

Oral Absorption of Middle-to-Large Molecules and Its Improvement, with a Focus on New Modality Drugs

Abstract
:1. Introduction
2. Technology to Improve Oral Absorption of Middle-to-Large Molecules
2.1. Chemical Modification to Acquire Membrane Permeability and Chameleonic Property
2.2. Utilization of Absorption Enhancers
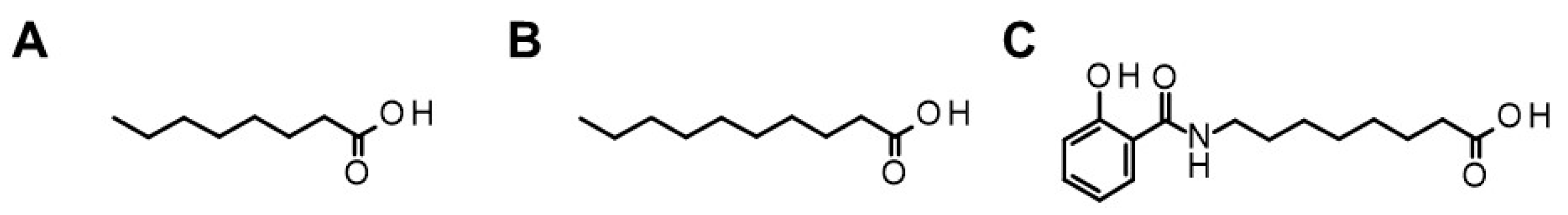
2.2.1. Fatty Acids (C8 and C10)
- Epocelin® suppositories (antibiotics prescribed in Japan [90]) contain C10 to enhance the rectal absorption of ceftizoxime sodium in humans.
- Krug et al. reported that C10 improved the rectal absorption of ampicillin in humans [91].
- Tuvia et al. reported that C8 enhanced the oral absorption of octreotide in humans [92].
- Leonard et al. reported that the oral administration of C10 at 1000 mg/kg for 7 days did not cause any side effects in dogs [99].
- Raoof et al. reported that the oral administration of C10 at 990 mg/body (as three ISIS104838-containing tablets) for 7 days was safe in dogs [100].
- Tuvia et al. reported that the oral administration of C8 (as octreotide-containing oily suspension) for 9 months was tolerated with minor toxicity in monkeys [96].
- Halberg et al. reported that the oral administration of C10 at 550 mg/body (as I338 tablets) for 8 weeks was well tolerated in humans [40].
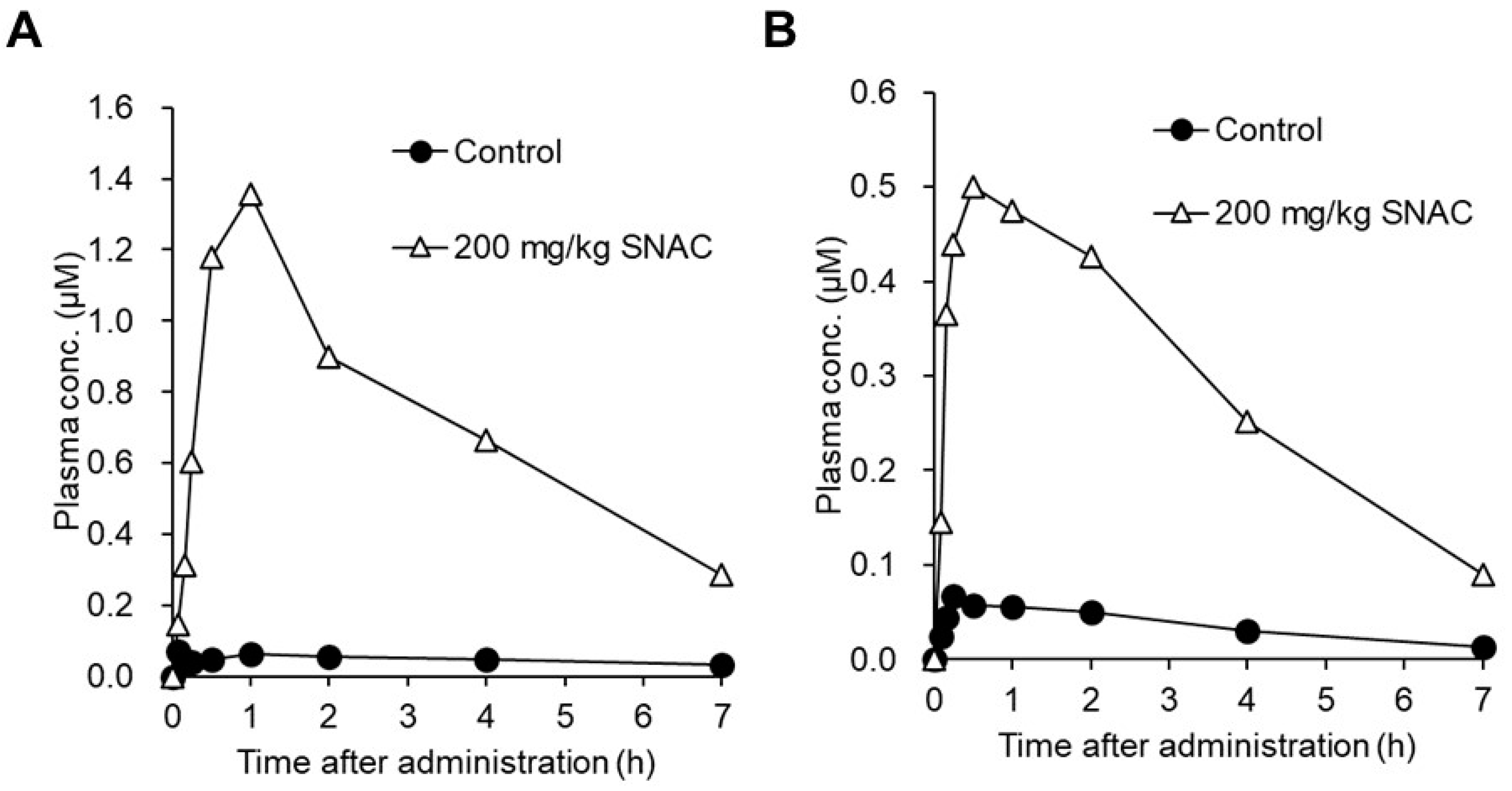
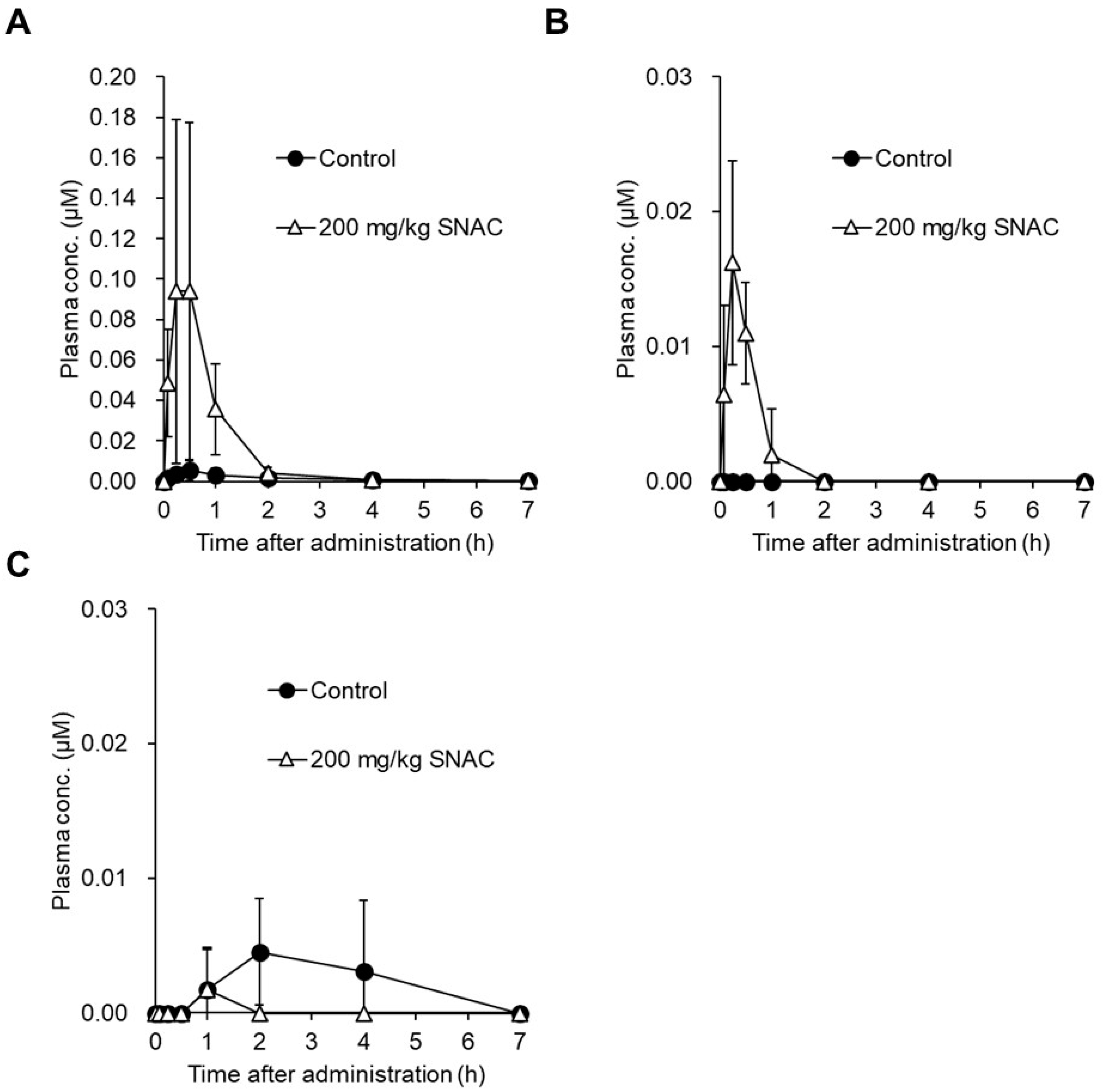
2.2.2. SNAC
- Is SNAC ineffective against intestinal permeation?
