
Accessing Mefenamic Acid Form II through High-Pressure Recrystallisation



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Compression Studies
2.2. Crystallisation at High Pressure
2.3. Single Crystal X-ray Diffraction
2.4. Infrared Spectroscopy
3. Results
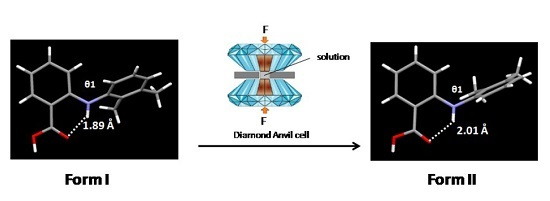
3.1. Direct Compression of a Single Crystal of Form I
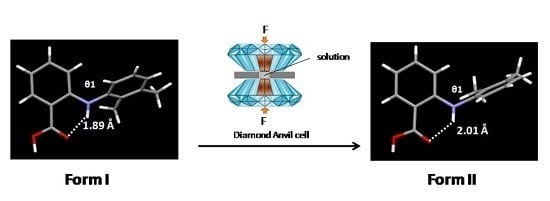
3.1.1. Crystallisation from Solution at High Pressure
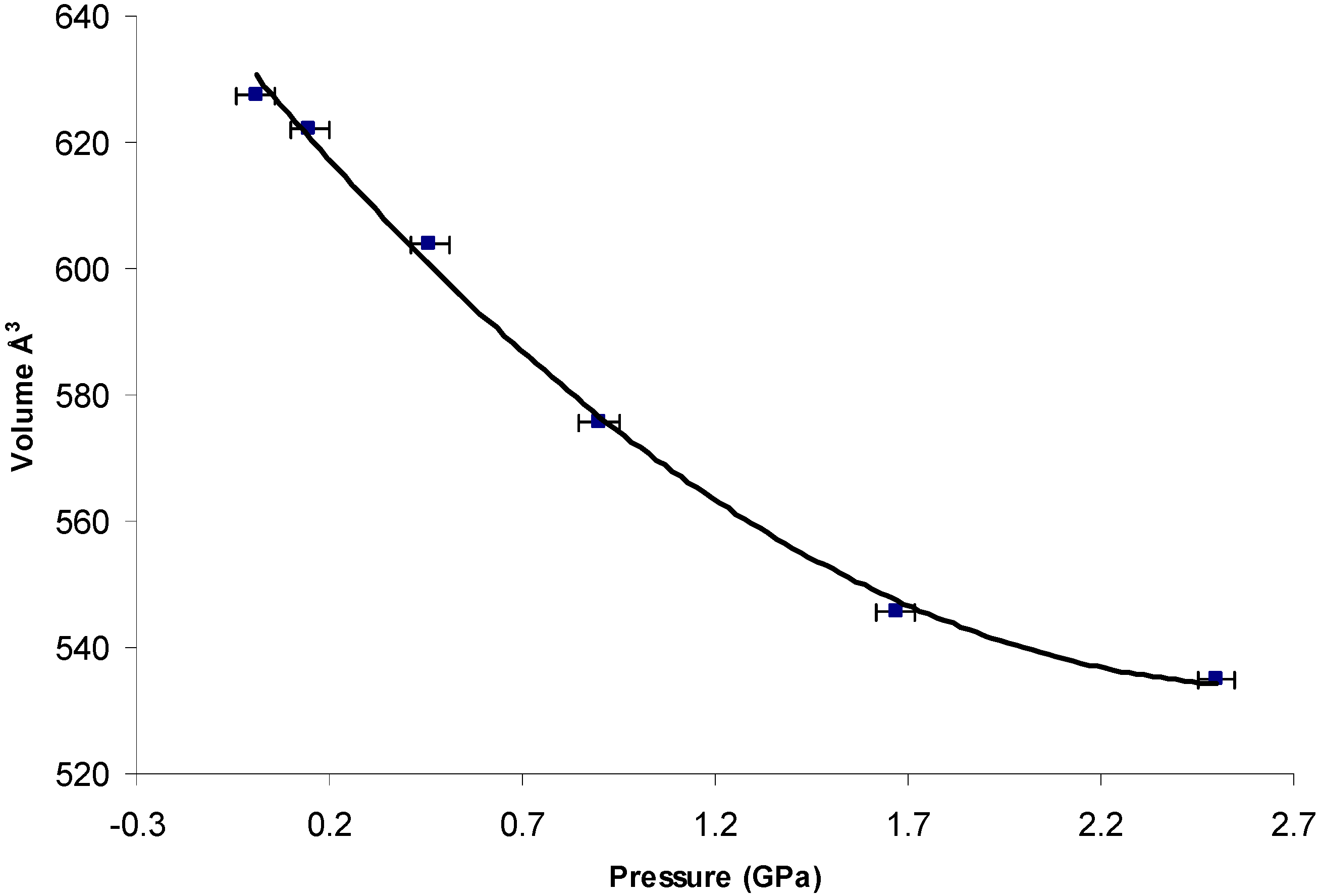
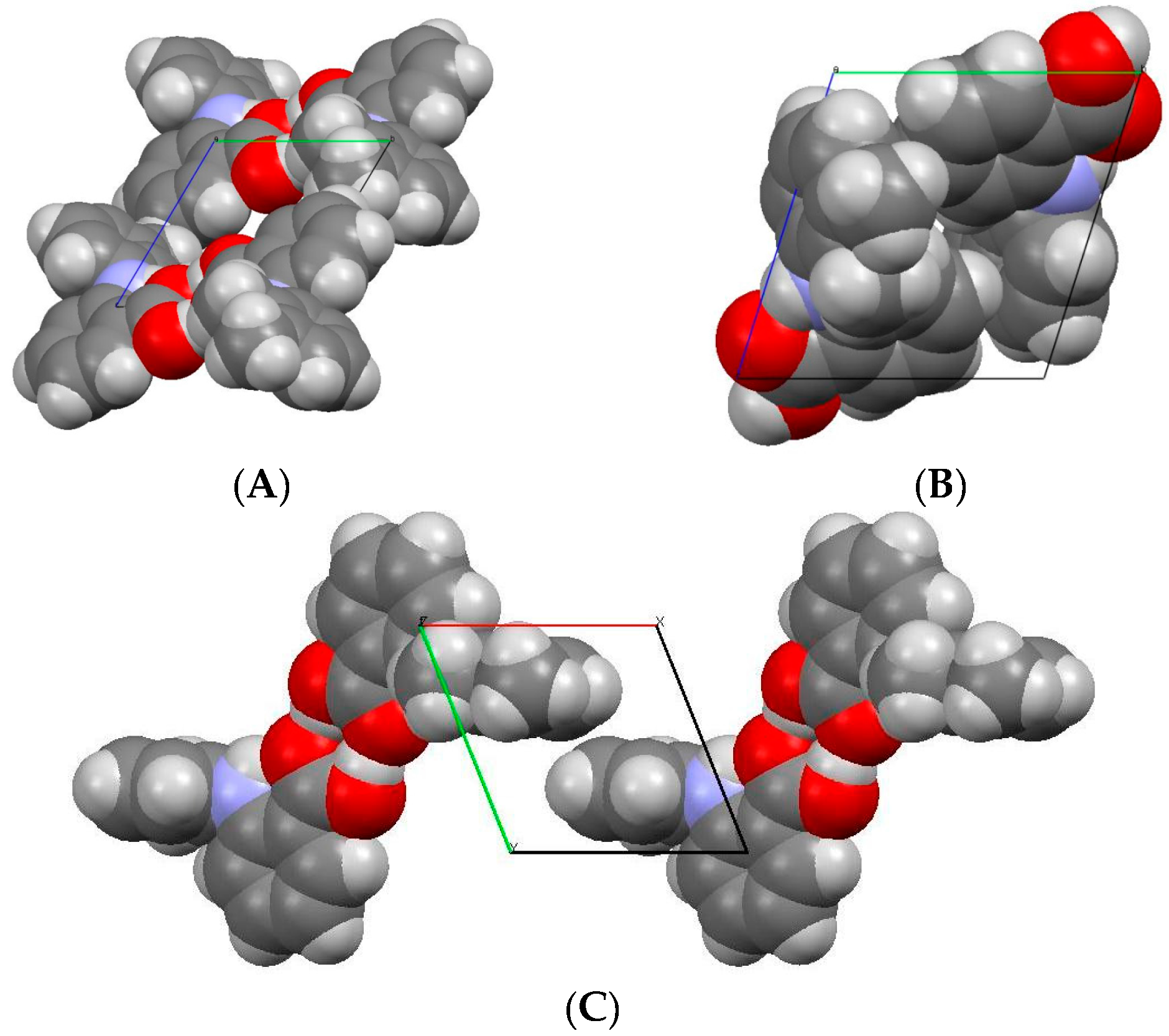
3.1.2. Comparison of Crystal Structure of High-Pressure Form II with Form I and Form III
3.1.3. Decompression Studies
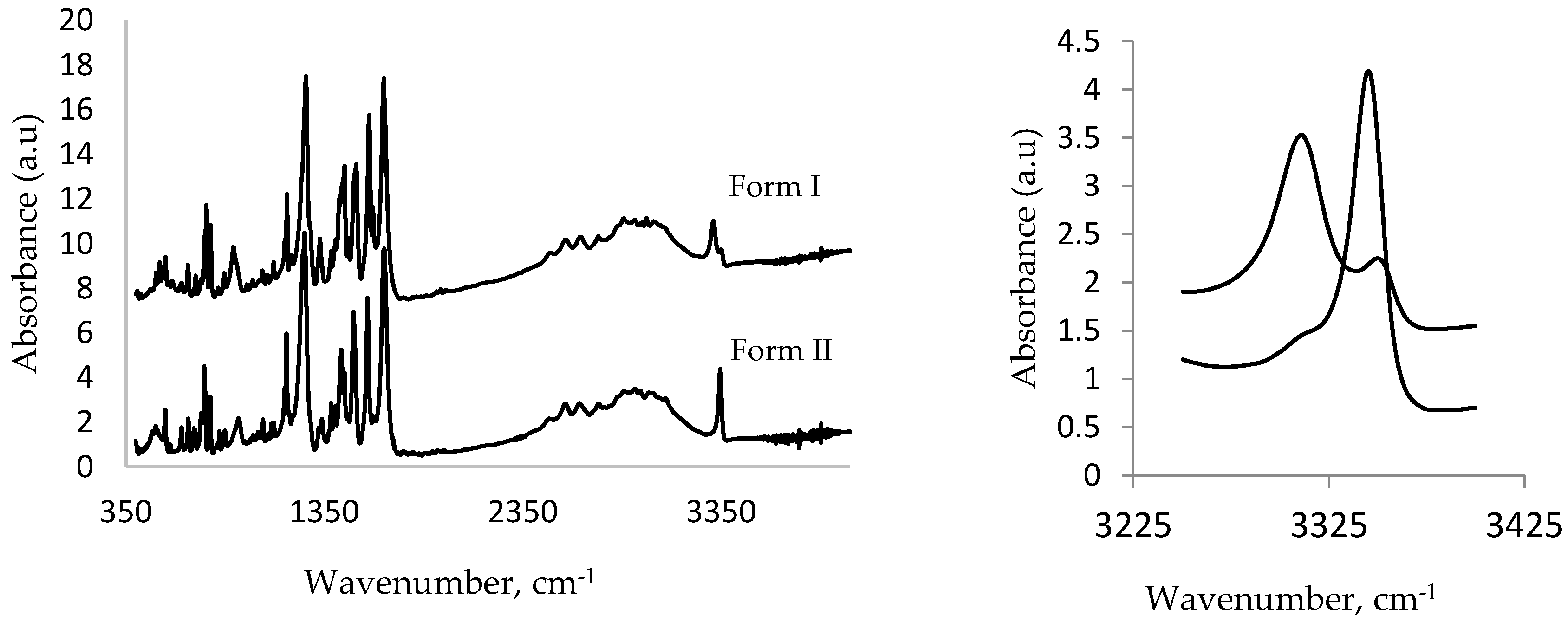
3.1.4. IR Studies
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Giron, D. Contribution of thermal methods and related techniques to the rational development of pharmaceuticals—Part 1. Pharm. Sci. Technol. Today 1998, 1, 191–199. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Chemburkar, S.R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W. Dealing with the impact of ritonavir polymorphs on the late stages of bulk drug process development. Org. Process Res. Dev. 2000, 4, 413–417. [Google Scholar] [CrossRef]
