Pharmacokinetic and Toxicodynamic Characterization of a Novel Doxorubicin Derivative

 , ,
, , 

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Synthesis of the DoxQ Conjugate
2.3. Physicochemical Properties
2.4. Analytical System and Conditions
2.4.1. Preparation of Standard Solutions
2.4.2. Calibration Curves
2.5. Animals and Surgical Procedures
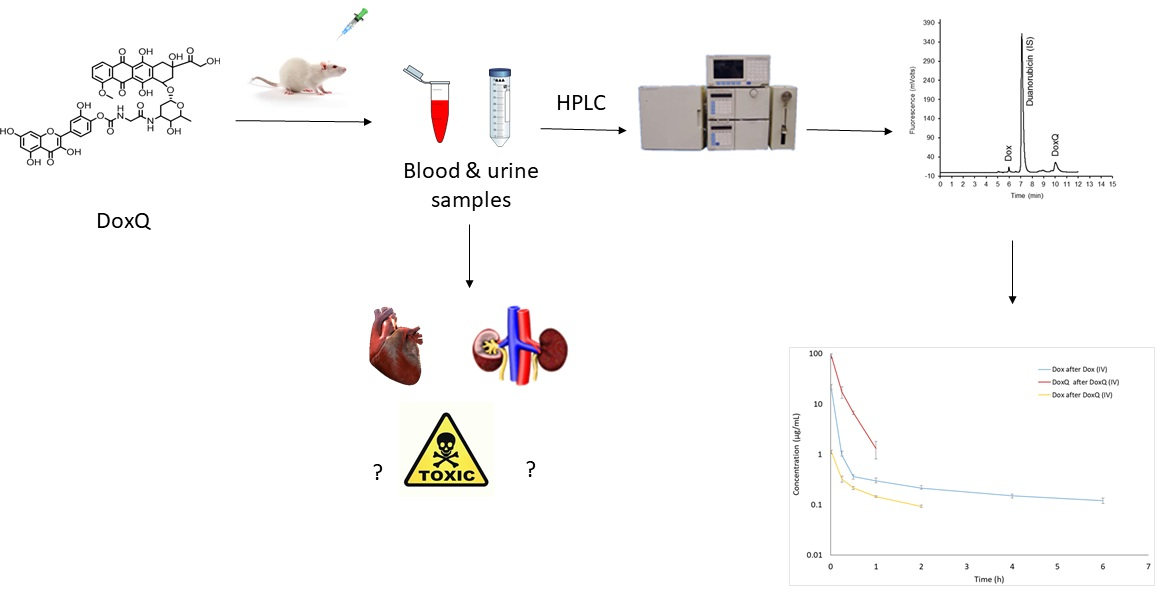
2.6. Pharmacokinetic Study
2.7. Intestinal Lymphatic Drug Delivery
2.7.1. Mesenteric Lymph Cannulation Surgery
2.7.2. Lymph Blockage by Cycloheximide
2.8. Treatment of Biological Samples for Analysis
2.8.1. Serum and Lymph Sample Preparation
2.8.2. Urine Sample Preparation
2.9. Pharmacokinetic Analysis
2.10. Assessment of Cardiac Toxicity of DoxQ and Dox
2.11. Assessment of Renal Toxicity of Dox and DoxQ
2.11.1. Urinary Output
2.11.2. β-N-Acetylglucosaminidase (NAG)
2.12. Statistical Analysis
3. Results
3.1. Physicochemical Properties
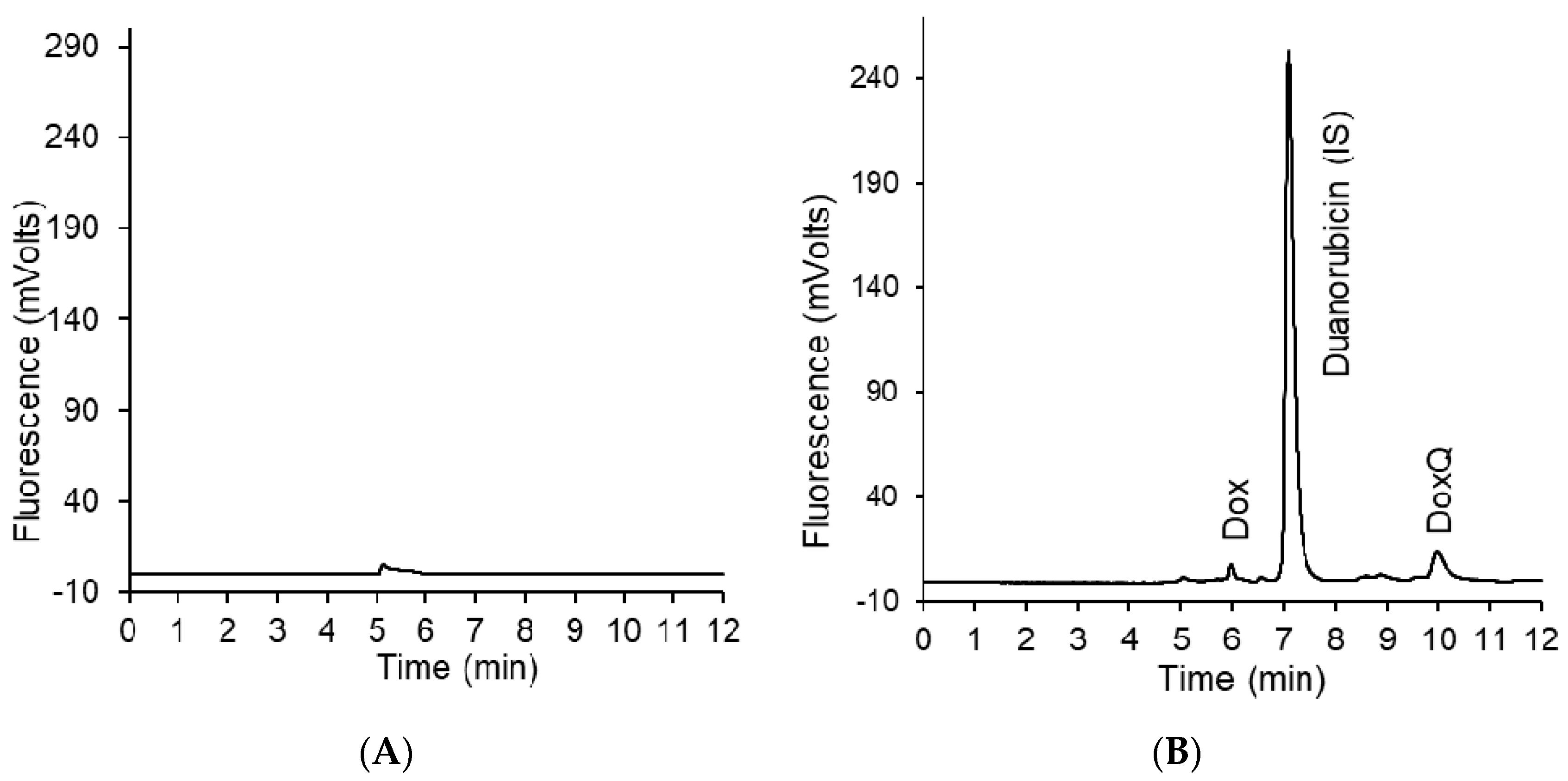
3.2. HPLC Analysis of Dox
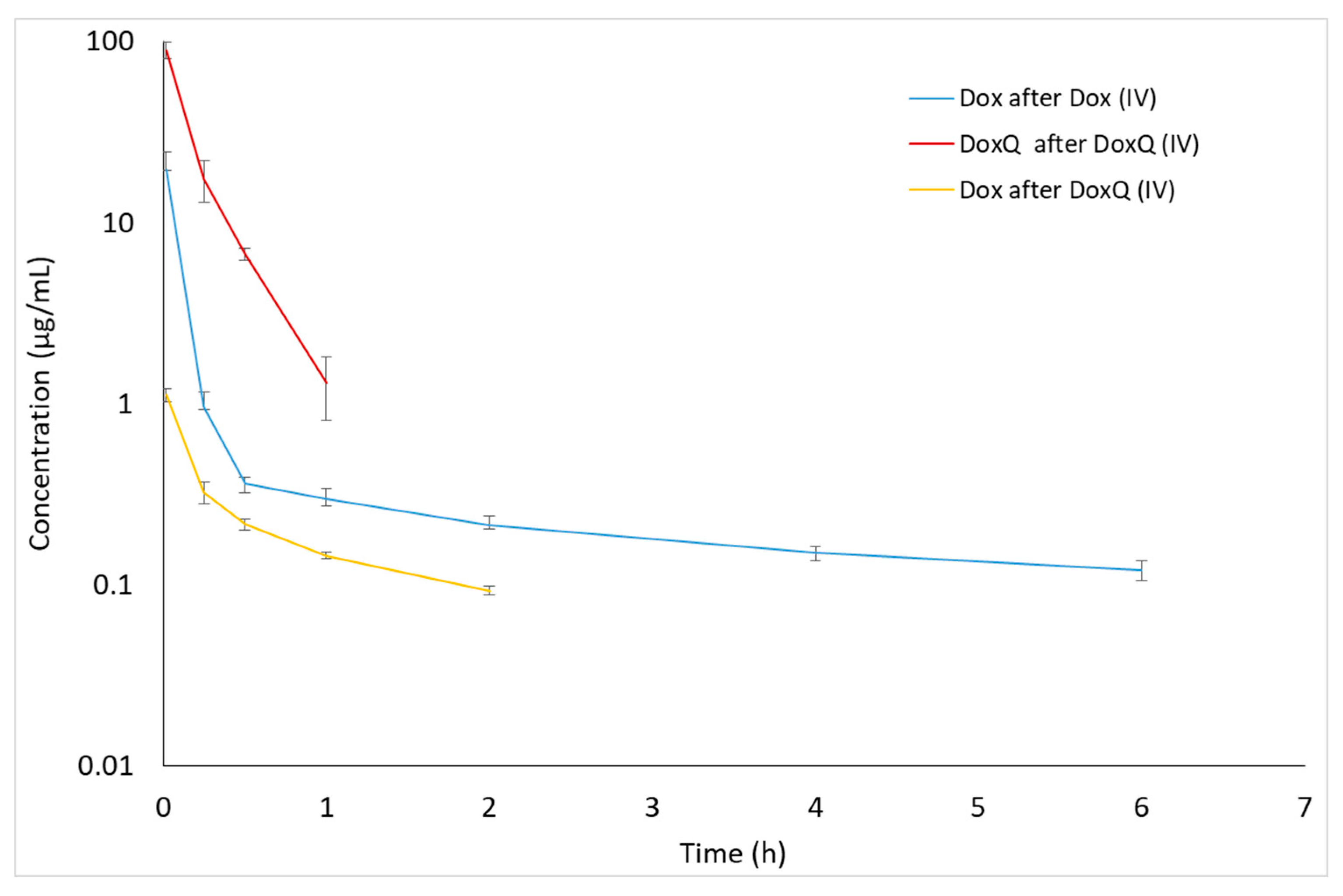
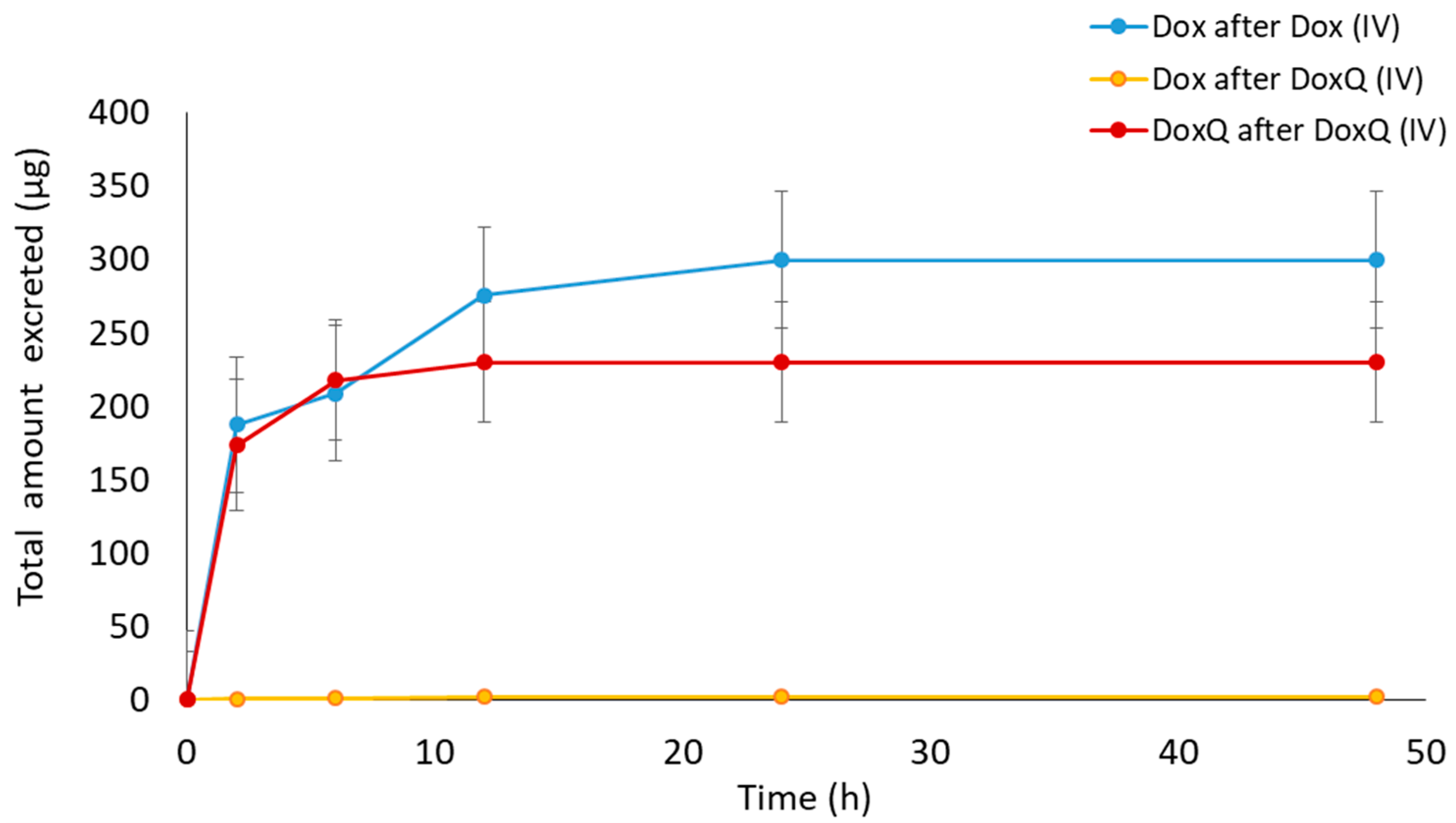
3.3. Pharmacokinetics of Dox and DoxQ
3.3.1. IV Administration
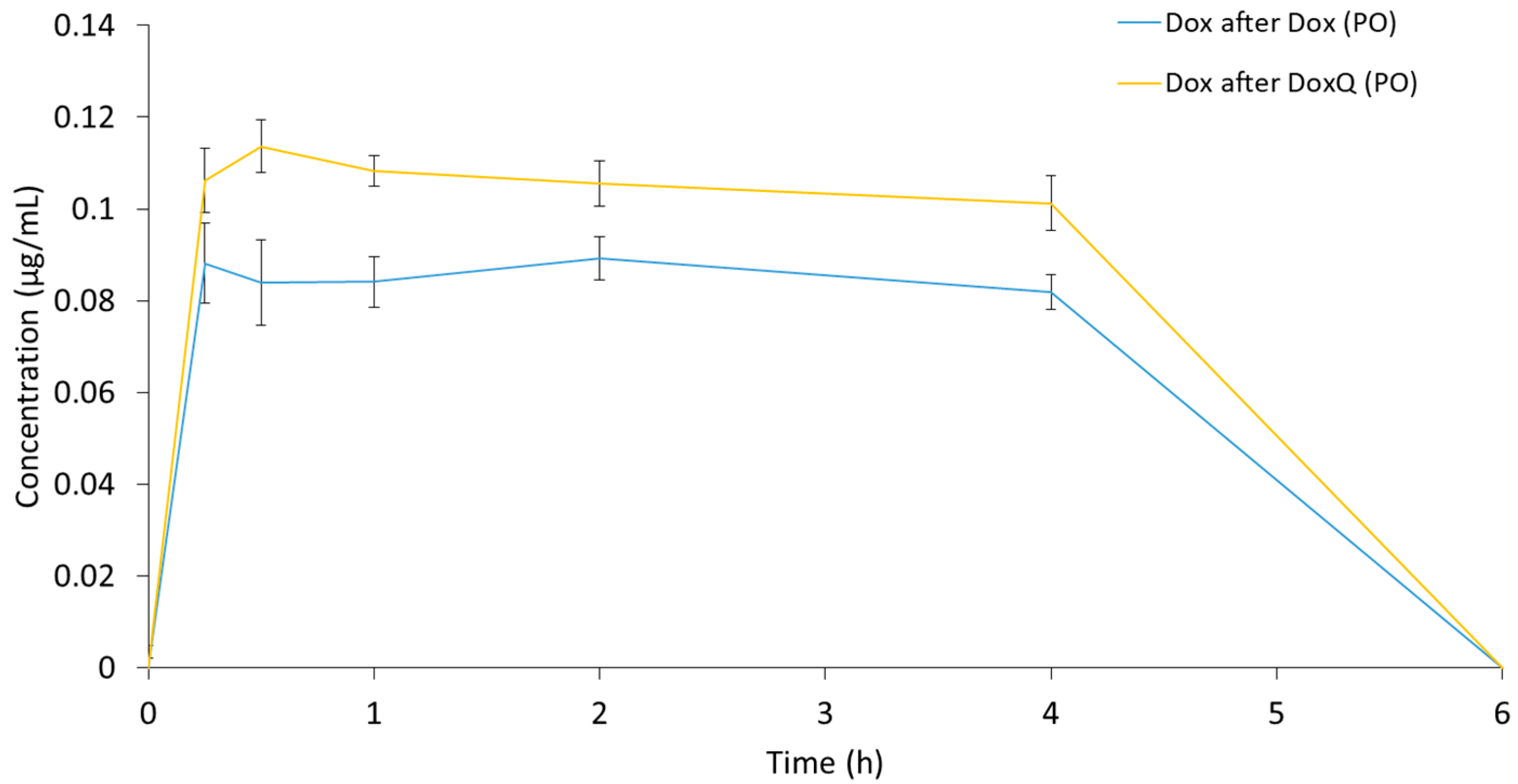
