Mikov, M. Bile Acids and Their Derivatives as Potential Modifiers of Drug Release and Pharmacokinetic. Profiles Front. Pharmacol. 2018, 9, 1283. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, S.J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Luo, Z.; Xiang, T.; Wang, K.; Li, J.; Wang, P. Dihydroartemisinin Induces Endoplasmic Reticulum Stress-Mediated Apoptosis in HepG2 Human Hepatoma Cells. Tumori J. 2011, 97, 771–780. [Google Scholar] [CrossRef]
- Im, E.; Yeo, C.; Lee, H.J.; Lee, E.O. Dihydroartemisinin induced caspase-dependent apoptosis through inhibiting the specificity protein 1 pathway in hepatocellular carcinoma SK-Hep-1 cells. Life Sci. 2018, 192, 286–292. [Google Scholar] [CrossRef]
- Qin, G.; Zhao, C.B.; Zhang, L.; Liu, H.; Quan, Y.; Chai, L.; Wu, S.; Wang, X.; Chen, T. Dihydroartemisinin induces apoptosis preferentially via a Bim-mediated intrinsic pathway in hepatocarcinoma cells. Apoptosis 2015, 20, 1072–1086. [Google Scholar] [CrossRef]
- Afzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef] [PubMed]
- Musiol, R. An overview of quinoline as a privileged scaffold in cancer drug discovery. Expert Opin. Drug Discov. 2017, 12, 583–597. [Google Scholar] [CrossRef]
- Nanda, A.K.; Ganguli, S.; Chakraborty, R. Antibacterial activity of some 3-(arylideneamino)-2-phenylquinazoline-4(3H)-ones: Synthesis and preliminary QSAR studies. Molecules 2007, 12, 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.P.; Wang, X.Y.; Song, B.A.; Yang, S.; Yan, K.; Xu, G.F.; Bhadury, P.S.; Liu, F.; Jin, L.H.; Hu, D.Y. Synthesis, antiviral and antifungal bioactivity of 2-cyano-acrylate derivatives containing phosphonyl moieties. Molecules 2007, 12, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, Y.; Wang, M.; Liu, Q.; Lei, X.; Wu, M.; Guo, S.; Yi, D.; Li, Q.; Ma, L.; et al. Quinoline and Quinazoline Derivatives Inhibit Viral RNA Synthesis by SARS-CoV-2 RdRp. ACS Infect. Dis. 2021, 7, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, Z.; Jin, L.; Song, B.; Liu, G.; Chen, J.; Chen, Z.; Hu, D.; Xue, W.; Xu, R. Synthesis and bioactivity of 4-alkyl(aryl)thioquinazoline derivatives. Bioorg. Med. Chem. Lett. 2007, 17, 2193–2196. [Google Scholar] [CrossRef]
- Chen, Y.; Jia, Y.; Song, W.; Zhang, L. Therapeutic Potential of Nitrogen Mustard Based Hybrid Molecules. Front. Pharmacol. 2018, 9, 1453. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Yu, E.; Yue, H.; Li, Q. Enhanced liver targeting of camptothecin via conjugation with deoxycholic acid. Molecules 2019, 24, 1179. [Google Scholar] [CrossRef]
- Liu, Y.-Q.; Li, W.-Q.; Morris-Natschke, S.L.; Qian, K.; Yang, L.; Zhu, G.-X.; Wu, X.-B.; Chen, A.-L.; Zhang, S.-Y.; Song, Z.-L.; et al. Perspectives on Biologically Active Camptothecin Derivatives. Med. Res. Rev. 2015, 35, 753–789. [Google Scholar] [CrossRef]
- Li, X.; Zhao, T.; Cheng, D.; Chu, C.; Tong, S.; Yan, J.; Li, Q.Y. Synthesis and biological activity of some bile acid-based camptothecin analogues. Molecules 2014, 19, 3761–3776. [Google Scholar] [CrossRef]
- Tan, X.; Zhou, H.; Wang, C.; Liu, X.; Yang, X.; Liu, W. GSH-responsive camptothecin prodrug-based hybrid micellar nanoparticles enable antitumor chemo-immunotherapy by PD-L1 knockdown. Nano Res. 2023, 16, 834–848. [Google Scholar] [CrossRef]