- Are other permeation enhancers (e.g., C8 and C10) effective against gastric permeation?
- Why do SNAC and other permeation enhancers have different sites of action, the stomach and the intestine, respectively?
2.3. Utilization of Special Formulations with an Absorption-Enhancing Effect
3. Recent Challenges of Orally Bioavailable Middle-to-Large Molecules
3.1. Application of Chemical Modification
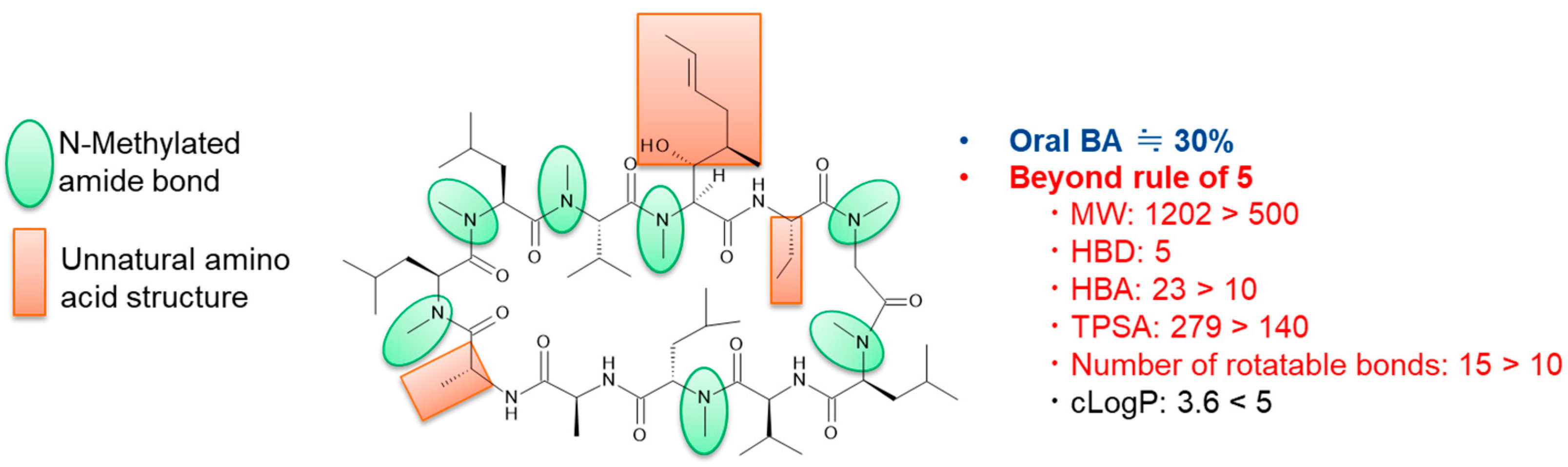
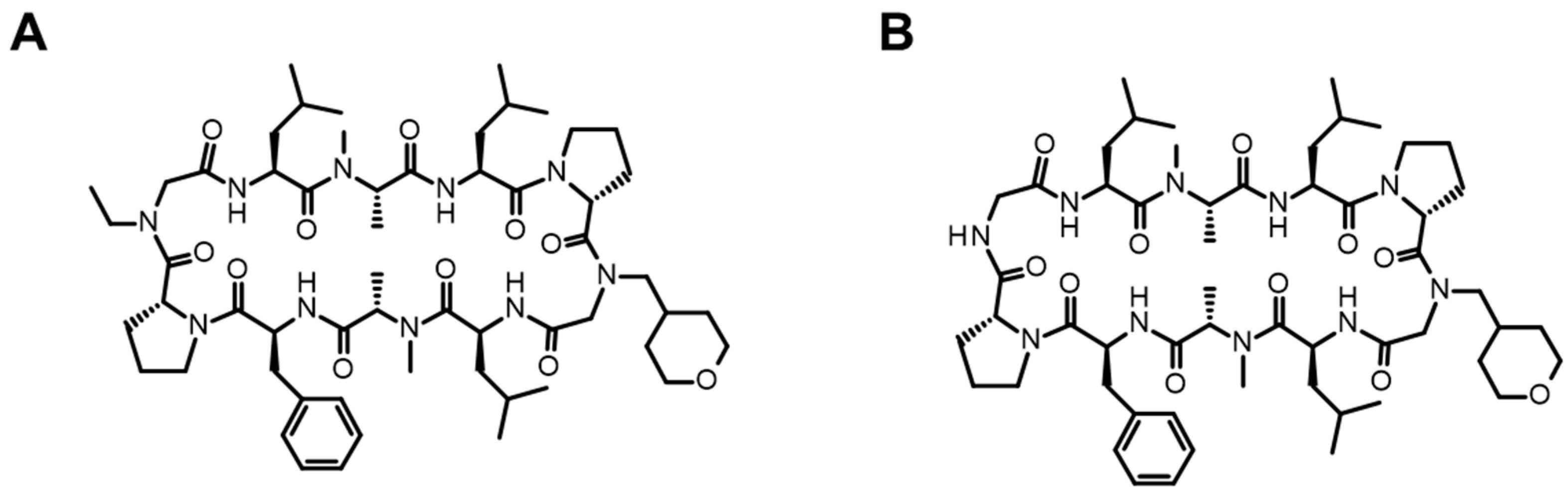
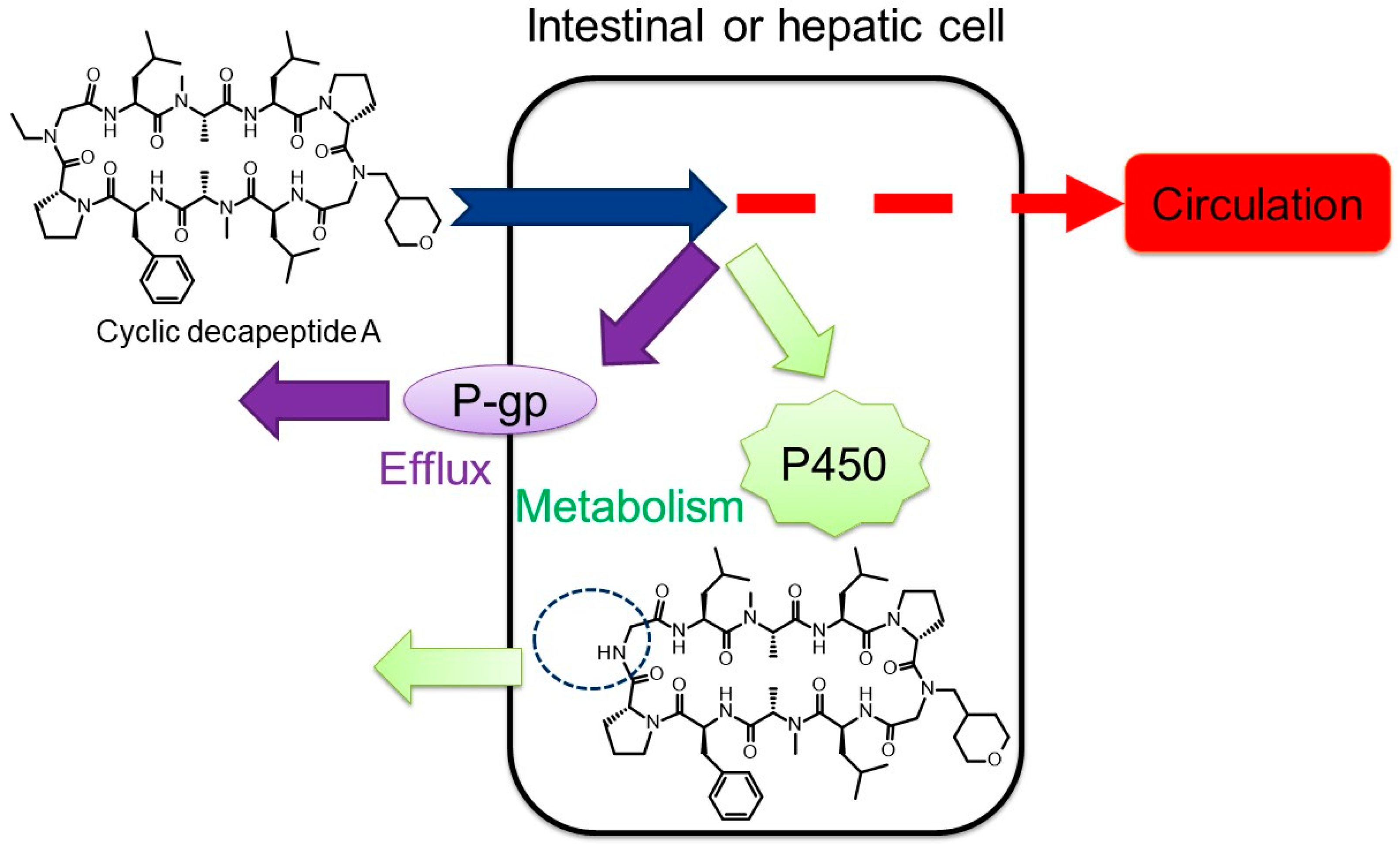
3.1.1. Cyclic Peptides
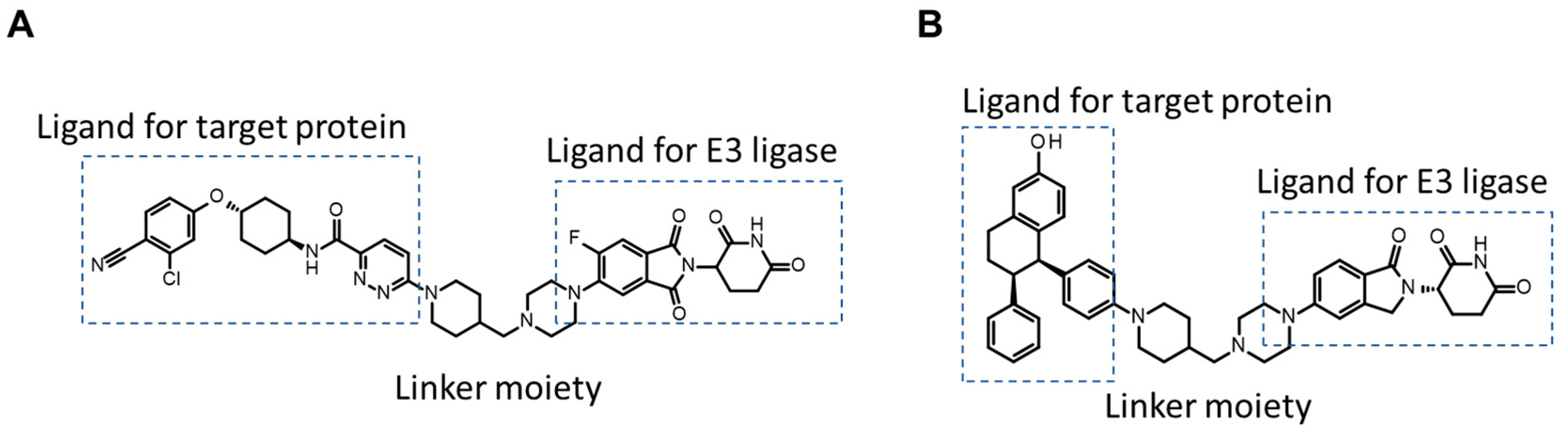
3.1.2. TPD
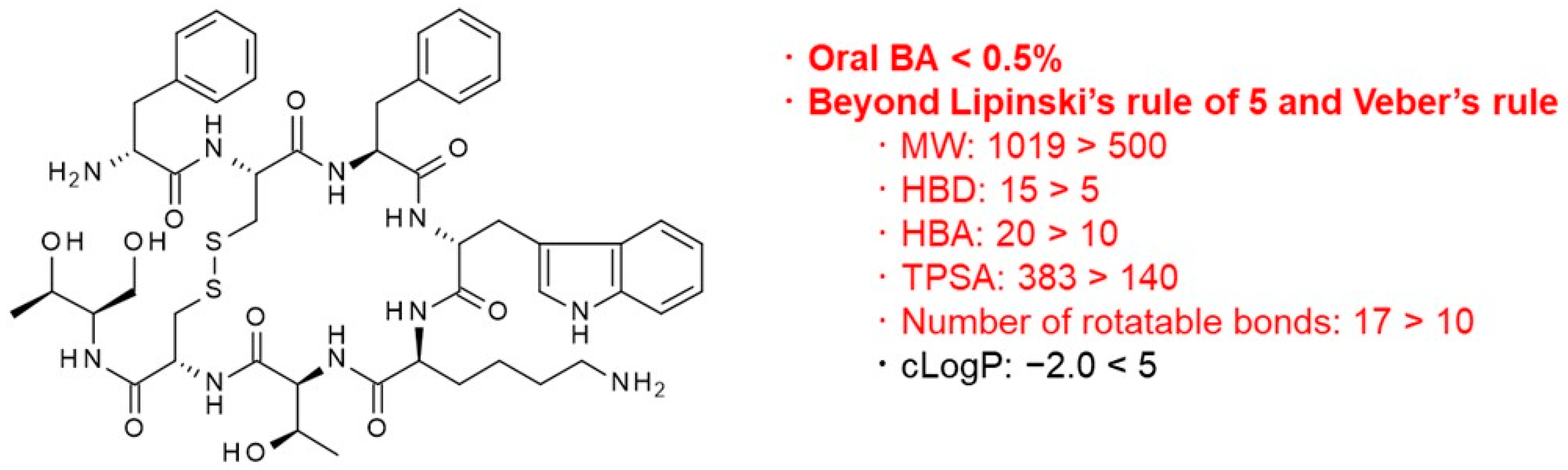
3.1.3. Other Middle-to-Large Molecules beyond the Rule of Five
3.2. Application of Absorption Enhancers and/or Special Formulations with an Absorption-Enhancing Effect
3.2.1. Peptides
3.2.2. Oligonucleotides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene (Name of Oligonucleotide) | MW | Type of Oligonucleotide | Formulation/Modification for Oral Delivery | Species | Bioavailability | Bioanalytical Method | Reference |
|---|---|---|---|---|---|---|---|
| TNF-α (ISIS104838) | ca. 7300 | PS-ASO 2′-MOE | C10 | Pig | IJ relative to IV: 1.7–2.8% in plasma | HPLC/UV | [150] |
| Dog | PO relative to IV: 1.1–1.7% in plasma 1.3–4.3% in tissues | HPLC/UV | [100] | ||||
| Human | PO relative to SC: 7.2–12.0% in plasma | hybridization ELISA | [93] | ||||
| NF-kB | Unknown | ASO, modification unspecified | biodegradable albumin polymer matrix | Rat | PO relative to IV: 70% in plasma | OliGreen fluorescence assay | [163] |
| PCSK9 (AZD8233, ION-863633) | ca. 6900 | PS-ASO GalNAc cET chemistry | C10 | Rat | IJ relative to SC: 5.3% in liver | hybridization ELISA | [162] |
| Dog | PO relative to SC: 1.3–1.8% in plasma 7.0–7.4% in liver 1.2–1.6% in kidney | ||||||
| TNF-α | Unknown | siRNA | fluorinated nanocapsules | Mouse | PO relative to IV: 20.4% in plasma | PCR-based method | [168] |
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| API | active pharmaceutical ingredient |
| ASO | antisense oligonucleotide |
| AUC | area under the curve |
| BA | bioavailability |
| C8 | caprylic acid, octanoic acid |
| C10 | capric acid, decanoic acid |
| COG | cost of goods |
| cEt | constrained ethyl |
| CPPs | cell-penetrating peptides |
| cLogD | calculated octanol-water distribution coefficient |
| cLogP | calculated octanol-water partition coefficient |
| EDTA | ethylenediaminetetraacetic acid |
| EPSA | experimental polar surface area |
| FDA | United States Food and Drug Administration |
| GalNAc | N-acetylgalactosamine |
| GRAS | generally recognized as safe |
| HBAs | number of hydrogen bond acceptors |
| HBDs | number of hydrogen bond donors |
| IJ | intrajejunal |
| IV | intravenous |
| MW | molecular weight |
| MPSA | molecular (3D) polar surface area in nonpolar environment |
| Ms | microsomes |
| NAr | number of aromatic rings |
| NMR | nuclear magnetic resonance |