- Raw, A.S.; Furness, M.S.; Gill, D.S.; Adams, R.C.; Holcombe, F.O.; Lawrence, X.Y. Regulatory considerations of pharmaceutical solid polymorphism in Abbreviated New Drug Applications (ANDAs). Adv. Drug Deliv. Rev. 2004, 56, 397–414. [Google Scholar] [CrossRef]
- Ismail, S.Z.; Anderton, C.L.; Copley, R.C.; Price, L.S.; Price, S.L. Evaluating a crystal energy landscape in the context of industrial polymorph screening. Cryst. Growth Des. 2013, 13, 2396–2406. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Bernstein, J. Disappearing polymorphs. Acc. Chem. Res. 1995, 28, 193–200. [Google Scholar] [CrossRef]
- Bernstein, J.; Henck, J.-O. Disappearing and reappearing polymorphs—An anathema to crystal engineering? Cryst. Eng. 1998, 1, 119–128. [Google Scholar] [CrossRef]
- Fabbiani, F.P.; Allan, D.R.; Marshall, W.G.; Parsons, S.; Pulham, C.R.; Smith, R.I. High-pressure recrystallisation—A route to new polymorphs and solvates of acetamide and parabanic acid. J. Cryst. Growth 2005, 275, 185–192. [Google Scholar] [CrossRef]
- Hemley, R.J.; Dera, P. Molecular crystals. Rev. Mineral. Geochem. 2000, 41, 335–419. [Google Scholar] [CrossRef]
- Boldyreva, E.V. High-pressure diffraction studies of molecular organic solids: A personal view. Acta Crystallogr. 2008, 64, 218–231. [Google Scholar] [CrossRef]
- Moggach, S.A.; Parsons, S.; Wood, P.A. High-pressure polymorphism in amino acids. Crystallogr. Rev. 2008, 14, 143–184. [Google Scholar] [CrossRef]