- Tabassum, S.; Norhayati Rosli, N.; Ichwan, S.J.A.; Mishra, P. Thymoquinone and its pharmacological perspective: A review. Pharmacol. Res.-Mod. Chin. Med. 2021, 1, 100020. [Google Scholar] [CrossRef]
- Sarkar, C.; Jamaddar, S.; Islam, T.; Mondal, M.; Islam, M.T.; Mubarak, M.S. Therapeutic perspectives of the black cumin component thymoquinone: A review. Food Funct. 2021, 12, 6167–6213. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, T.; Lungu, C.N. Anticancer Activity of Natural and Synthetic Chalcones. Int. J. Mol. Sci. 2021, 22, 11306. [Google Scholar] [CrossRef]
- Bahare, S.; Quispe, C.; Chamkhi, I.; El Omari, N.; Balahbib, A.; Sharifi-Rad, J.; Bouyahya, A.; Akram, A.; Iqbal, A.; Docea1, O.; et al. Pharmacological Properties of Chalcones: A Review of Preclinical Including Molecular Mechanisms and Clinical Evidence. Front. Pharmacol. 2021, 11, 592654. [Google Scholar] [CrossRef]
- Ouyang, Y.; Li, J.; Chen, X.; Fu, X.; Sun, S.; Wu, Q. Chalcone Derivatives: Role in Anticancer Therapy. Biomolecules 2021, 11, 894. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Gaur, R.; Pathania, A.S.; Malik, F.A.; Bhakuni, R.S.; Verma, R.K. Synthesis of a series of novel dihydroartemisinin monomers and dimers containing chalcone as a linker and their anticancer activity. Eur. J. Med. Chem. 2016, 122, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, F.; Pinna, C.; Dallavalle, S.; Tamborini, L.; Pinto, A. An Overview of Coumarin as a Versatile and Readily Accessible Scaffold with Broad-Ranging Biological Activities. Int. J. Mol. Sci. 2020, 21, 4618. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; Cruz-Martins, N.; Lόpez-Jornet, P.; Pons-Fuster Lopez, E.; Harun, N.; Yeskaliyeva, B.; Beyatli, A.; Sytar, O.; Shaheen, S.; Sharopov, F.; et al. Natural Coumarins: Exploring the Pharmacological Complexity and Underlying Molecular Mechanisms. Oxid. Med. Cell. Longev. 2021, 2021, 6492346. [Google Scholar] [CrossRef] [PubMed]
- Akkol, E.K.; Gen, Y.; Büsra Karpuz, B.; Sobarzo-Sanchez, E.; Capasso, R. Coumarins and Coumarin-Related Compounds in Pharmacotherapy of Cancer. Cancers 2020, 12, 1959. [Google Scholar] [CrossRef]
- Lu, W.J.; Desta, Z.; Flockhart, D.A. Tamoxifen metabolites as active inhibitors of aromatase in the treatment of breast cancer. Breast Cancer Res. Treat. 2012, 131, 473–481. [Google Scholar] [CrossRef]
- Ekholm, M.; Bendahl, P.O.; Fernö, M.; Nordenskjöld, B.; Stål, O.; Rydén, L. Effects of adjuvant tamoxifen over three decades on breast cancer–free and distant recurrence–free interval among premenopausal women with oestrogen receptor–positive breast cancer randomised in the Swedish SBII:2pre trial. Eur. J. Cancer 2019, 110, 53–61. [Google Scholar] [CrossRef]
- Dey, P.; Kundu, A.; Chakraborty, H.J.; Kar, B.; Choi, W.S.; Lee, B.M.; Bhakta, T.; Atanasov, A.G.; Kim, H.S. Therapeutic value of steroidal alkaloids in cancer: Current trends and future perspectives. Int. J. Cancer 2019, 145, 1731–1744. [Google Scholar] [CrossRef]