| LD50 | median lethal dose |
| LPE | lipophilic permeability efficiency |
| NF-kB | nuclear factor kappa B |
| NOAEL | no-observed-adverse-effect level |
| P450 | cytochrome P450 |
| P-gp | P-glycoprotein |
| PO | per oral |
| TPSA | topological polar surface area |
| SC | subcutaneous |
| siRNA | small interfering RNA |
| SNAC | salcaprozate sodium |
| TEER | transepithelial electrical resistance |
| TNF-α | tumor necrosis factor α |
| TPDs | target protein degraders |
References
- Blanco, M.J.; Gardinier, K.M. New Chemical Modalities and Strategic Thinking in Early Drug Discovery. ACS Med. Chem. Lett. 2020, 11, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.J.; Gardinier, K.M.; Namchuk, M.N. Advancing New Chemical Modalities into Clinical Studies. ACS Med. Chem. Lett. 2022, 13, 1691–1698. [Google Scholar] [CrossRef]
- Kansy, M.; Caron, G. New therapeutic modalities in drug discovery and development: Insights & opportunities. ADMET DMPK 2021, 9, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Pinto, P.C. The Potential Impact of New Drug and Therapeutic Modalities on Drug Resistance to Renal Cell Carcinoma. Anticancer Res. 2023, 43, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Roth, A. New Drug Modalities Demand a Refined Preclinical Safety Assessment: A Call for Patient-Relevant Tissue Models. Toxicol. Sci. 2022, 189, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Aube, J.; Lindsley, C.W.; Muller, C.E. Virtual Special Issue: New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science. ACS Med. Chem. Lett. 2023, 14, 867. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef]
- Naylor, M.R.; Bockus, A.T.; Blanco, M.J.; Lokey, R.S. Cyclic peptide natural products chart the frontier of oral bioavailability in the pursuit of undruggable targets. Curr. Opin. Chem. Biol. 2017, 38, 141–147. [Google Scholar] [CrossRef]
- Villar, E.A.; Beglov, D.; Chennamadhavuni, S.; Porco, J.A., Jr.; Kozakov, D.; Vajda, S.; Whitty, A. How proteins bind macrocycles. Nat. Chem. Biol. 2014, 10, 723–731. [Google Scholar] [CrossRef]
- Doak, B.C.; Zheng, J.; Dobritzsch, D.; Kihlberg, J. How Beyond Rule of 5 Drugs and Clinical Candidates Bind to Their Targets. J. Med. Chem. 2016, 59, 2312–2327. [Google Scholar] [CrossRef]
- Chen, J.; Yuan, Z.; Tu, Y.; Hu, W.; Xie, C.; Ye, L. Experimental and computational models to investigate intestinal drug permeability and metabolism. Xenobiotica 2023, 53, 25–45. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Chagas, C.M.; Moss, S.; Alisaraie, L. Drug metabolites and their effects on the development of adverse reactions: Revisiting Lipinski’s Rule of Five. Int. J. Pharm. 2018, 549, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Rybelsus Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/213051s000lbl.pdf (accessed on 18 August 2023).
- Mycapssa Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208232s000lbl.pdf (accessed on 18 August 2023).