3.3.2. Oral Administration
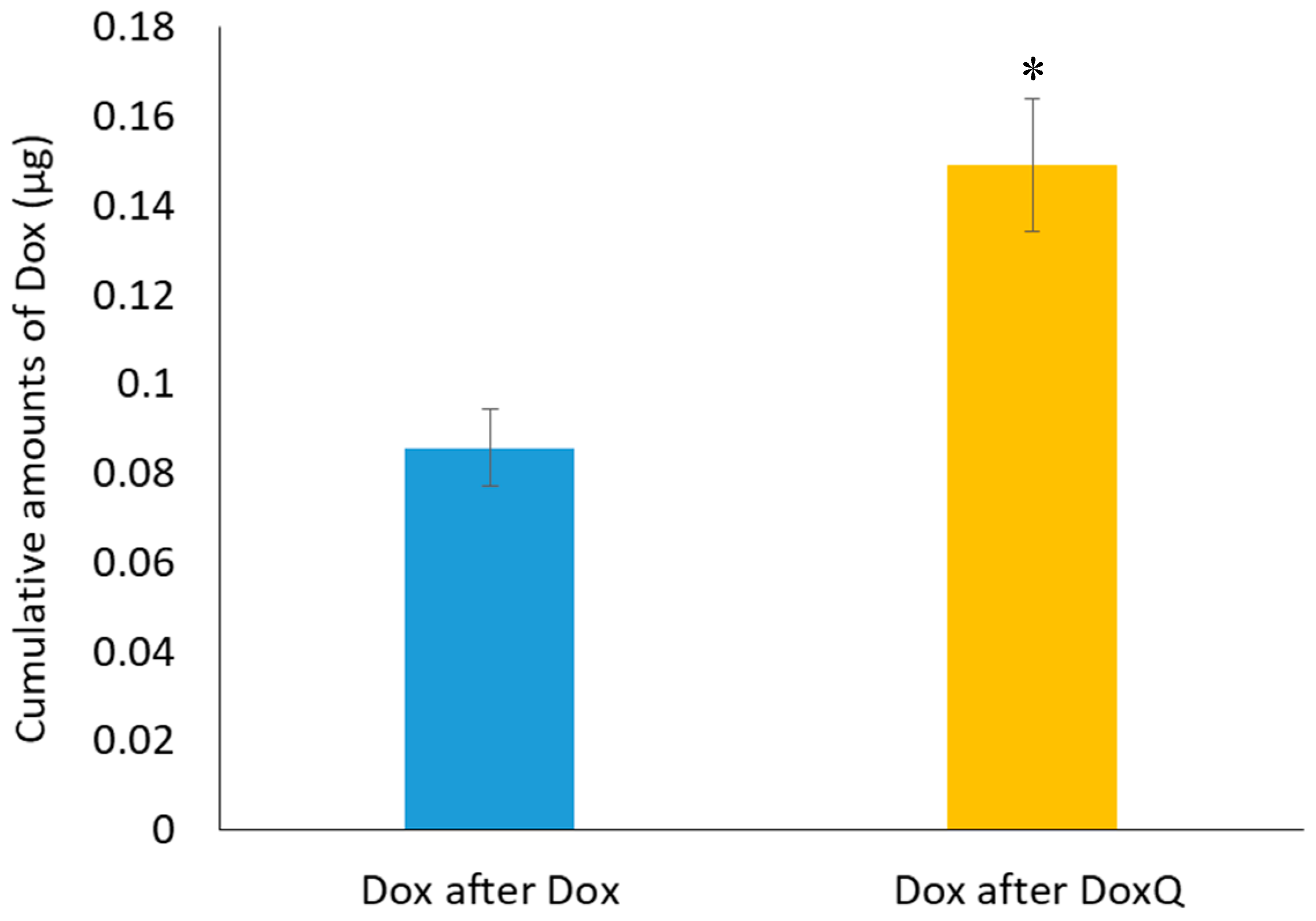
3.4. Intestinal Lymphatic Drug Delivery
3.4.1. Mesenteric Lymph Duct Cannulation
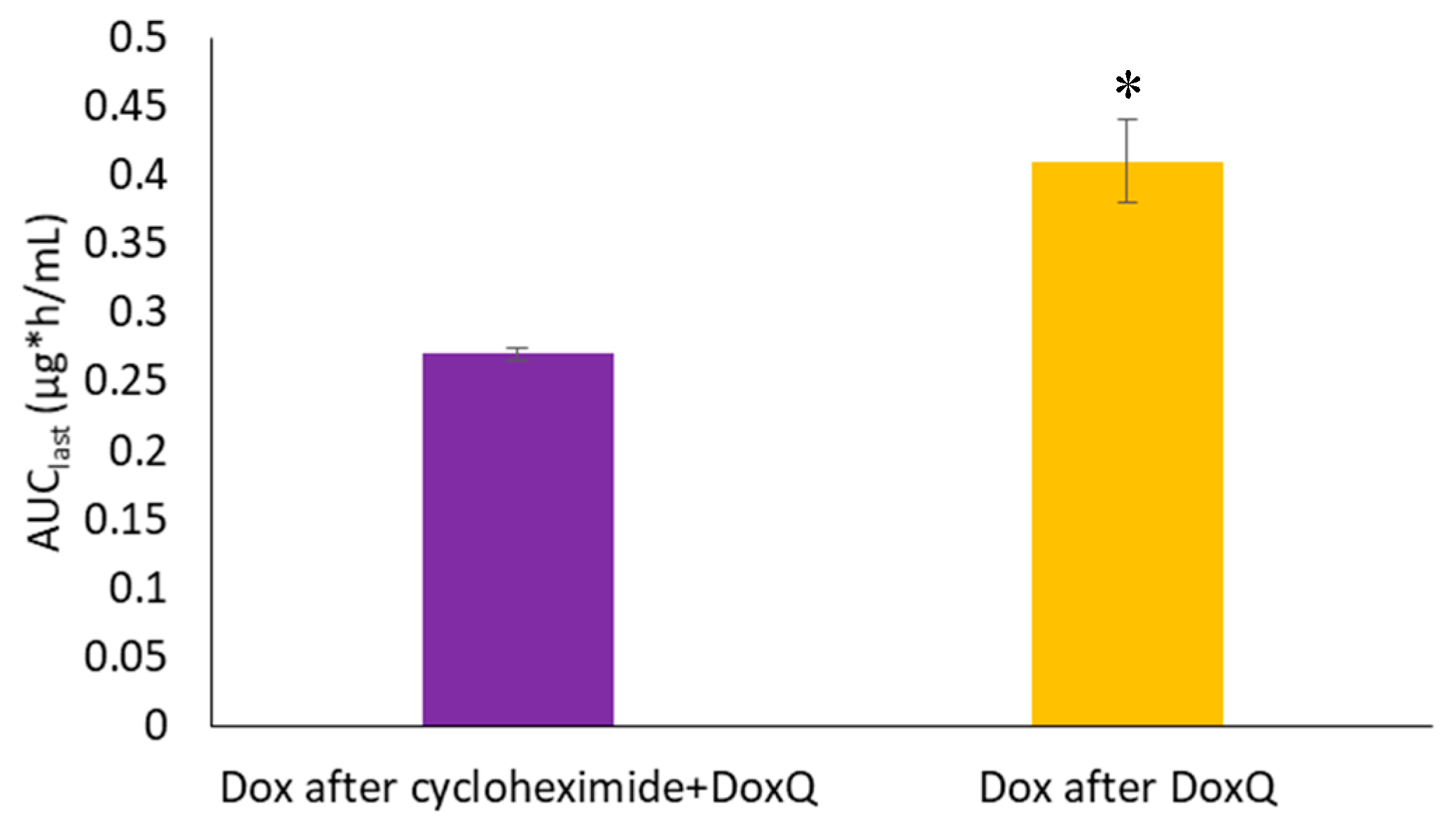
3.4.2. Lymph Blockage by Cycloheximide
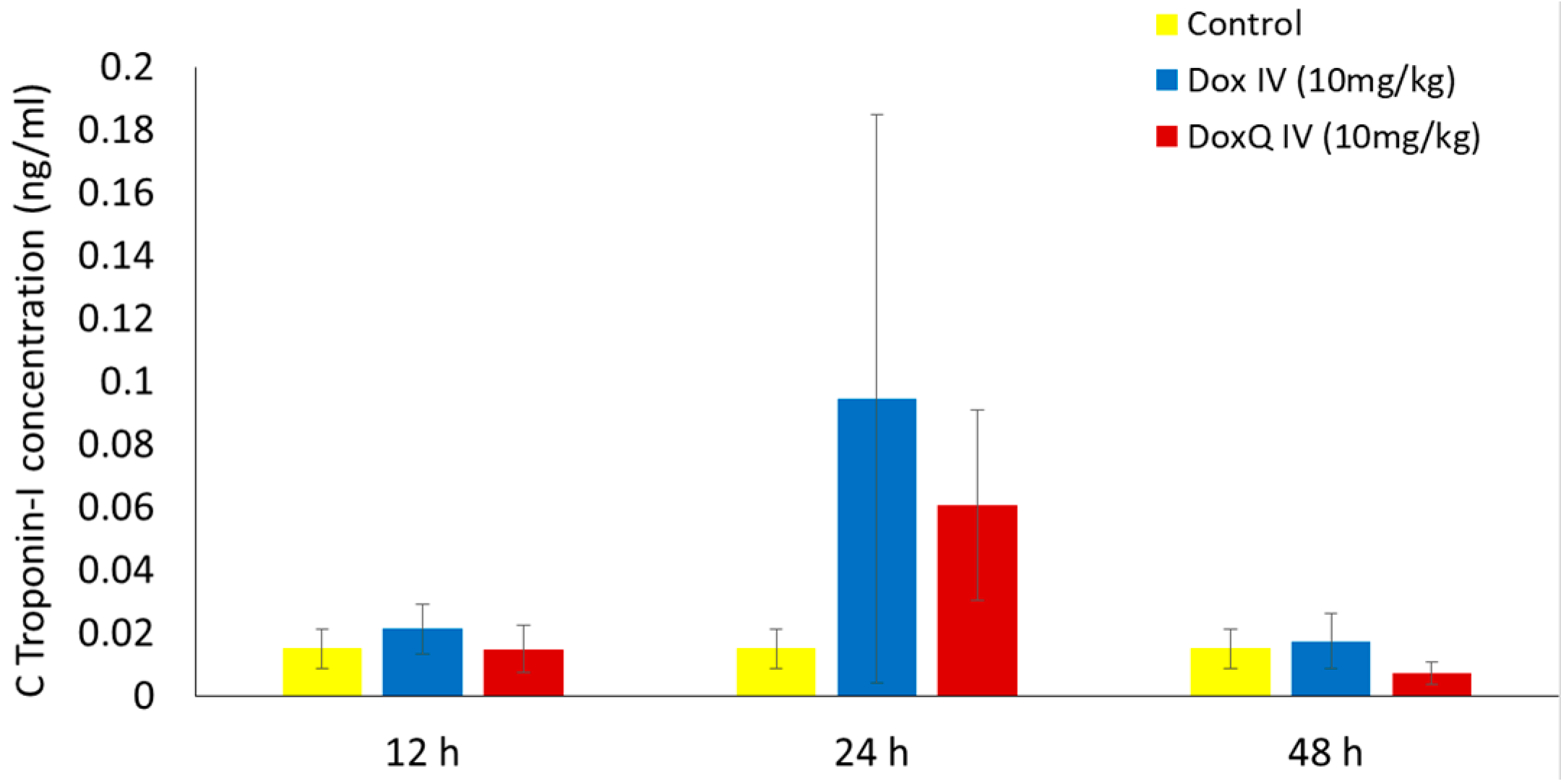
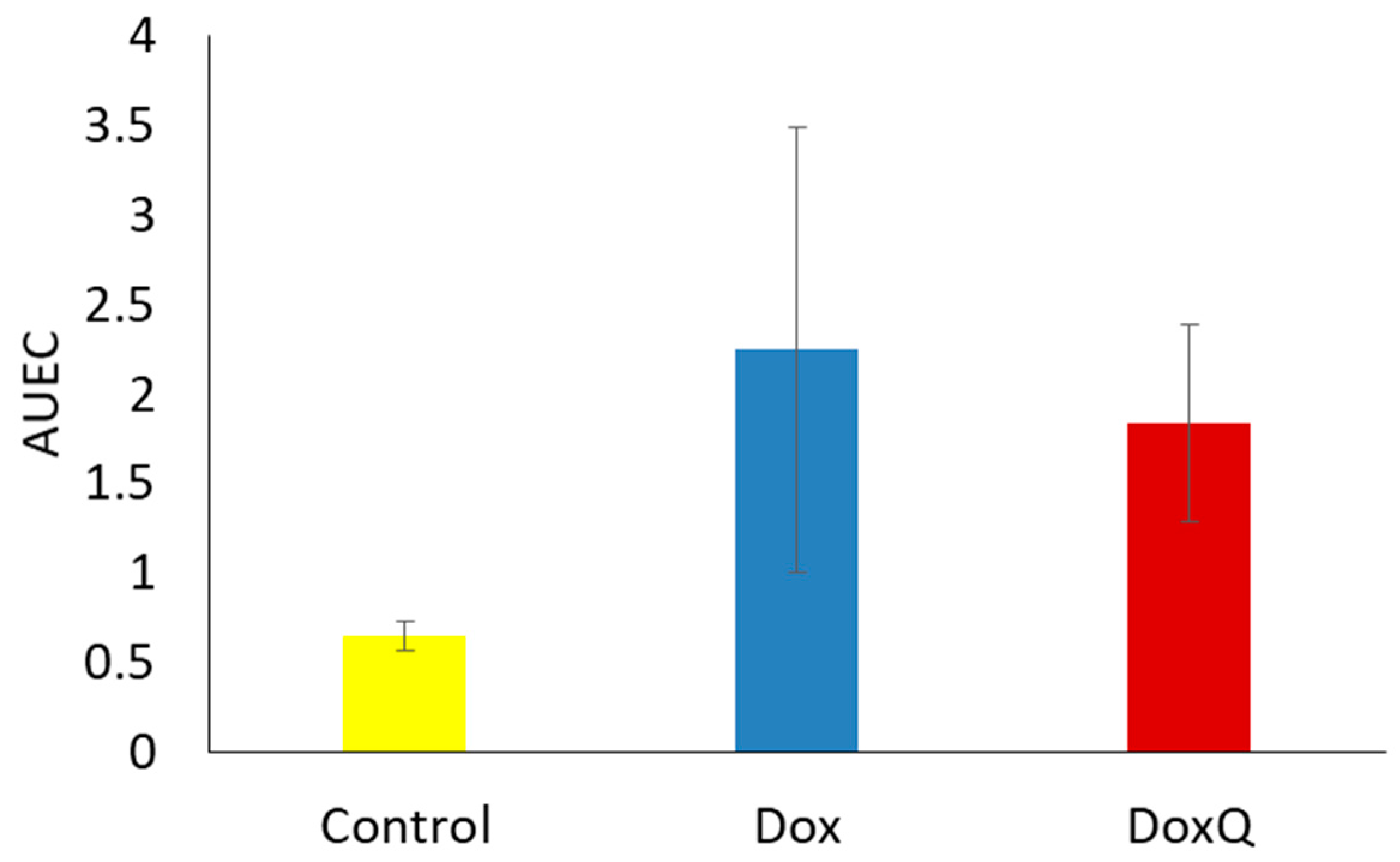
3.5. Cardiotoxicity of Dox and Dox
3.6. Renal Toxicity of Dox and DoxQ
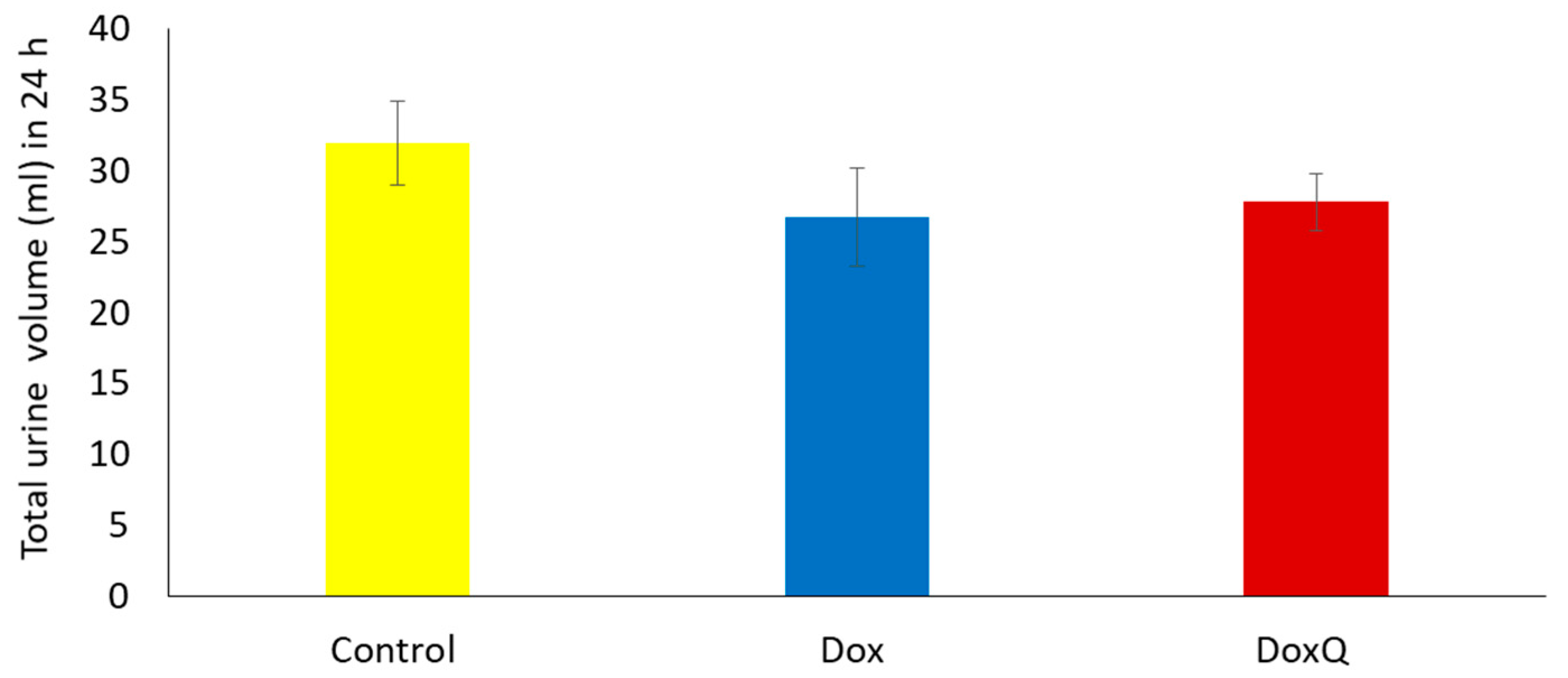
3.6.1. Urinary Output over 24 h
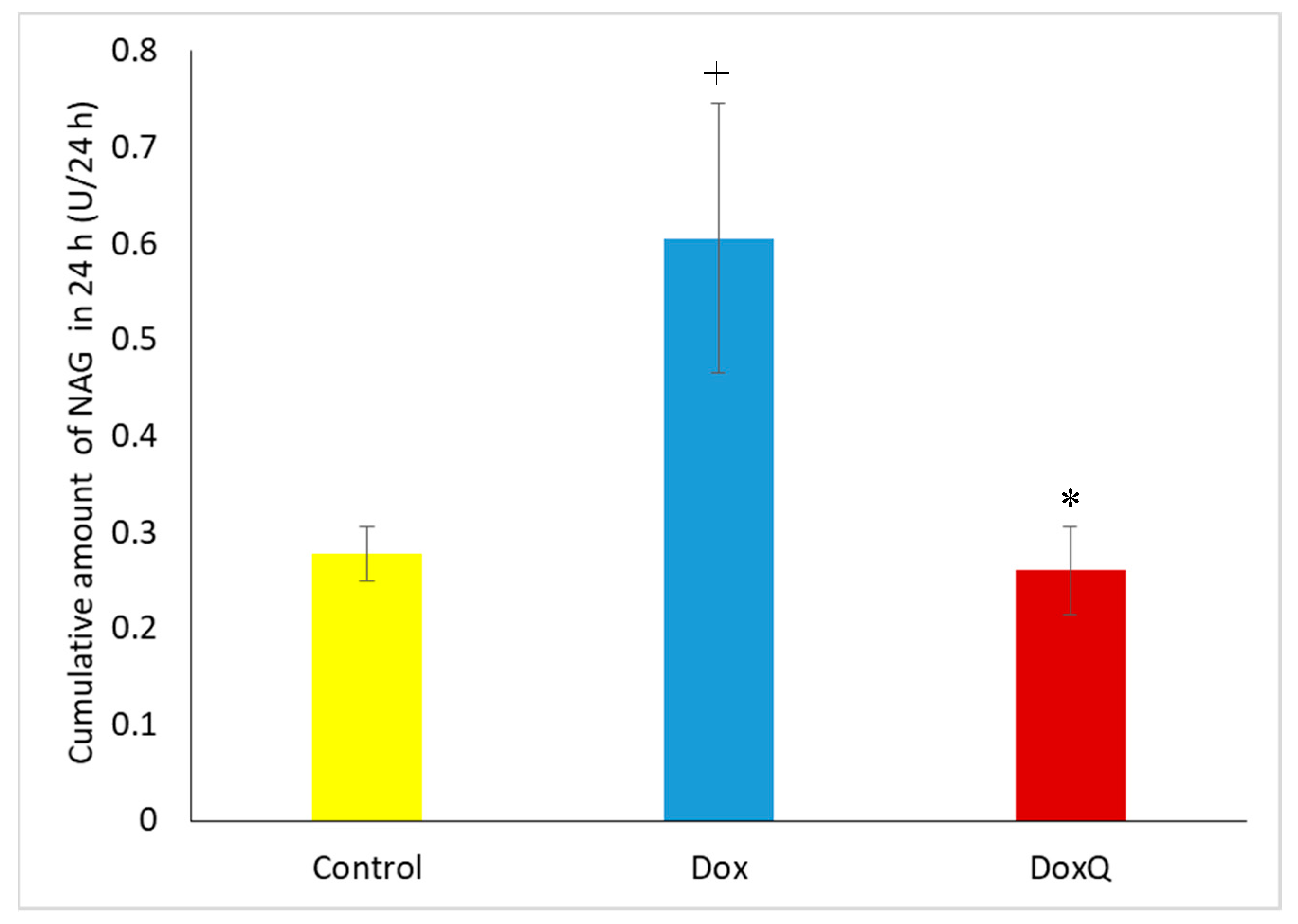
3.6.2. β-N-Acetylglucosaminidase (NAG)
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Funes, H.; Coronado, C. Role of anthracyclines in the era of targeted therapy. Cardiovasc. Toxicol. 2007, 7, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.D.; Pyle, L.; Allen, M.; Vaughan, M.; Webb, A.; Johnston, S.R.; Gore, M.E. A phase I dose-finding study of a combination of pegylated liposomal doxorubicin (Doxil), carboplatin and paclitaxel in ovarian cancer. Br. J. Cancer 2002, 86, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Dombernowsky, P.; Sledge, G.; Martin, M.; Amadori, D.; Arbuck, S.G.; Ravdin, P.; Brown, M.; Messina, M.; Tuck, D.; et al. Cardiac function following combination therapy with paclitaxel and doxorubicin: An analysis of 657 women with advanced breast cancer. Ann. Oncol. 