- Potter, B.V.L. Steroid sulphatase inhibition via aryl sulphamates: Clinical progress, mechanism and future prospects. J. Mol. Endocrinol. 2018, 61, T233–T252. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.D. Natural cinnamic acids, synthetic derivatives and hybrids with antimicrobial activity. Molecules 2014, 19, 19292–19349. [Google Scholar] [CrossRef]
- Liu, L.; Hudgins, W.R.; Shack, S.; Yin, M.Q.; Samid, D. Cinnamic acid: A natural product with potential use in cancer intervention. Int. J. Cancer 1995, 62, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Gensicka-Kowalewska, M.; Cholewiński, G.; Dzierzbicka, K. Recent developments in the synthesis and biological activity of acridine/acridone analogues. RSC Adv. 2017, 7, 15776–15804. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, X. Current Scenario of Acridine Hybrids with Anticancer Potential. Curr. Top. Med. Chem. 2021, 21, 1773–1786. [Google Scholar] [CrossRef] [PubMed]
- Ferraz de Paiva, R.E.; Vieira, E.G.; Rodrigues da Silva, D.; Wegermann, C.A.; Costa Ferreira, A.M. Anticancer Compounds Based on Isatin-Derivatives: Strategies to Ameliorate Selectivity and Efficiency. Front. Mol. Biosci. 2021, 7, 627272. [Google Scholar] [CrossRef]
- Zhang, M.Z.; Chen, Q.; Yang, G.F. A review on recent developments of indole-containing antiviral agents. Eur. J. Med. Chem. 2015, 89, 421–441. [Google Scholar] [CrossRef]
- Bharathi Dileepan, A.G.; Daniel Prakash, T.; Ganesh Kumar, A.; Shameela Rajam, P.; Violet Dhayabaran, V.; Rajaram, R. Isatin based macrocyclic Schiff base ligands as novel candidates for antimicrobial and antioxidant drug design: In vitro DNA binding and biological studies. J. Photochem. Photobiol. B Biol. 2018, 183, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Guo, H. Isatin derivatives and their anti-bacterial activities. Eur. J. Med. Chem. 2019, 164, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A. Oxindole inhibitors of cyclin-dependent kinase as anti-tumor agents. In Inhibitors of Cyclin-Dependent Kinases as Anti-Tumor Agents; Smith, P.J., Yue, E.W., Eds.; CRC Press: Boca Raton, FL, USA; Taylor& Francis Group: Abingdon, UK, 2007; pp. 265–281. [Google Scholar]
- Prakash, C.R.; Theivendren, P.; Raja, S. Indolin-2-Ones in Clinical Trials as Potential Kinase Inhibitors: A Review. Pharmacol. Pharm. 2012, 3, 62–71. [Google Scholar] [CrossRef]
- Roth, G.J.; Binder, R.; Colbatzky, F.; Dallinger, C.; Schlenker-Herceg, R.; Hilberg, F.; Wollin, S.L.; Kaiser, R. Nintedanib: From discovery to the clinic. J. Med. Chem. 2015, 58, 1053–1063. [Google Scholar] [CrossRef]
- Izzedine, H.; Buhaescu, I.; Rixe, O.; Deray, G. Sunitinib malate. Cancer Chemother. Pharmacol. 2007, 60, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewé, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; Van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef] [PubMed]
- Harbord, M.; Eliakim, R.; Bettenworth, D.; Karmiris, K.; Katsanos, K.; Kopylov, U.; Kucharzik, T.; Molnár, T.; Raine, T.; Sebastian, S.; et al. Third European evidence-based consensus on diagnosis and management of ulcerative colitis. Part 2: Current management. J. Crohn’s Colitis 2017, 11, 769–784. [Google Scholar] [CrossRef]
- Sehm, T.; Fan, Z.; Ghoochani, A.; Rauh, M.; Engelhorn, T.; Minakaki, G.; Dörfler, A.; Klucken, J.; Buchfelder, M.; Eyüpoglu, I.Y.; et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget 2016, 7, 36021–36033. [Google Scholar] [CrossRef]