- Olejniczak, A.; Ostrowska, K.; Katrusiak, A. H-bond breaking in high-pressure urea. J. Phys. Chem. C 2009, 113, 15761–15767. [Google Scholar] [CrossRef]
- Fabbiani, F.P.; Allan, D.R.; Dawson, A.; David, W.I.; McGregor, P.A.; Oswald, I.D.H.; Parsons, S.; Pulham, C.R. Pressure-induced formation of a solvate of paracetamol. Chem. Commun. 2003, 3004–3005. [Google Scholar] [CrossRef]
- Flower, R.J. Drugs which inhibit prostaglandin biosynthesis. Pharmacol. Rev. 1974, 26, 33–67. [Google Scholar] [PubMed]
- SeethaLekshmi, S.; Guru Row, T.N. Conformational polymorphism in a non-steroidal anti-inflammatory drug, mefenamic acid. Cryst. Growth Des. 2012, 12, 4283–4289. [Google Scholar] [CrossRef]
- Aguiar, A.J.; Zelmer, J.E. Dissolution behavior of polymorphs of chloramphenicol palmitate and mefenamic acid. J. Pharm. Sci. 1969, 58, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Romero, S.; Escalera, B.; Bustamante, P. Solubility behavior of polymorphs I and II of mefenamic acid in solvent mixtures. Int. J. Pharm. 1999, 178, 193–202. [Google Scholar] [CrossRef]
- McConnell, J.; Company, F.Z. N-(2,3-xylyl) anthranilic acid, C15H15NO2. Mefenamic acid. Cryst. Struct. Commun. 1976, 5, 861–864. [Google Scholar]