2001, 12, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- McBride, N.C.; Cavenagh, J.D.; Ward, M.C.; Grant, I.; Schey, S.; Gray, A.; Hughes, A.; Mills, M.J.; Cervi, P.; Newland, A.C.; et al. Liposomal daunorubicin (DaunoXome) in combination with cyclophosphamide, vincristine and prednisolone (COP-X) as salvage therapy in poor-prognosis non-Hodgkins lymphoma. Leuk. Lymphoma 2001, 42, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Soloman, R.; Gabizon, A.A. Clinical pharmacology of liposomal anthracyclines: Focus on pegylated liposomal Doxorubicin. Clin. Lymphoma Myeloma 2008, 8, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, D.; Stefano, A.D.; Carone, V.; Fagotti, A.; Pisconti, S.; Scambia, G. Pegylated liposomal doxorubicin-related palmar-plantar erythrodysesthesia (‘hand-foot’ syndrome). Ann. Oncol. 2007, 18, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Thati, S.; Bagby, T.R.; Diab, H.M.; Davies, N.M.; Cohen, M.S.; Forrest, M.L. Localized doxorubicin chemotherapy with a biopolymeric nanocarrier improves survival and reduces toxicity in xenografts of human breast cancer. J. Control Release 2010, 146, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Hamaguchi, T.; Ura, T.; Muro, K.; Yamada, Y.; Shimada, Y.; Shirao, K.; Okusaka, T.; Ueno, H.; Ikeda, M.; et al. Phase I clinical trial and pharmacokinetic evaluation of NK911, a micelle-encapsulated doxorubicin. Br. J. Cancer 2004, 91, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Ríhová, B.; Strohalm, J.; Prausová, J.; Kubácková, K.; Jelínková, M.; Rozprimová, L.; Sírová, M.; Plocová, D.; Etrych, T.; Subr, V.; et al. Cytostatic and immunomobilizing activities of polymer-bound drugs: Experimental and first clinical data. J. Control Release 2003, 91, 1–16. [Google Scholar] [PubMed]
- Tolcher, A.W.; Sugarman, S.; Gelmon, K.A.; Cohen, R.; Saleh, M.; Isaacs, C.; Young, L.; Healey, D.; Onetto, N.; Slichenmyer, W. Randomized phase II study of BR96-doxorubicin conjugate in patients with metastatic breast cancer. J. Clin. Oncol. 1999, 17, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.L.; Merz, A.L.; Long, M.E. Pharmacokinetics of combined doxorubicin and paclitaxel in mice. Cancer Lett. 2005, 220, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lee, M.G. Effects of dexamethasone on the pharmacokinetics of adriamycin after intravenous administration to rats. Res. Commun. Mol. Pathol. Pharmacol. 