- Sleire, L.; Skeie, B.S.; Netland, I.A.; Førde, H.E.; Dodoo, E.; Selheim, F.; Leiss, L.; Heggdal, J.I.; Pedersen, P.H.; Wang, J.; et al. Drug repurposing: Sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene 2015, 34, 5951–5959. [Google Scholar] [CrossRef]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x-c cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef]
- Lo, M.; Ling, V.; Low, C.; Wang, Y.Z.; Gout, P.W. Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr. Oncol. 2010, 17, 9–16. [Google Scholar] [CrossRef]
- Nagane, M.; Kanai, E.; Shibata, Y.; Shimizu, T.; Yoshioka, C.; Maruo, T.; Yamashita, T. Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma. PLoS ONE 2018, 13, e0195151. [Google Scholar] [CrossRef]
- Awasthi, S.; Sharma, R.; Singhal, S.S.; Herzog, N.K.; Chaubey, M.; Awasthi, Y.C. Modulation of cisplatin cytotoxicity by sulphasalazine. Br. J. Cancer 1994, 70, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.Z.; Chen, G.; Wang, P.; Lu, W.H.; Zhu, C.F.; Song, M.; Yang, J.; Wen, S.; Xu, R.H.; Hu, Y.; et al. Xc- inhibitor sulfasalazine sensitizes colorectal cancer to cisplatin by a GSH-dependent mechanism. Cancer Lett. 2015, 368, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Narang, V.S.; Pauletti, G.M.; Gout, P.W.; Buckley, D.J.; Buckley, A.R. Sulfasalazine-induced reduction of glutathione levels in breast cancer cells: Enhancement of growth-inhibitory activity of doxorubicin. Chemotherapy 2007, 53, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Müerköster, S.; Arlt, A.; Witt, M.; Gehrz, A.; Haye, S.; March, C.; Grohmann, F.; Wegehenkel, K.; Kalthoff, H.; Fölsch, U.R.; et al. Usage of the NF-κB inhibitor sulfasalazine as sensitizing agent in combined chemotherapy of pancreatic cancer. Int. J. Cancer 2003, 104, 469–476. [Google Scholar] [CrossRef] [PubMed]
































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrid | Hybridization Chemistry | Cancer Cell Type | Ref. | Comments |
|---|---|---|---|---|
| Artemisinin–bile acid | Ester | Leukemia, B-cell lymphomas and hepatocellular carcinoma | [44,45,53] |  Cytotoxic activity Cytotoxic activity Apoptotic cell death + Safety profile |
| Triazole click | Leukemia and hepatocellular carcinoma | [44,46] | ||
| Ether | Lung, ovarian, prostatic, cervical and renal cell carcinoma | [54] | Cytotoxic activity In vivo tumor growth In vivo tumor growthImmunotherapy effects | |
| Amide | Sensitive and multidrug-resistant leukemia | [55] | Cytotoxic activity | |
| Artemisinin–quinoline | Amide | Leukemia, oral squamous carcinoma, lung and colon | [56,57,58] | Cytotoxic activity + Safety profile |
| Hydrazone | Breast | [59,60] | More potent than reference drugs Apoptotic cell death | |
| Diamine/piperazine | Renal, melanoma and breast | [61,62] | More potent than reference drugs | |
| Artemisinin–quinazoline | Amide | Sensitive and multidrug-resistant leukemia | [63] | Cytotoxic activity |
| Ether/ester/chiral amide | Colon, melanoma | [64] | Cytotoxic activityIn vivo tumor growth | |