- Lee, E.H.; Byrn, S.R.; Carvajal, M.T. Additive-induced metastable single crystal of mefenamic acid. Pharm. Res. 2006, 23, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.L.; Craik, D.J. NMR conformational studies of fenamate non-steroidal anti-inflammatory drugs. Magn. Reson. Chem. 1994, 32, 335–342. [Google Scholar] [CrossRef]
- Alvarez, A.J.; Singh, A.; Myerson, A.S. Polymorph screening: Comparing a semi-automated approach with a high throughput method. Cryst. Growth Des. 2009, 9, 4181–4188. [Google Scholar] [CrossRef]
- Merrill, L.; Bassett, W.A. Miniature diamond anvil pressure cell for single crystal X-ray diffraction studies. Rev. Sci. Instrum. 1974, 45, 290–294. [Google Scholar] [CrossRef]
- Moggach, S.A.; Allan, D.R.; Parsons, S.; Warren, J.E. Incorporation of a new design of backing seat and anvil in a Merrill–Bassett diamond anvil cell. J. Appl. Crystallogr. 2008, 41, 249–251. [Google Scholar] [CrossRef]
- Piermarini, G.J.; Block, S.; Barnett, J.; Forman, R. Calibration of the pressure dependence of the R1 ruby fluorescence line to 195 kbar. J. Appl. Phys. 1975, 46, 2774–2780. [Google Scholar] [CrossRef]
- Piermarini, G.; Block, S.; Barnett, J. Hydrostatic limits in liquids and solids to 100 kbar. J. Appl. Phys. 1973, 44, 5377–5382. [Google Scholar] [CrossRef]
- Sheldrick, G.M. CELL NOW, program for unit cell determination. University of Göttingen: Germany, 2003. Available online: http://xray.tamu.edu/pdf/manuals/cell_now.pdf (accessed on 12 May 2017).