- Overgaard, R.V.; Navarria, A.; Ingwersen, S.H.; Baekdal, T.A.; Kildemoes, R.J. Clinical Pharmacokinetics of Oral Semaglutide: Analyses of Data from Clinical Pharmacology Trials. Clin. Pharmacokinet. 2021, 60, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Pieber, T.R.; Hartoft-Nielsen, M.L.; Hansen, O.K.H.; Jabbour, S.; Rosenstock, J. Effect of Oral Semaglutide Compared With Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Clinical Trial. JAMA 2017, 318, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Brayden, D.J.; Maher, S. Transient Permeation Enhancer(R) (TPE(R)) technology for oral delivery of octreotide: A technological evaluation. Expert Opin. Drug Deliv. 2021, 18, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Brayden, D.J. Formulation strategies to improve the efficacy of intestinal permeation enhancers. Adv. Drug Deliv. Rev. 2021, 177, 113925. [Google Scholar] [CrossRef]
- Zizzari, A.T.; Pliatsika, D.; Gall, F.M.; Fischer, T.; Riedl, R. New perspectives in oral peptide delivery. Drug Discov. Today 2021, 26, 1097–1105. [Google Scholar] [CrossRef]
- Chen, G.; Kang, W.; Li, W.; Chen, S.; Gao, Y. Oral delivery of protein and peptide drugs: From non-specific formulation approaches to intestinal cell targeting strategies. Theranostics 2022, 12, 1419–1439. [Google Scholar] [CrossRef]
- Maher, S.; Geoghegan, C.; Brayden, D.J. Intestinal permeation enhancers to improve oral bioavailability of macromolecules: Reasons for low efficacy in humans. Expert Opin. Drug Deliv. 2021, 18, 273–300. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Park, E.J.; Na, D.H. Gastrointestinal Permeation Enhancers for the Development of Oral Peptide Pharmaceuticals. Pharmaceuticals 2022, 15, 1585. [Google Scholar] [CrossRef] [PubMed]
- Twarog, C.; Fattah, S.; Heade, J.; Maher, S.; Fattal, E.; Brayden, D.J. Intestinal Permeation Enhancers for Oral Delivery of Macromolecules: A Comparison between Salcaprozate Sodium (SNAC) and Sodium Caprate (C(10)). Pharmaceutics 2019, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, T.A.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.S.; Alonso, M.J.; Brayden, D.J. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106, 223–241. [Google Scholar] [CrossRef]
- Liu, S.; Wen, X.; Zhang, X.; Mao, S. Oral delivery of biomacromolecules by overcoming biological barriers in the gastrointestinal tract: An update. Expert Opin. Drug Deliv. 2023, 20, 1333–1347. [Google Scholar] [CrossRef] [PubMed]
- Spoorthi Shetty, S.; Halagali, P.; Johnson, A.P.; Spandana, K.M.A.; Gangadharappa, H.V. Oral insulin delivery: Barriers, strategies, and formulation approaches: A comprehensive review. Int. J. Biol. Macromol. 2023, 242, 125114. [Google Scholar] [CrossRef] [PubMed]
- Kommineni, N.; Sainaga Jyothi, V.G.S.; Butreddy, A.; Raju, S.; Shapira, T.; Khan, W.; Angsantikul, P.; Domb, A.J. SNAC for Enhanced Oral Bioavailability: An Updated Review. Pharm. Res. 2023, 40, 633–650. [Google Scholar] [CrossRef] [PubMed]
- Berg, S.; Edlund, H.; Goundry, W.R.F.; Bergström, C.A.S.; Davies, N.M. Considerations in the developability of peptides for oral administration when formulated together with transient permeation enhancers. Int. J. Pharm. 2022, 628, 122238. [Google Scholar] [CrossRef]
- Verma, S.; Goand, U.K.; Husain, A.; Katekar, R.A.; Garg, R.; Gayen, J.R. Challenges of peptide and protein drug delivery by oral route: Current strategies to improve the bioavailability. Drug Dev. Res. 2021, 82, 927–944. [Google Scholar] [CrossRef]
- Dan, N.; Samanta, K.; Almoazen, H. An Update on Pharmaceutical Strategies for Oral Delivery of Therapeutic Peptides and Proteins in Adults and Pediatrics. Children 2020, 7, 307. [Google Scholar] [CrossRef]
- Yamamoto, A.; Ukai, H.; Morishita, M.; Katsumi, H. Approaches to improve intestinal and transmucosal absorption of peptide and protein drugs. Pharmacol. Ther. 2020, 211, 107537. [Google Scholar] [CrossRef] [PubMed]
- Brayden, D.J.; Hill, T.A.; Fairlie, D.P.; Maher, S.; Mrsny, R.J. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv. Drug Deliv. Rev. 2020, 157, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Muheem, A.; Shakeel, F.; Jahangir, M.A.; Anwar, M.; Mallick, N.; Jain, G.K.; Warsi, M.H.; Ahmad, F.J. A review on the strategies for oral delivery of proteins and peptides and their clinical perspectives. Saudi Pharm. J. 2016, 24, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Fouche, M.; Schafer, M.; Berghausen, J.; Desrayaud, S.; Blatter, M.; Piechon, P.; Dix, I.; Martin Garcia, A.; Roth, H.J. Design and Development of a Cyclic Decapeptide Scaffold with Suitable Properties for Bioavailability and Oral Exposure. ChemMedChem 2016, 11, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Whitty, A.; Zhong, M.; Viarengo, L.; Beglov, D.; Hall, D.R.; Vajda, S. Quantifying the chameleonic properties of macrocycles and other high-molecular-weight drugs. Drug Discov. Today 2016, 21, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Pye, C.R.; Hewitt, W.M.; Schwochert, J.; Haddad, T.D.; Townsend, C.E.; Etienne, L.; Lao, Y.; Limberakis, C.; Furukawa, A.; Mathiowetz, A.M.; et al. Nonclassical Size Dependence of Permeation Defines Bounds for Passive Adsorption of Large Drug Molecules. J. Med. Chem. 2017, 60, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Pollak, R.; Wong, R.L.; Chang, C.T. Cyclosporine bioavailability of Neoral and Sandimmune in white and black de novo renal transplant recipients. Neoral Study Group. Ther. Drug Monit. 1999, 21, 661–663. [Google Scholar] [CrossRef]
- Halberg, I.B.; Lyby, K.; Wassermann, K.; Heise, T.; Zijlstra, E.; Plum-Morschel, L. Efficacy and safety of oral basal insulin versus subcutaneous insulin glargine in type 2 diabetes: A randomised, double-blind, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 179–188. [Google Scholar] [CrossRef]
- Jin, M.; Shimada, T.; Shintani, M.; Yokogawa, K.; Nomura, M.; Miyamoto, K. Long-term levothyroxine treatment decreases the oral bioavailability of cyclosporin A by inducing P-glycoprotein in small intestine. Drug Metab. Pharmacokinet. 2005, 20, 324–330. [Google Scholar] [CrossRef]
- Witek, J.; Keller, B.G.; Blatter, M.; Meissner, A.; Wagner, T.; Riniker, S. Kinetic Models of Cyclosporin A in Polar and Apolar Environments Reveal Multiple Congruent Conformational States. J. Chem. Inf. Model. 2016, 56, 1547–1562. [Google Scholar] [CrossRef]
- Rezai, T.; Yu, B.; Millhauser, G.L.; Jacobson, M.P.; Lokey, R.S. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J. Am. Chem. Soc. 2006, 128, 2510–2511. [Google Scholar] [CrossRef] [PubMed]
- Naylor, M.R.; Ly, A.M.; Handford, M.J.; Ramos, D.P.; Pye, C.R.; Furukawa, A.; Klein, V.G.; Noland, R.P.; Edmondson, Q.; Turmon, A.C.; et al. Lipophilic Permeability Efficiency Reconciles the Opposing Roles of Lipophilicity in Membrane Permeability and Aqueous Solubility. J. Med. Chem. 2018, 61, 11169–11182. [Google Scholar] [CrossRef] [PubMed]
- Goetz, G.H.; Farrell, W.; Shalaeva, M.; Sciabola, S.; Anderson, D.; Yan, J.; Philippe, L.; Shapiro, M.J. High throughput method for the indirect detection of intramolecular hydrogen bonding. J. Med. Chem. 2014, 57, 2920–2929. [Google Scholar] [CrossRef] [PubMed]
- Goetz, G.H.; Philippe, L.; Shapiro, M.J. EPSA: A Novel Supercritical Fluid Chromatography Technique Enabling the Design of Permeable Cyclic Peptides. ACS Med. Chem. Lett. 2014, 5, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.; Northfield, S.E.; Colless, B.; Chaousis, S.; Hamernig, I.; Lohman, R.J.; Nielsen, D.S.; Schroeder, C.I.; Liras, S.; Price, D.A.; et al. Rational design and synthesis of an orally bioavailable peptide guided by NMR amide temperature coefficients. Proc. Natl. Acad. Sci. USA 2014, 111, 17504–17509. [Google Scholar] [CrossRef] [PubMed]
- White, T.R.; Renzelman, C.M.; Rand, A.C.; Rezai, T.; McEwen, C.M.; Gelev, V.M.; Turner, R.A.; Linington, R.G.; Leung, S.S.; Kalgutkar, A.S.; et al. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol. 2011, 7, 810–817. [Google Scholar] [CrossRef]
- Shalaeva, M.; Caron, G.; Abramov, Y.A.; O’Connell, T.N.; Plummer, M.S.; Yalamanchi, G.; Farley, K.A.; Goetz, G.H.; Philippe, L.; Shapiro, M.J. Integrating intramolecular hydrogen bonding (IMHB) considerations in drug discovery using DeltalogP as a tool. J. Med. Chem. 2013, 56, 4870–4879. [Google Scholar] [CrossRef]
- David, L.; Wenlock, M.; Barton, P.; Ritzen, A. Prediction of Chameleonic Efficiency. ChemMedChem 2021, 16, 2669–2685. [Google Scholar] [CrossRef]
- Caron, G.; Vallaro, M.; Ermondi, G. High throughput methods to measure the propensity of compounds to form intramolecular hydrogen bonding. Medchemcomm 2017, 8, 1143–1151. [Google Scholar] [CrossRef]
- Sethio, D.; Poongavanam, V.; Xiong, R.; Tyagi, M.; Duy Vo, D.; Lindh, R.; Kihlberg, J. Simulation Reveals the Chameleonic Behavior of Macrocycles. J. Chem. Inf. Model. 2023, 63, 138–146. [Google Scholar] [CrossRef]