1999, 105, 87–96. [Google Scholar] [PubMed]
- Alrushaid, S.; Zhao, Y.; Sayre, C.L.; Maayah, Z.H.; Forrest, M.L.; Senadheera, S.N.; Chaboyer, K.; Anderson, H.D.; El-Kadi, A.; Davies, N.M. Mechanistically Elucidating the In Vitro Safety and Efficacy of a Novel Doxorubicin Derivative. Drug Deliv. Transl. Res. 2017, 7, 582–597. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Piao, Y.J.; Kang, K.W. Effects of quercetin on the bioavailability of doxorubicin in rats: Role of CYP3A4 and P-gp inhibition by quercetin. Arch Pharm. Res. 2011, 34, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.L.; Tsai, Y.J.; Huang, C.M.; Tsai, T.H. Lymphatic absorption of quercetin and rutin in rat and their pharmacokinetics in systemic plasma. J. Agric. Food Chem. 2010, 58, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Murota, K.; Cermak, R.; Terao, J.; Wolffram, S. Influence of fatty acid patterns on the intestinal absorption pathway of quercetin in thoracic lymph duct-cannulated rats. Br. J. Nutr. 2013, 109, 2147–2153. [Google Scholar] [CrossRef] [PubMed]
- Murota, K.; Terao, J. Quercetin appears in the lymph of unanesthetized rats as its phase II metabolites after administered into the stomach. FEBS Lett. 2005, 579, 5343–5346. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, N.L.; Kaminskas, L.M.; Porter, C.J. From sewer to saviour—Targeting the lymphatic system to promote drug exposure and activity. Nat. Rev. Drug Discov. 2015, 14, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, N.L.; Hu, L.; Caliph, S.M.; Han, S.; Porter, C.J. The mesenteric lymph duct cannulated rat model: Application to the assessment of intestinal lymphatic drug transport. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, J.A.; Wang, S.W.; Knemeyer, I.W.; Wirth, M.A.; Alton, K.B. Intestinal lymphatic transport for drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 923–942. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.J.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory—Design and description. J. Comput. Aid. Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Virtual Computational Chemistry Laboratory. Available online: http://www.vcclab.org (accessed on 24 August 2017).