- Dawson, A.; Allan, D.R.; Parsons, S.; Ruf, M. Use of a CCD diffractometer in crystal structure determinations at high pressure. J. Appl. Crystallogr. 2004, 37, 410–416. [Google Scholar] [CrossRef]
- Bruker, A.P.E.X. 2004 Search PubMed GM Sheldrick. Acta Crystallogr. 2008, 64, 112. [Google Scholar]
- Parsons, S. SHADE: Program for empirical absorption corrections to high pressure data. Ph.D. Thesis, University of Edinburgh, Edinburgh, UK, 2004. [Google Scholar]
- Sheldrick, G.M. SADABS program for area detector adsorption correction. Ph.D. Thesis, University of Göttingen, Göttingen, Germany, 2006. [Google Scholar]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Crystallogr. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Dhanaraj, V.; Vijayan, M. Structural studies of analgesics and their interactions. XII. Structure and interactions of anti-inflammatory fenamates. A concerted crystallographic and theoretical conformational study. Acta Crystallogr. 1988, 44, 406–412. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.L. Patterns in hydrogen bonding: Functionality and graph set analysis in crystals. Angew. Chem. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Motherwell, W.S.; Shields, G.P.; Allen, F.H. Visualization and characterization of non-covalent networks in molecular crystals: Automated assignment of graph-set descriptors for asymmetric molecules. Acta Crystallogr. 1999, 55, 1044–1056. [Google Scholar] [CrossRef]
- Fabbiani, F.P.; Allan, D.R.; David, W.I.; Moggach, S.A.; Parsons, S.; Pulham, C.R. High-pressure recrystallization—A route to new polymorphs and solvates. CrystEngComm 2004, 6, 505–511. [Google Scholar] [CrossRef]
- Lee, I.S.; Lee, A.Y.; Myerson, A.S. Concomitant polymorphism in confined environment. Pharm. Res. 2008, 25, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Mukuta, T.; Lee, A.Y.; Kawakami, T.; Myerson, A.S. Influence of impurities on the solution-mediated phase transformation of an active pharmaceutical ingredient. Cryst. Growth Des. 2005, 5, 1429–1436. [Google Scholar] [CrossRef]
- Bernstein, J.; Davey, R.J.; Henck, J.O. Concomitant polymorphs. Angew. Chem. 1999, 38, 3440–3461. [Google Scholar] [CrossRef]
- Iwasaki, T.; Takahara, M.; Sonoda, R.; Watano, S. Dry grinding of mefenamic acid particles for enhancement of its water dissolution rate. Part. Part. Syst. Charact. 2007, 24, 236–241. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pressure | Ambient | 0.18 GPa | 0.49 Gpa | 0.90 GPa | 1.67 GPa | 2.50 GPa |
|---|---|---|---|---|---|---|
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group | P1̅ | P1̅ | P1̅ | P1̅ | P1̅ | P1̅ |
| a (Å) | 6.7582(14) | 6.775(2) | 6.7120(13) | 6.6432(3) | 6.5642(13) | 6.5153(13) |