- Ono, S.; Naylor, M.R.; Townsend, C.E.; Okumura, C.; Okada, O.; Lokey, R.S. Conformation and Permeability: Cyclic Hexapeptide Diastereomers. J. Chem. Inf. Model. 2019, 59, 2952–2963. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; Nielsen, H.M. Cell-Penetrating Peptides as Carriers for Oral Delivery of Biopharmaceuticals. Basic Clin. Pharmacol. Toxicol. 2016, 118, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Kondoh, M.; Hanada, T.; Takahashi, A.; Hamakubo, T.; Yagi, K. A claudin-4 modulator enhances the mucosal absorption of a biologically active peptide. Biochem. Pharmacol. 2010, 79, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Tomita, M.; Hayashi, M.; Awazu, S. Absorption-enhancing mechanism of EDTA, caprate, and decanoylcarnitine in Caco-2 cells. J. Pharm. Sci. 1996, 85, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Imai, T.; Ohtake, H.; Azuma, H.; Otagiri, M. Effects of absorption enhancers on the transport of model compounds in Caco-2 cell monolayers: Assessment by confocal laser scanning microscopy. J. Pharm. Sci. 1997, 86, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Ito, S.; Kurogi-Hirayama, M.; Ohtsuki, S. Identification of cyclic peptides for facilitation of transcellular transport of phages across intestinal epithelium in vitro and in vivo. J. Control. Release 2017, 262, 232–238. [Google Scholar] [CrossRef]
- Ito, S.; Torii, Y.; Chikamatsu, S.; Harada, T.; Yamaguchi, S.; Ogata, S.; Sonoda, K.; Wakayama, T.; Masuda, T.; Ohtsuki, S. Oral Coadministration of Zn-Insulin with d-Form Small Intestine-Permeable Cyclic Peptide Enhances Its Blood Glucose-Lowering Effect in Mice. Mol. Pharm. 2021, 18, 1593–1603. [Google Scholar] [CrossRef]
- Yamada, Y.; Onda, T.; Hamada, K.; Kikkawa, Y.; Nomizu, M. Octa-arginine and Octa-lysine Promote Cell Adhesion through Heparan Sulfate Proteoglycans and Integrins. Biol. Pharm. Bull. 2022, 45, 207–212. [Google Scholar] [CrossRef]
- Malkov, D.; Angelo, R.; Wang, H.Z.; Flanders, E.; Tang, H.; Gomez-Orellana, I. Oral delivery of insulin with the eligen technology: Mechanistic studies. Curr. Drug Deliv. 2005, 2, 191–197. [Google Scholar] [CrossRef]
- Kapitza, C.; Zijlstra, E.; Heinemann, L.; Castelli, M.C.; Riley, G.; Heise, T. Oral insulin: A comparison with subcutaneous regular human insulin in patients with type 2 diabetes. Diabetes Care 2010, 33, 1288–1290. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Byrjalsen, I.; Henriksen, K.; Riis, B.J.; Lau, E.M.; Arnold, M.; Christiansen, C. The effect of oral salmon calcitonin delivered with 5-CNAC on bone and cartilage degradation in osteoarthritic patients: A 14-day randomized study. Osteoarthr. Cartil. 2010, 18, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Rybelsus Non-Clinical Reviews. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/213051Orig1s000PharmR.pdf (accessed on 18 August 2023).
- Brayden, D.J.; Gleeson, J.; Walsh, E.G. A head-to-head multi-parametric high content analysis of a series of medium chain fatty acid intestinal permeation enhancers in Caco-2 cells. Eur. J. Pharm. Biopharm. 2014, 88, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Dimitrijevic, D.; Shaw, A.J.; Florence, A.T. Effects of some non-ionic surfactants on transepithelial permeability in Caco-2 cells. J. Pharm. Pharmacol. 2000, 52, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Leung, H.W.; Paustenbach, D.J. Organic acids and bases: Review of toxicological studies. Am. J. Ind. Med. 1990, 18, 717–735. [Google Scholar] [CrossRef] [PubMed]
- Smyth, H.F., Jr.; Carpenter, C.P.; Weil, C.S.; Pozzani, U.C.; Striegel, J.A. Range-finding toxicity data: List VI. Am. Ind. Hyg. Assoc. J. 1962, 23, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Khafagy, E.S.; Hirose, J.; Takeda-Morishita, M. Potential of single cationic amino acid molecule “Arginine” for stimulating oral absorption of insulin. Int. J. Pharm. 2017, 521, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Tamiwa, H.; Miyata, M.; Haruna, Y.; Matsumura, K.; Ogino, H.; Hirano, S.; Higashiyama, K.; Takeda-Morishita, M. Hydrophobic Amino Acid Tryptophan Shows Promise as a Potential Absorption Enhancer for Oral Delivery of Biopharmaceuticals. Pharmaceutics 2018, 10, 182. [Google Scholar] [CrossRef]
- EFSA. Opinion of the Panel on additives and products or substances used in animal feed (FEEDAP) on the safety and efficacy of the product containing L-arginine produced by fermentation from Corynebacterium glutamicum (ATCC-13870) for all animal species. EFSA J. 2007, 5, 473. [Google Scholar]
- Moehn, S.; Pencharz, P.B.; Ball, R.O. Lessons learned regarding symptoms of tryptophan deficiency and excess from animal requirement studies. J. Nutr. 2012, 142, 2231S–2235S. [Google Scholar] [CrossRef]
- Duizer, E.; van der Wulp, C.; Versantvoort, C.H.; Groten, J.P. Absorption enhancement, structural changes in tight junctions and cytotoxicity caused by palmitoyl carnitine in Caco-2 and IEC-18 cells. J. Pharmacol. Exp. Ther. 1998, 287, 395–402. [Google Scholar]
- Salzman, A.L.; Menconi, M.J.; Unno, N.; Ezzell, R.M.; Casey, D.M.; Gonzalez, P.K.; Fink, M.P. Nitric oxide dilates tight junctions and depletes ATP in cultured Caco-2BBe intestinal epithelial monolayers. Am. J. Physiol. 1995, 268, G361–G373. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Su, Z.; Li, S.; Sun, M.; Xiao, Y.; Ping, Q.; Deng, Y. Oral absorption enhancement of salmon calcitonin by using both N-trimethyl chitosan chloride and oligoarginines-modified liposomes as the carriers. Drug Deliv. 2014, 21, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Fein, K.C.; Lamson, N.G.; Whitehead, K.A. Structure-Function Analysis of Phenylpiperazine Derivatives as Intestinal Permeation Enhancers. Pharm. Res. 2017, 34, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Ucuncu, H.; Ertekin, M.V.; Yoruk, O.; Sezen, O.; Ozkan, A.; Erdogan, F.; Kiziltunc, A.; Gundogdu, C. Vitamin E and L-carnitine, separately or in combination, in the prevention of radiation-induced oral mucositis and myelosuppression: A controlled study in a rat model. J. Radiat. Res. 2006, 47, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Scientific Committee on Toxicity, Ecotoxicity and the Environment (CSTEE) Opinion on the Results of the Risk Assessment of: Tetrasodium Ethylenediamine Tetraacetate (NA4EDTA). Available online: https://ec.europa.eu/health/ph_risk/committees/sct/documents/out191_en.pdf (accessed on 2 December 2023).
- Compound Summary of Deoxycholic Acid in ChemIDplus-Datenbank of United States National Library of Medicine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Deoxycholic%20acid (accessed on 2 December 2023).
- Qorpak. Material Safety Data Sheet Sodium Nitroprusside, ACS. Available online: https://www.qorpak.com/msds/375452.pdf (accessed on 2 December 2023).
- Hirano, S. Chitin biotechnology applications. Biotechnol. Annu. Rev. 1996, 2, 237–258. [Google Scholar] [CrossRef] [PubMed]
- Compound Summary of 1-Phenylpiperazine in ChemIDplus-Datenbank of United States National Library of Medicine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/7096 (accessed on 2 December 2023).
- Maggio, E.T.; Grasso, P. Oral delivery of octreotide acetate in Intravail(R) improves uptake, half-life, and bioavailability over subcutaneous administration in male Swiss webster mice. Regul. Pept. 2011, 167, 233–238. [Google Scholar] [CrossRef]
- Rabinowicz, A.L.; Carrazana, E.; Maggio, E.T. Improvement of Intranasal Drug Delivery with Intravail((R)) Alkylsaccharide Excipient as a Mucosal Absorption Enhancer Aiding in the Treatment of Conditions of the Central Nervous System. Drugs R&D 2021, 21, 361–369. [Google Scholar] [CrossRef]
- Maggio, E.T.; Pillion, D.J. High efficiency intranasal drug delivery using Intravail(R) alkylsaccharide absorption enhancers. Drug Deliv. Transl. Res. 2013, 3, 16–25. [Google Scholar] [CrossRef]
- Welling, S.H.; Hubalek, F.; Jacobsen, J.; Brayden, D.J.; Rahbek, U.L.; Buckley, S.T. The role of citric acid in oral peptide and protein formulations: Relationship between calcium chelation and proteolysis inhibition. Eur. J. Pharm. Biopharm. 2014, 86, 544–551. [Google Scholar] [CrossRef]
- Compound Summary of Citric Acid in ChemIDplus-Datenbank of United States National Library of Medicine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/311 (accessed on 2 December 2023).
- van Hoogdalem, E.J.; Hardens, M.A.; de Boer, A.G.; Breimer, D.D. Absorption enhancement of rectally infused cefoxitin sodium by medium-chain fatty acids in conscious rats: Concentration-effect relationship. Pharm. Res. 1988, 5, 453–456. [Google Scholar] [CrossRef]
- Paszczyk, B. Cheese and Butter as a Source of Health-Promoting Fatty Acids in the Human Diet. Animals 2022, 12, 3424. [Google Scholar] [CrossRef] [PubMed]
- EPOCELIN Label. Available online: https://www.pmda.go.jp/PmdaSearch/iyakuDetail/ResultDataSetPDF/450064_6132700J1022_4_05 (accessed on 18 August 2023).