| Artemisinin–nitrogen mustard | Ester | Leukemia | [65] | More potent than reference drugs Apoptotic cell death |
| Amide | Ovarian | [66] | Cytotoxic activity More potent than reference drugs Apoptotic cell death Migration inhibition | |
| Artemisinin–tyrosol | Ester | Cervical, melanoma | [67,68] | Cytotoxic activity + Safety profile |
| Artemisinin–camptothecin | Ester | Melanoma | [69] | More potent than reference drugs |
| Artemisinin–thymoquinone | Ester/ether | Colon, sensitive and multidrug-resistant leukemia | [70,71] | Cytotoxic activity + Safety profile |
| Ferrocene–ester | Sensitive and multidrug-resistant leukemia | [72] | Cytotoxic activity | |
| Artemisinin–chalcone | Ether | Leukemia, pancreatic, prostate, hepatocellular carcinoma, colon, lung, breast and cervical | [69,73,74] | Cytotoxic activity Apoptotic cell death |
| Ester | Renal, melanoma and breast | [75] | + Safety profile | |
| Artemisinin–coumarin | Triazole click/ether | Colon and breast | [76,77,78] | Cytotoxic activity Apoptotic and ferroptotic cell death Migration inhibition + Safety profile |
| Artemisinin–tamoxifen | Ester/amide | Prostate and breast | [79] | Cytotoxic activity |
| Artemisinin–steroid | Ester | Prostate, breast and cervical | [79,80] | Cytotoxic activity More potent than reference drugs |
| Amide/triazole click | Breast and cervical | [80] | Cytotoxic activity More potent than reference drugs | |
| Artemisinin–cinnamic acid | Ester | Prostate, gastric, lung and melanoma | [81] | More potent than reference drugs + Safety profile Apoptotic cell death |
| Artemisinin–acridine | Amide | Leukemia, colon and breast | [82] | Cytotoxic activity Apoptotic cell death |
| Ether | Cervical | [83] | More potent than reference drugs | |
| Artemisinin–isatin | Triazole click | Lung | [84] | Cytotoxic activity More potent than reference drugs |
| Ether | Lung, breast | [85,86,87] | Cytotoxic activity + Safety profile | |
| Ester | Breast | [88,89] | Cytotoxic activity | |
| Artemisinin–sulfasalazine | Ester | Glioma | [90] | Apoptotic cell death Migration inhibition |
symbol marks the cytotoxic effect of hybrids with respect to artemisinins. The symbol marks the tumor suppressor effect. The + symbol marks the safety profile of hybrids in healthy cells.Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchesi, E.; Perrone, D.; Navacchia, M.L. Molecular Hybridization as a Strategy for Developing Artemisinin-Derived Anticancer Candidates. Pharmaceutics 2023, 15, 2185. https://doi.org/10.3390/pharmaceutics15092185
Marchesi E, Perrone D, Navacchia ML. Molecular Hybridization as a Strategy for Developing Artemisinin-Derived Anticancer Candidates. Pharmaceutics. 2023; 15(9):2185. https://doi.org/10.3390/pharmaceutics15092185
Chicago/Turabian StyleMarchesi, Elena, Daniela Perrone, and Maria Luisa Navacchia. 2023. "Molecular Hybridization as a Strategy for Developing Artemisinin-Derived Anticancer Candidates" Pharmaceutics 15, no. 9: 2185. https://doi.org/10.3390/pharmaceutics15092185
APA StyleMarchesi, E., Perrone, D., & Navacchia, M. L. (2023). Molecular Hybridization as a Strategy for Developing Artemisinin-Derived Anticancer Candidates. Pharmaceutics, 15(9), 2185. https://doi.org/10.3390/pharmaceutics15092185