- Daeihamed, M.; Haeri, A.; Dadashzadeh, S. A Simple and Sensitive HPLC Method for Fluorescence Quantitation of Doxorubicin in Micro-volume Plasma: Applications to Pharmacokinetic Studies in Rats. Iran J. Pharm. Res. 2015, 14 (Suppl. 1), 33–42. [Google Scholar] [PubMed]
- Dahan, A.; Hoffman, A. Evaluation of a chylomicron flow blocking to investigate the intestinal lymphatic transport of lipophilic drugs. Eur. J. Pharm. Sci. 2005, 24, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Bhalekar, M.R.; Upadhaya, P.G.; Madgulkar, A.R.; Kshirsagar, S.J.; Dube, A.; Bartakke, U.S. In-Vivo bioavailability and lymphatic uptake evaluation of lipid nanoparticulates of darunavir. Drug Deliv. 2016, 23, 2581–2586. [Google Scholar] [PubMed]
- Fu, Q.; Sun, J.; Ai, X.; Zhang, P.; Li, M.; Wang, Y.; Liu, X.; Sun, Y.; Sui, X.; Sun, L.; et al. Nimodipine nanocrystals for oral bioavailability improvement: Role of mesenteric lymph transport in the oral absorption. Int. J. Pharm. 2013, 448, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Attili-Qadri, S.; Karra, N.; Nemirovski, A.; Schwob, O.; Talmon, Y.; Nassar, T.; Benita, S. Oral delivery system prolongs blood circulation of docetaxel nanocapsules via lymphatic absorption. Proc. Natl. Acad. Sci. USA 2013, 110, 17498–17503. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhai, X.; Xue, K.; Hu, L.; Yang, X.; Li, G.; Si, L. Intestinal absorption and intestinal lymphatic transport of sirolimus from self-microemulsifying drug delivery systems assessed using the single-passintestinal perfusion (SPIP) technique and a chylomicron flow blocking approach: Linear correlation with oral bioavailabilities in rats. Eur. J. Pharm. Sci. 2011, 43, 132–140. [Google Scholar] [PubMed]
- Lind, M.L.; Jacobsen, J.; Holm, R.; Müllertz, A. Intestinal lymphatic transport of halofantrine in rats assessed using a chylomicron flow blocking approach: The influence of polysorbate 60 and 80. Eur. J. Pharm. Sci. 2008, 35, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhang, Z.; Bu, H.; Huang, Y.; Gao, Z.; Shen, J.; Zhao, C.; Li, Y. Nanoemulsion improves the oral absorption of candesartan cilexetil in rats:Performance and mechanism. J. Control Release 2011, 149, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Vuddanda, P.R.; Singh, S. Intestinal Lymphatic Delivery of Praziquantel by Solid Lipid Nanoparticles: Formulation Design, In Vitro and In Vivo Studies. J. Nanotechnol. vol. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Makwana, V.; Jain, R.; Patel, K.; Nivsarkar, M.; Joshi, A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int. J. Pharm. 2015, 495, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.J.; Tsai, T.H. Mesenteric lymphatic absorption and the pharmacokinetics of naringin and naringenin in the rat. J. Agric. Food Chem. 2012, 60, 12435–12442. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.; Tozer, T.N. Metabolites and drug response. In Clinical Pharmacokinetics and Pharmacodynamics: Concepts and applications, 4th ed.; Tozer, R.M., Thomas, N., Eds.; Lippincott Williams & Wilkins: Baltimore, MD, USA, 2011; pp. 603–631. [Google Scholar]
- Mehvar, R.; Jamali, F.; Watson, M.W.; Skelton, D. Pharmacokinetics of tetrabenazine and its major metabolite in man and rat. Bioavailability and dose dependency studies. Drug Metab. Dispos. 1987, 15, 250–255. [Google Scholar] [PubMed]
- Hanafy, S.; Dagenais, N.J.; Dryden, W.F.; Jamali, F. Effects of angiotensin II blockade on inflammation-induced alterations of pharmacokinetics and pharmacodynamics of calcium channel blockers. Br. J. Pharmacol. 2008, 153, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Sattari, S.; Dryden, W.F.; Eliot, L.A.; Jamali, F. Despite increased plasma concentration, inflammation reduces potency of calcium channel antagonists due to lower binding to the rat heart. Br. J. Pharmacol. 2003, 139, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.P.; Ganguli, A.; Prakash, A. Drug-induced kidney diseases. J. Assoc. Physic. India 2003, 51, 970–979. [Google Scholar]
- Naughton, C.A. Drug-induced nephrotoxicity. Am. Fam. Physic. 2008, 78, 743–750. [Google Scholar]
- Okuda, S.; Oh, Y.; Tsuruda, H.; Onoyama, K.; Fujimi, S.; Fujishima, M. Adriamycin-induced nephropathy as a model of chronic progressive glomerular disease. Kidney Int. 1986, 29, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Le, J.M.; Han, Y.H.; Choi, S.J.; Park, J.S.; Jang, J.J.; Bae, R.J.; Lee, M.J.; Kim, M.J.; Lee, Y.H.; Kim, D.; et al. Variation of nephrotoxicity biomarkers by urinary storage condition in rats. Toxicol. Res. 2014, 30, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.E.; Davies, N.M. Enantiospecific pharmacokinetics of isoxanthohumol and its metabolite 8-prenylnaringenin in the rat. Mol. Nutr. Food Res. 2015, 59, 1674–1689. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.L.; Martinez, S.E.; Nagabushnam, K.; Majeed, M.; Alrushaid, S.; Sayre, C.L.; Davies, N.M. Calebin A: Analytical Development for Pharmacokinetics Study, Elucidation of Pharmacological Activities and Content Analysis of Natural Health Products. J. Pharm. Pharm. Sci. 2015, 18, 494–514. [Google Scholar] [CrossRef] [PubMed]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5280343#section=Entrez-Crosslink (accessed on 24 August 2017).