- Lindmark, T.; Soderholm, J.D.; Olaison, G.; Alvan, G.; Ocklind, G.; Artursson, P. Mechanism of absorption enhancement in humans after rectal administration of ampicillin in suppositories containing sodium caprate. Pharm. Res. 1997, 14, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Tuvia, S.; Atsmon, J.; Teichman, S.L.; Katz, S.; Salama, P.; Pelled, D.; Landau, I.; Karmeli, I.; Bidlingmaier, M.; Strasburger, C.J.; et al. Oral octreotide absorption in human subjects: Comparable pharmacokinetics to parenteral octreotide and effective growth hormone suppression. J. Clin. Endocrinol. Metab. 2012, 97, 2362–2369. [Google Scholar] [CrossRef] [PubMed]
- Tillman, L.G.; Geary, R.S.; Hardee, G.E. Oral delivery of antisense oligonucleotides in man. J. Pharm. Sci. 2008, 97, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Tomita, M.; Hayashi, M.; Awazu, S. Absorption-enhancing mechanism of sodium caprate and decanoylcarnitine in Caco-2 cells. J. Pharmacol. Exp. Ther. 1995, 272, 739–743. [Google Scholar] [PubMed]
- Krug, S.M.; Amasheh, M.; Dittmann, I.; Christoffel, I.; Fromm, M.; Amasheh, S. Sodium caprate as an enhancer of macromolecule permeation across tricellular tight junctions of intestinal cells. Biomaterials 2013, 34, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Tuvia, S.; Pelled, D.; Marom, K.; Salama, P.; Levin-Arama, M.; Karmeli, I.; Idelson, G.H.; Landau, I.; Mamluk, R. A novel suspension formulation enhances intestinal absorption of macromolecules via transient and reversible transport mechanisms. Pharm. Res. 2014, 31, 2010–2021. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Heade, J.; McCartney, F.; Waters, S.; Bleiel, S.B.; Brayden, D.J. Effects of surfactant-based permeation enhancers on mannitol permeability, histology, and electrogenic ion transport responses in excised rat colonic mucosae. Int. J. Pharm. 2018, 539, 11–22. [Google Scholar] [CrossRef]
- Kajii, H.; Horie, T.; Hayashi, M.; Awazu, S. Fluorescence study of the membrane-perturbing action of sodium caprylate as related to promotion of drug absorption. J. Pharm. Sci. 1988, 77, 390–392. [Google Scholar] [CrossRef]
- Leonard, T.W.; Lynch, J.; McKenna, M.J.; Brayden, D.J. Promoting absorption of drugs in humans using medium-chain fatty acid-based solid dosage forms: GIPET. Expert Opin. Drug Deliv. 2006, 3, 685–692. [Google Scholar] [CrossRef]
- Raoof, A.A.; Chiu, P.; Ramtoola, Z.; Cumming, I.K.; Teng, C.; Weinbach, S.P.; Hardee, G.E.; Levin, A.A.; Geary, R.S. Oral bioavailability and multiple dose tolerability of an antisense oligonucleotide tablet formulated with sodium caprate. J. Pharm. Sci. 2004, 93, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Santiago, N.; Milstein, S.; Rivera, T.; Garcia, E.; Zaidi, T.; Hong, H.; Bucher, D. Oral immunization of rats with proteinoid microspheres encapsulating influenza virus antigens. Pharm. Res. 1993, 10, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Leone-Bay, A.; Santiago, N.; Achan, D.; Chaudhary, K.; DeMorin, F.; Falzarano, L.; Haas, S.; Kalbag, S.; Kaplan, D.; Leipold, H.; et al. N-acylated alpha-amino acids as novel oral delivery agents for proteins. J. Med. Chem. 1995, 38, 4263–4269. [Google Scholar] [CrossRef] [PubMed]
- Leone-Bay, A.; Ho, K.K.; Agarwal, R.; Baughman, R.A.; Chaudhary, K.; DeMorin, F.; Genoble, L.; McInnes, C.; Lercara, C.; Milstein, S.; et al. 4-[4-[(2-Hydroxybenzoyl)amino]phenyl]butyric acid as a novel oral delivery agent for recombinant human growth hormone. J. Med. Chem. 1996, 39, 2571–2578. [Google Scholar] [CrossRef] [PubMed]
- Brayden, D.; Creed, E.; O’Connell, A.; Leipold, H.; Agarwal, R.; Leone-Bay, A. Heparin absorption across the intestine: Effects of sodium N-[8-(2-hydroxybenzoyl)amino]caprylate in rat in situ intestinal instillations and in Caco-2 monolayers. Pharm. Res. 1997, 14, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- Fattah, S.; Ismaiel, M.; Murphy, B.; Rulikowska, A.; Frias, J.M.; Winter, D.C.; Brayden, D.J. Salcaprozate sodium (SNAC) enhances permeability of octreotide across isolated rat and human intestinal epithelial mucosae in Ussing chambers. Eur. J. Pharm. Sci. 2020, 154, 105509. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Rath, P.; Angelo, R.; Stringfellow, T.; Flanders, E.; Dinh, S.; Gomez-Orellana, I.; Robinson, J.R. Oral absorption enhancement of cromolyn sodium through noncovalent complexation. Pharm. Res. 2004, 21, 2196–2206. [Google Scholar] [CrossRef]
- Hess, S.; Rotshild, V.; Hoffman, A. Investigation of the enhancing mechanism of sodium N-[8-(2-hydroxybenzoyl)amino]caprylate effect on the intestinal permeability of polar molecules utilizing a voltage clamp method. Eur. J. Pharm. Sci. 2005, 25, 307–312. [Google Scholar] [CrossRef]
- Buckley, S.T.; Baekdal, T.A.; Vegge, A.; Maarbjerg, S.J.; Pyke, C.; Ahnfelt-Ronne, J.; Madsen, K.G.; Scheele, S.G.; Alanentalo, T.; Kirk, R.K.; et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Riley, M.G.; Castelli, M.C.; Paehler, E.A. Subchronic oral toxicity of salcaprozate sodium (SNAC) in Sprague-Dawley and Wistar rats. Int. J. Toxicol. 2009, 28, 278–293. [Google Scholar] [CrossRef]
- Castelli, M.C.; Wong, D.F.; Friedman, K.; Riley, M.G. Pharmacokinetics of oral cyanocobalamin formulated with sodium N-[8-(2-hydroxybenzoyl)amino]caprylate (SNAC): An open-label, randomized, single-dose, parallel-group study in healthy male subjects. Clin. Ther. 2011, 33, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Sager, M.; Grimm, M.; Aude, P.; Schick, P.; Merdivan, S.; Hasan, M.; Kromrey, M.L.; Sivert, A.; Benameur, H.; Koziolek, M.; et al. In vivo characterization of enTRinsic drug delivery technology capsule after intake in fed state: A cross-validation approach using salivary tracer technique in comparison to MRI. J. Control. Release 2019, 313, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Amory, J.K.; Leonard, T.W.; Page, S.T.; O’Toole, E.; McKenna, M.J.; Bremner, W.J. Oral administration of the GnRH antagonist acyline, in a GIPET-enhanced tablet form, acutely suppresses serum testosterone in normal men: Single-dose pharmacokinetics and pharmacodynamics. Cancer Chemother. Pharmacol. 2009, 64, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Eldor, R.; Francis, B.H.; Fleming, A.; Neutel, J.; Homer, K.; Kidron, M.; Rosenstock, J. Oral insulin (ORMD-0801) in type 2 diabetes mellitus: A dose-finding 12-week randomized placebo-controlled study. Diabetes Obes. Metab. 2023, 25, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Pipeline-Oramed Pharmaceuticals. Available online: https://oramed.com/pipeline/ (accessed on 3 December 2023).
- New, R.R.C.; Ramanujam, S.; Chaudhari, V.; Bogus, M.; Travers, G.N.; Namjoshi, G. Safety and efficacy of an oral insulin (Capsulin) in patients with early-stage type 2 diabetes: A dose-ranging phase 2b study. Diabetes Obes. Metab. 2023, 25, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, T.A.; Rosa, M.; Coulter, I.S.; Brayden, D.J. In vitro and in vivo preclinical evaluation of a minisphere emulsion-based formulation (SmPill(R)) of salmon calcitonin. Eur. J. Pharm. Sci. 2015, 79, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Products of Sigmoidpharma. Available online: http://sigmoidpharma.com/products (accessed on 3 December 2023).
- Geho, W.B.; Geho, H.C.; Lau, J.R.; Gana, T.J. Hepatic-directed vesicle insulin: A review of formulation development and preclinical evaluation. J. Diabetes Sci. Technol. 2009, 3, 1451–1459. [Google Scholar] [CrossRef]
- Our Story of Diasome. Available online: https://www.diasome.com/ (accessed on 3 December 2023).