| b (Å) | 7.3391(15) | 7.288(2) | 7.2369(14) | 7.1516(6) | 7.1406(14) | 7.0188(14) |
| c (Å) | 14.3127(29) | 14.303(15) | 14.0762(28) | 13.7098(8) | 13.4047(27) | 13.1342(26) |
| α (°) | 76.69(3) | 76.72(5) | 77.02(3) | 77.724(6) | 77.888(3) | 78.38(26) |
| β (°) | 79.83(3) | 79.11(5) | 79.43(3) | 78.962(4) | 77.426(3) | 77.11(3) |
| γ (°) | 65.70(3) | 65.55(2) | 65.72(3) | 65.606(6) | 63.846(3) | 62.82(3) |
| V (Å3) | 626.94(31) | 622.1(7) | 604.1(31) | 575.69(6) | 545.78(2) | 535.0(17) |
| Z | 2 | 2 | 2 | 2 | 2 | 2 |
| T (K) | 298 | 298 | 298 | 298 | 298 | 298 |
| Parameter | MA II (high-pressure form) a | MA II (CSD:XYANAC04) b | MA III (CSD:XYANAC03) b | MA I (CSD:XYANAC) c |
|---|---|---|---|---|
| Chemical formula | C15H15NO2 | C15H15NO2 | C15H15NO2 | C15H15NO2 |
| Formula weight | 241.29 | 241.29 | 241.29 | 241.29 |
| Crystal system | P1̅ | P1̅ | P1̅ | P1̅ |
| Space group | Triclinic | Triclinic | Triclinic | Triclinic |
| a (Å) | 7.7900(15) | 7.7584(5) | 7.723(2) | 14.556 |
| b (Å) | 9.1890(18) | 9.2772(6) | 7.9340(10) | 6.811 |
| c (Å) | 9.4120(19) | 9.3991(4) | 11.2320(10) | 7.657 |
| α (°) | 106.751(10) | 106.308(5) | 83.590(10) | 119.57 |
| β (°) | 92.287(12) | 91.847(4) | 80.940(10) | 103.93 |
| γ (°) | 101.377(11) | 101.856(5) | 67.510(10) | 91.30 |
| V (Å3) | 629.1(2) | 632.52(6) | 626.96) | 631.766 |
| Z | 2 | 2 | 2 | 2 |
| T (K) | 298(2) | 298(2) | 298(2) | 298 |
| R (Fo) | 0.095 | 0.089 | 0.042 | 0.045 |
| Rw (F2o) | 0.095 | 0.302 | 0.109 | - |
| Programme used | CRYSTALS | SHELX97 | SHELX97 | MULTAN |
| Torsional Angle | MA I [18] | MA II (This Study) | MA II [15] | MA III [15] | |
|---|---|---|---|---|---|
| a a | b b | ||||
| θ1 | 178.60 | 177.45 | −177.43 | −177.43 | −177.38 |
| θ2 | −119.99 | −85.18 | −68.20 | −71.01 | −80.82 |
| θ3 | −179.34 | −171.50 | −176.32 | −168.41 | −179.55 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbas, N.; Oswald, I.D.H.; Pulham, C.R. Accessing Mefenamic Acid Form II through High-Pressure Recrystallisation. Pharmaceutics 2017, 9, 16. https://doi.org/10.3390/pharmaceutics9020016
Abbas N, Oswald IDH, Pulham CR. Accessing Mefenamic Acid Form II through High-Pressure Recrystallisation. Pharmaceutics. 2017; 9(2):16. https://doi.org/10.3390/pharmaceutics9020016
Chicago/Turabian StyleAbbas, Nasir, Iain D. H. Oswald, and Colin R. Pulham. 2017. "Accessing Mefenamic Acid Form II through High-Pressure Recrystallisation" Pharmaceutics 9, no. 2: 16. https://doi.org/10.3390/pharmaceutics9020016
APA StyleAbbas, N., Oswald, I. D. H., & Pulham, C. R. (2017). Accessing Mefenamic Acid Form II through High-Pressure Recrystallisation. Pharmaceutics, 9(2), 16. https://doi.org/10.3390/pharmaceutics9020016