- Nielsen, D.S.; Shepherd, N.E.; Xu, W.; Lucke, A.J.; Stoermer, M.J.; Fairlie, D.P. Orally Absorbed Cyclic Peptides. Chem. Rev. 2017, 117, 8094–8128. [Google Scholar] [CrossRef]
- Asano, D. Experimental approach to evaluating the oral bioavailability of peptides, with a focus on membrane permeability, P-glycoprotein mediated efflux and cytochrome P450 metabolism. In Abstracts of Annual meeting of Japanese Society for the Study of Xenobiotics, Proceedings of the 36th JSSX Annual Meeting, Online, 19 November 2021; The Japanese Society for the Study of Xenobiotics: Tokyo, Japan, 2022. [Google Scholar]
- van Waterschoot, R.A.; Lagas, J.S.; Wagenaar, E.; van der Kruijssen, C.M.; van Herwaarden, A.E.; Song, J.Y.; Rooswinkel, R.W.; van Tellingen, O.; Rosing, H.; Beijnen, J.H.; et al. Absence of both cytochrome P450 3A and P-glycoprotein dramatically increases docetaxel oral bioavailability and risk of intestinal toxicity. Cancer Res. 2009, 69, 8996–9002. [Google Scholar] [CrossRef]
- van Waterschoot, R.A.; Lagas, J.S.; Wagenaar, E.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. Individual and combined roles of CYP3A, P-glycoprotein (MDR1/ABCB1) and MRP2 (ABCC2) in the pharmacokinetics of docetaxel. Int. J. Cancer 2010, 127, 2959–2964. [Google Scholar] [CrossRef]
- Matsson, P.; Doak, B.C.; Over, B.; Kihlberg, J. Cell permeability beyond the rule of 5. Adv. Drug Deliv. Rev. 2016, 101, 42–61. [Google Scholar] [CrossRef] [PubMed]
- Marelli, U.K.; Bezencon, J.; Puig, E.; Ernst, B.; Kessler, H. Enantiomeric cyclic peptides with different Caco-2 permeability suggest carrier-mediated transport. Chemistry 2015, 21, 8023–8027. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Hashimoto, S.; Takeyama, R.; Tamiya, M.; Kato, T.; Muraoka, T.; Kage, M.; Nii, K.; Kotake, K.; Iida, S.; et al. Broadly Applicable and Comprehensive Synthetic Method for N-Alkyl-Rich Drug-like Cyclic Peptides. J. Med. Chem. 2022, 65, 13401–13412. [Google Scholar] [CrossRef] [PubMed]
- Tanada, M.; Tamiya, M.; Matsuo, A.; Chiyoda, A.; Takano, K.; Ito, T.; Irie, M.; Kotake, T.; Takeyama, R.; Kawada, H.; et al. Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor. J. Am. Chem. Soc. 2023, 145, 16610–16620. [Google Scholar] [CrossRef] [PubMed]
- Kusumoto, Y.; Hayashi, K.; Sato, S.; Yamada, T.; Kozono, I.; Nakata, Z.; Asada, N.; Mitsuki, S.; Watanabe, A.; Wakasa-Morimoto, C.; et al. Highly Potent and Oral Macrocyclic Peptides as a HIV-1 Protease Inhibitor: mRNA Display-Derived Hit-to-Lead Optimization. ACS Med. Chem. Lett. 2022, 13, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Banka, P.; Mendez, G.; Garcia, R.; Rosenstock, J.; Rodgers, A.; Mendizabal, G.; Mitchel, Y.; Catapano, A.L. Phase 2b Randomized Trial of the Oral PCSK9 Inhibitor MK-0616. J. Am. Coll. Cardiol. 2023, 81, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Johns, D.G.; Campeau, L.C.; Banka, P.; Bautmans, A.; Bueters, T.; Bianchi, E.; Branca, D.; Bulger, P.G.; Crevecoeur, I.; Ding, F.X.; et al. Orally Bioavailable Macrocyclic Peptide That Inhibits Binding of PCSK9 to the Low Density Lipoprotein Receptor. Circulation 2023, 148, 144–158. [Google Scholar] [CrossRef] [PubMed]
- PeptiDream Affiliated Company, PeptiAID Inc., Completes Preclinical Studies of PA-001 Candidate Compound for COVID-19 Therapeutics and Announces Future Plans. Available online: https://contents.xj-storage.jp/xcontents/45870/bfc69946/cf52/42a1/ab2f/c8908a52f8f8/20211111150641447s.pdf (accessed on 18 August 2023).
- PeptiDream Affiliated Company, PeptiAID Inc., Announces Initiation of Clinical Research and the Progress on the Omicron Variant of PA-001, Candidate Compound for COVID-19 Therapeutics. Available online: https://contents.xj-storage.jp/xcontents/45870/c764c946/8a18/466d/a7ae/4cc1eb196b55/20220204184301276s.pdf (accessed on 18 August 2023).
- Qin, L.; Dai, H.; Wang, J. Key Considerations in Targeted Protein Degradation Drug Discovery and Development. Front. Chem. 2022, 10, 934337. [Google Scholar] [CrossRef]
- Troup, R.I.; Fallan, C.; Baud, M.G.J. Current strategies for the design of PROTAC linkers: A critical review. Explor. Target. Anti-Tumor Ther. 2020, 1, 273–312. [Google Scholar] [CrossRef]
- Pike, A.; Williamson, B.; Harlfinger, S.; Martin, S.; McGinnity, D.F. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: A drug metabolism and pharmacokinetics perspective. Drug Discov. Today 2020, 25, 1793–1800. [Google Scholar] [CrossRef]
- Bekes, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Cantrill, C.; Chaturvedi, P.; Rynn, C.; Petrig Schaffland, J.; Walter, I.; Wittwer, M.B. Fundamental aspects of DMPK optimization of targeted protein degraders. Drug Discov. Today 2020, 25, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.E.; Gao, Y.; Tan, L.; Donovan, K.A.; Nowak, R.P.; Loehr, A.; Bahcall, M.; Fischer, E.S.; Janne, P.A.; George, R.E.; et al. Chemically Induced Degradation of Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2018, 61, 4249–4255. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hu, M.; Yang, Y.; Du, C.; Zhou, H.; Liu, C.; Chen, Y.; Fan, L.; Ma, H.; Gong, Y.; et al. An overview of PROTACs: A promising drug discovery paradigm. Mol. Biomed. 2022, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- ARV-110 Phase 1/2 Dose Escalation: Interim Update. Available online: https://ir.arvinas.com/static-files/8f6f9c9b-e738-4ea5-8655-0f90a2fb5faa (accessed on 18 August 2023).
- The Discovery of ARV-471, an Orally Bioavailable Estrogen Receptor Degrading PROTAC® for the Treatment of Patients with Breast Cancer. Available online: https://www.arvinas.com/wp-content/uploads/2022/09/AACR21_ARV471_Structure_Disclosure_2021-08-06-014540_nkje.pdf (accessed on 18 August 2023).
- Hornberger, K.R.; Araujo, E.M.V. Physicochemical Property Determinants of Oral Absorption for PROTAC Protein Degraders. J. Med. Chem. 2023, 66, 8281–8287. [Google Scholar] [CrossRef] [PubMed]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [PubMed]
- Viarengo-Baker, L.A.; Brown, L.E.; Rzepiela, A.A.; Whitty, A. Defining and navigating macrocycle chemical space. Chem. Sci. 2021, 12, 4309–4328. [Google Scholar] [CrossRef]
- Garcia Jimenez, D.; Poongavanam, V.; Kihlberg, J. Macrocycles in Drug Discovery horizontal line Learning from the Past for the Future. J. Med. Chem. 2023, 66, 5377–5396. [Google Scholar] [CrossRef]
- Danelius, E.; Poongavanam, V.; Peintner, S.; Wieske, L.H.E.; Erdelyi, M.; Kihlberg, J. Solution Conformations Explain the Chameleonic Behaviour of Macrocyclic Drugs. Chemistry 2020, 26, 5231–5244. [Google Scholar] [CrossRef]
- DeGoey, D.A.; Chen, H.J.; Cox, P.B.; Wendt, M.D. Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J. Med. Chem. 2018, 61, 2636–2651. [Google Scholar] [CrossRef]
- Asano, D. Utilization of SNAC to improve an oral absorption of peptide drugs. In Abstracts of Annual meeting of Japanese Society for the Study of Xenobiotics, Proceedings of the 36th JSSX Annual Meeting, Online, 18 November 2021; The Japanese Society for the Study of Xenobiotics: Tokyo, Japan, 2022. [Google Scholar]
- Leone-Bay, A.; Leipold, H.; Sarubbi, D.; Variano, B.; Rivera, T.; Baughman, R.A. Oral delivery of sodium cromolyn: Preliminary studies in vivo and in vitro. Pharm. Res. 1996, 13, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Raoof, A.A.; Ramtoola, Z.; McKenna, B.; Yu, R.Z.; Hardee, G.; Geary, R.S. Effect of sodium caprate on the intestinal absorption of two modified antisense oligonucleotides in pigs. Eur. J. Pharm. Sci. 2002, 17, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Yonebayashi, S.; Yoshida, M.; Shimizu, K.; Aotsuka, T.; Takayama, K. Improvement in the bioavailability of poorly absorbed glycyrrhizin via various non-vascular administration routes in rats. Int. J. Pharm. 2003, 265, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chow, M.S.; Zuo, Z. Effect of sodium caprate on the oral absorptions of danshensu and salvianolic acid B. Int. J. Pharm. 2009, 379, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Mamluk, R.; Teichman, S.L. Method of Treating Diseases. U.S. Patent 10,695,397 B2, 30 June 2020. [Google Scholar]
- Narasipura, E.A.; VanKeulen-Miller, R.; Ma, Y.; Fenton, O.S. Ongoing Clinical Trials of Nonviral siRNA Therapeutics. Bioconjugate Chem. 2023, 34, 1177–1197. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X.H. Antisense technology: An overview and prospectus. Nat. Rev. Drug Discov. 2021, 20, 427–453. [Google Scholar] [CrossRef]
- Crooke, S.T.; Liang, X.H.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef]
- Ranasinghe, P.; Addison, M.L.; Dear, J.W.; Webb, D.J. Small interfering RNA: Discovery, pharmacology and clinical development—An introductory review. Br. J. Pharmacol. 2023, 180, 2697–2720. [Google Scholar] [CrossRef]
- Migliorati, J.M.; Liu, S.; Liu, A.; Gogate, A.; Nair, S.; Bahal, R.; Rasmussen, T.P.; Manautou, J.E.; Zhong, X.B. Absorption, Distribution, Metabolism, and Excretion of US Food and Drug Administration-Approved Antisense Oligonucleotide Drugs. Drug Metab. Dispos. 2022, 50, 888–897. [Google Scholar] [CrossRef]
- Takakusa, H.; Iwazaki, N.; Nishikawa, M.; Yoshida, T.; Obika, S.; Inoue, T. Drug Metabolism and Pharmacokinetics of Antisense Oligonucleotide Therapeutics: Typical Profiles, Evaluation Approaches, and Points to Consider Compared with Small Molecule Drugs. Nucleic Acid Ther. 2023, 33, 83–94. [Google Scholar] [CrossRef]
- Shadid, M.; Badawi, M.; Abulrob, A. Antisense oligonucleotides: Absorption, distribution, metabolism, and excretion. Expert Opin. Drug Metab. Toxicol. 2021, 17, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- McDougall, R.; Ramsden, D.; Agarwal, S.; Agarwal, S.; Aluri, K.; Arciprete, M.; Brown, C.; Castellanos-Rizaldos, E.; Charisse, K.; Chong, S.; et al. The Nonclinical Disposition and Pharmacokinetic/Pharmacodynamic Properties of N-Acetylgalactosamine-Conjugated Small Interfering RNA Are Highly Predictable and Build Confidence in Translation to Human. Drug Metab. Dispos. 2022, 50, 781–797. [Google Scholar] [CrossRef] [PubMed]
- Gennemark, P.; Walter, K.; Clemmensen, N.; Rekic, D.; Nilsson, C.A.M.; Knochel, J.; Holtta, M.; Wernevik, L.; Rosengren, B.; Kakol-Palm, D.; et al. An oral antisense oligonucleotide for PCSK9 inhibition. Sci. Transl. Med. 2021, 13, eabe9117. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.N.; Patel, N.J.; Bhowmik, T.; D’Souza, B.; Akalkotkar, A.; Etzlar, F.; Oettinger, C.W.; D’Souza, M. Enhanced bioavailability of orally administered antisense oligonucleotide to nuclear factor kappa B mRNA after microencapsulation with albumin. J. Drug Target. 2013, 21, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Iacob, A.T.; Lupascu, F.G.; Apotrosoaei, M.; Vasincu, I.M.; Tauser, R.G.; Lupascu, D.; Giusca, S.E.; Caruntu, I.D.; Profire, L. Recent Biomedical Approaches for Chitosan Based Materials as Drug Delivery Nanocarriers. Pharmaceutics 2021, 13, 587. [Google Scholar] [CrossRef] [PubMed]
- Subhan, M.A.; Torchilin, V.P. Biopolymer-Based Nanosystems for siRNA Drug Delivery to Solid Tumors including Breast Cancer. Pharmaceutics 2023, 15, 153. [Google Scholar] [CrossRef] [PubMed]
- Ballarin-Gonzalez, B.; Dagnaes-Hansen, F.; Fenton, R.A.; Gao, S.; Hein, S.; Dong, M.; Kjems, J.; Howard, K.A. Protection and Systemic Translocation of siRNA Following Oral Administration of Chitosan/siRNA Nanoparticles. Mol. Ther. Nucleic Acids 2013, 2, e76. [Google Scholar] [CrossRef]
- Han, L.; Tang, C.; Yin, C. Oral delivery of shRNA and siRNA via multifunctional polymeric nanoparticles for synergistic cancer therapy. Biomaterials 2014, 35, 4589–4600. [Google Scholar] [CrossRef]
- Wei, Y.; Li, X.; Lin, J.; Zhou, Y.; Yang, J.; Hou, M.; Wu, F.; Yan, J.; Ge, C.; Hu, D.; et al. Oral Delivery of siRNA Using Fluorinated, Small-Sized Nanocapsules toward Anti-Inflammation Treatment. Adv. Mater. 2023, 35, e2206821. [Google Scholar] [CrossRef]
















| Advantages | Limitations | |
|---|---|---|
| Chemical modifications |
|
|
| Absorption enhancers |
|
|
| Evaluation Method | Detail | Reference |
|---|---|---|
| Partition coefficient in octanol-water (LogPoct) and toluene/water (LogPtol) | The difference between LogPoct and LogPtol (∆LogP) correlates with the presence or absence of intramolecular hydrogen bonding. | [49] |
| Molecular (3D) polar surface area in nonpolar environments (MPSA) and topological polar surface area (TPSA) | TPSA is a polar surface area calculated as a sum of fragment-based contributions. MPSA is the minimal solvent-accessible polar surface area in 3D conformations. If the value of TPSA minus MPSA (∆PSA) is larger than 0.2 × molecular weight—140 Å2 or TPSA—140 Å2, the evaluated middle-to-large molecules would possess chameleonic property. | [37] |
| Lipophilic permeability efficiency (LPE) | LPE is an index of the membrane permeability of middle-to-large molecules. It can be calculated as follows: LPE = distribution coefficient in decadiene-water at pH 7.4 − mlipo (scaling factor) × calculated LogPoct + bscaffold (scaling factor). | [44] |
| Experimental polar surface area (EPSA) | EPSA is an index of the membrane permeability with consideration of intramolecular hydrogen bonding. It can be measured by supercritical fluid chromatography. | [45,46,50,51] |
| Nuclear magnetic resonance (NMR) analysis | Amide temperature coefficients and H/D exchange study measured by NMR indicate the presence or absence of intramolecular hydrogen bonding. | [47,48] |
| X-ray analysis | Three-dimensional structure of middle-to-large molecules can be elucidated by X-ray crystallography, indicating the presence or absence of intramolecular hydrogen bonding. | [36] |
| In silico structural simulation | Molecular dynamic method can predict chameleonic property or membrane permeability. | [52,53] |
| Absorption Enhancer | Mechanism | Available Safety Information |
|---|---|---|
| SNAC and related compounds (4-CNAB and 5-CNAC) | Enhancing transcellular permeation [61,62,63] | NOAEL of SNAC: 500 and 500 mg/kg/day in male and female mice, 500 and 75 mg/kg/day in male and female rats, and 300 and 300 mg/kg/day in male and female monkeys, respectively [64]. |
| C8, C10, fatty acids, and surfactants | Opening tight junctions and/or causing membrane perturbation [65,66] | LD50 of C8 and C10: 1280–10,080 mg/kg [67] and 3730 mg/kg [68] in rats, respectively. |
| Amino acids (arginine and tryptophan) | Unclear (possible involvement of receptor- or transporter-mediated uptake) [69,70] | NOAEL of arginine and LD50 of tryptophan: 3131 mg/kg in rats [71] and 5000 mg/kg in mice [72], respectively. |
| Acylcarnitines, EDTA, bile acid, NO, chitosan (polysaccharide), claudin modulator, 1-phenylpiperazine | Opening tight junctions [55,56,57,73,74,75,76] | LD50 of carnitine, EDTA, deoxycholic acid, nitroprusside, chitosan, claudin modulator, and 1-phenylpiperazine: 19.2 g/kg in mice [77], 2 g/kg [78], 1 g/kg in mice and rats [79], 43 mg/kg in mice [80], 16 g/kg in mice [81], unknown, and 210 mg/kg in rats [82], respectively. |
| TAT, octa-arginine, and related peptides (cell-penetrating peptides: CPPs) | Inducing macropinocytosis [54,60] | Unknown |
| Cyclic DNP peptide (CPPs) | Inducing macropinocytosis [58,59] | Unknown |
| Intravail® (alkylsaccharide excipient) | Opening tight junctions and enhancing transcellular permeation [83,84] | LD50 of Intravail®: 2000 mg/kg in rats [85]. |
| Citric acid and protease inhibitors | Protecting peptides and proteins from digestive enzymes [86] | LD50 of citric acid: 5040 and 3000 mg/kg in mice and rats, respectively [87]. |
| Formulation | Composition and Design | API | Marketed | Reference |
|---|---|---|---|---|
| Rybelsus® and Eligen® B12 | Immediate-release tablet with SNAC | Semaglutide (MW: ca. 4100) and vitamin B12 (MW: 1355) | Yes | [17,18,20,21,35,108,110] |
| enTRinsic™ | Enteric-coated capsule composed of cellulose acetate phthalate | Esomeprazole (MW: 345) | No | [20,111] |
| GIPET™ | Enteric-coated tablet with various additives (C10, etc.) | Heparin (MW: ca. 1000–35,000), I338 (MW: ca. 6400), acyline (MW: ca. 1500), and GLP-1 (MW: ca. 3000–4000) | No | [20,24,112] |
| POD™ (Protein Oral Delivery) | Enteric-coated capsule with various additives (SNAC, EDTA, aprotinin, fatty acid, trypsin inhibitor, etc.) | Insulin (MW: ca. 5800) and exenatide (MW: ca. 4200) | No | [20,113,114] |
| Peptelligence™ and Ovarest® | Enteric-coated tablet with various additives (acylcarnitine, citric acid, etc.) | Salmon calcitonin (MW: ca. 3400), leuprolide (MW: ca. 1200), and difelikefalin (MW: ca. 680) | No | [20,21] |
| TPE™ and Mycapssa® | Enteric-coated capsule containing oily suspension of C8 and additives | Octreotide (MW: ca. 1000) | Yes | [19,20,21,35] |
| NodlinTM | Enteric-coated nanoparticle | Insulin (MW: ca. 5800) | No | [26] |
| Capsulin™ | Enteric-coated capsule with bile salt and antioxidant | Insulin (MW: ca. 5800) | No | [35,115] |
| SmPill® | Emulsion-based formulation containing various absorption enhancers (sodium taurodeoxycholate, C10, etc.) | Salmon calcitonin (MW: ca. 3400) and cyclosporin (MW: 1202) | No | [116,117] |
| Oraldel™ | Cyanocobalamin-coated nanoparticle consisting of carbohydrate-based sugar | Insulin (MW: ca. 5800) | No | [35] |
| HDV (hepatocyte-directed vesicle) and other liposomes | Liposome composed of hepatocyte-targeting molecule (disofenin, etc.), various phospholipids and/or cholesterol | Insulin (MW: ca. 5800) | No | [35,118,119] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asano, D.; Takakusa, H.; Nakai, D. Oral Absorption of Middle-to-Large Molecules and Its Improvement, with a Focus on New Modality Drugs. Pharmaceutics 2024, 16, 47. https://doi.org/10.3390/pharmaceutics16010047
Asano D, Takakusa H, Nakai D. Oral Absorption of Middle-to-Large Molecules and Its Improvement, with a Focus on New Modality Drugs. Pharmaceutics. 2024; 16(1):47. https://doi.org/10.3390/pharmaceutics16010047
Chicago/Turabian StyleAsano, Daigo, Hideo Takakusa, and Daisuke Nakai. 2024. "Oral Absorption of Middle-to-Large Molecules and Its Improvement, with a Focus on New Modality Drugs" Pharmaceutics 16, no. 1: 47. https://doi.org/10.3390/pharmaceutics16010047
APA StyleAsano, D., Takakusa, H., & Nakai, D. (2024). Oral Absorption of Middle-to-Large Molecules and Its Improvement, with a Focus on New Modality Drugs. Pharmaceutics, 16(1), 47. https://doi.org/10.3390/pharmaceutics16010047