- Tian, S.; Hirshfield, K.M.; Jabbour, S.K.; Toppmeyer, D.; Haffty, B.G.; Khan, A.J.; Goyal, S. Serum biomarkers for the detection of cardiac toxicity after chemotherapy and radiation therapy in breast cancer patients. Front. Oncol. 2014, 4, 277. [Google Scholar] [CrossRef] [PubMed]
- Hadi, N.; Yousif, N.G.; Al-amran, F.G.; Huntei, N.K.; Mohammad, B.I.; Ali, S.J. Vitamin E and telmisartan attenuates doxorubicin induced cardiac injury in rat through down regulation of inflammatory response. BMC Cardiovasc. Disord. 2012, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Adamcova, M.; Sterba, M.; Simunek, T.; Potacova, A.; Popelova, O.; Mazurova, Y.; Gersl, V. Troponin as a marker of myocardiac damage in drug-induced cardiotoxicity. Expert Opin. Drug Saf. 2005, 4, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Babuin, L.; Jaffe, A.S. Troponin: The biomarker of choice for the detection of cardiac injury. CMAJ 2005, 173, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.W.; Abu-Mellal, A.K.; Davies, N.M. Formulation dependent pharmacokinetics, bioavailability and renal toxicity of a selective cyclooxygenase-1 inhibitor SC-560 in the rat. J. Pharm. Pharm. Sci. 2003, 6, 205–210. [Google Scholar] [PubMed]
- Burke, J.F.; Laucius, J.F.; Brodovsky, H.S.; Soriano, R.Z. Doxorubicin Hydrochloride-Associated Renal Failure. Arch Int. Med. 1977, 137, 385–388. [Google Scholar] [CrossRef]
- Luo, R.; Li, Y.; He, M.; Zhang, H.; Yuan, H.; Johnson, M.; Palmisano, M.; Zhou, S.; Sun, D. Distinct biodistribution of doxorubicin and the altered dispositions mediated by different liposomal formulations. Int. J. Pharm. 2017, 519, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, K.; Venkateswarlu, V. Pharmacokinetics, tissue distribution and bioavailability of clozapine solid nanoparticles after intravenous and intraduodenal administration. J. Control Release 2005, 107, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.E.; Sayre, C.L.; Davies, N.M. Pharmacometrics of 3-Methoxypterostilbene: A Component of Traditional Chinese Medicinal Plants. Evid.-Based Complement. Altern. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Schulze, J.D.; Waddington, W.A.; Eli, P.J.; Parsons, G.E.; Coffin, M.D.; Basit, A.W. Concentration-dependent effects of polyethylene glycol 400 on gastrointestinal transit and drug absorption. Pharm. Res. 2003, 20, 1984–1988. [Google Scholar] [CrossRef] [PubMed]
- Basit, A.W.; Newton, J.M.; Short, M.D.; Waddington, W.A.; Ell, P.J.; Lacey, L.F. The effect of polyethylene glycol 400 on gastrointestinal transit: Implications for the formulation of poorly-water soluble drugs. Pharm. Res. 2001, 18, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.L.; Yang, Y.; Dai, Y.; Li, Q.; Lin, G.; Ma, Y.M. Polyethylene glycol 400 (PEG400) affects the systemic exposure of oral drugs based on multiple mechanisms: Taking berberine as an example. RSC Adv. 2017, 7, 2435–2442. [Google Scholar] [CrossRef]
- Olson, R.D.; Mushlin, P.S. Doxorubicin cardiotoxicity: Analysis of prevailing hypotheses. FASEB J. 1990, 4, 3076–3086. [Google Scholar] [PubMed]
- Cote, B.; Carlson, L.J.; Rao, D.A.; Alani, A.W.G. Combinatorial resveratrol and quercetin polymeric micelles mitigate doxorubicin induced cardiotoxicity in vitro and in vivo. J. Control Release 2015, 213, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Yagmurca, M.; Yasar, Z.; Bas, O. Effects of quercetin on kidney injury induced by doxorubicin. Bratisl. Lekárske Listy 2015, 116, 486–489. [Google Scholar] [CrossRef]
- Heeba, G.H.; Mahmoud, M.E. Dual effects of quercetin in doxorubicin-induced nephrotoxicity in rats and its modulation of the cytotoxic activity of doxorubicin on human carcinoma cells. Environ. Toxicol. 2016, 31, 624–636. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Doxorubicin (Free Base) | Quercetin | DoxQ |
|---|---|---|---|
| Structure |  |  |  |
| Molecular Weight (g/mol) | 543.53 | 302.238 | 928.82 |
| Formula | C27H29NO11 | C15H10O7 | C45H40N2O20 |
| pKa (MarvinSketch) | 8.00, 9.17, 9.93, 12.67, 13.49, 14.10 | 6.38, 7.85, 8.63, 10.29, 12.82 | 6.37, 7.72, 7.94, 8.97, 9.51, 10.21, 12.53, 13.10, 13.57, 14.06, 14.77 |
| pKa (GastroPlus) | 6.77, 8.43, 9.5 | 7.24, 8.15, 9.12, 10.25, 11.35 | 7.21, 8.08, 8.78, 9.38, 9.91, 10.64, 11.26 |
| pKa (GastroPlus, after fitting solubility) | 6.974, 10.08 | 6.582, 8.15, 10.25, 11.35 | 7.978, 8.08, 8.78, 9.38, 10.64, 11.26 |
| logP (MarvinSketch) | 1.30 | 1.75 | 2.60 |
| logP (neutral, GastroPlus) | 0.49 | 1.96 | 2.61 |
| logP (VCCLAB) | 1.3 | 1.44 ± 0.55 | 3.8 ± 1.5 |
| logD7.4 (MarvinSketch) | 0.097 | 1.00 | 2.407 |
| Intrinsic solubility (MarvinSketch) | −4.05 logS | −2.49 logS | −6.47 logS |
| Solubility at pH 7.4 (MarvinSketch) | −3.27 logS | −1.42 logS | −5.34 logS |
| Solubility at pH 7.4 (MarvinSketch) | 0.243 mg/mL | 15.15 mg/mL | 0.006 mg/mL |
| logS (VCCLAB) | 2.7 | 2.78 | 3.43 |
| Melting point (experimental) | 242 °C | 316.5 °C * | 175 °C |
| Pharmacokinetic Parameter | Dox Administered | DoxQ Administered | |
|---|---|---|---|
| Dox | DoxQ 1 | Dox | |
| C0 (μg/mL) | 24.7 ± 14.2 | 108 ± 26.4 * | 1.23 ± 0.11 + |
| kel (h−1) | 0.16 ± 0.02 | 4.59 ± 0.78 * | 0.75 ± 0.076 + |
| t1/2 (h) | 4.69 ± 0.8 @ | 0.16 ± 0.3 * | 0.87 ± 0.07 |
| Clast (μg/mL) | 0.12 ± 0.03 | 1.11 ± 0.17 * | 0.09 ± 0.01 + |
| Tlast (h) 2 | 6 | 1 | 2 |
| AUClast (μg * h/mL) | 3.97 ± 0.7 @ | 18.6 ± 1.98 * | 0.46 ± 0.04 + |
| AUCinf (μg * h/mL) | 4.79 ± 1.83 | NC | 0.62 ± 0.03 |
| Vss (L/kg) | 6.35 ± 2.11 | 0.08 ± 0.015 | NA |
| CLrenal (L/h/kg) 1 | 0.28 ± 0.84 | 0.02 ± 0.005 | NA |
| CLhepatic (L/h/kg) 1 | 2.35 ± 0.36 | 0.51 ± 0.06* | NA |
| CLtotal (L/h/kg) 1 | 2.63 ± 0.39 | 0.53 ± 0.01 * | NA |
| fe (%) | 10.73 ± 3.14 | 4.32 ± 1.005 | NA |
| fm (%) | NA | NA | 11.66 ±0.86 |
| Pharmacokinetic Parameter | Dox Administered | DoxQ Administered | Cycloheximide + DoxQ Administered |
|---|---|---|---|
| Dox 1 | Dox | Dox | |
| Cmax (μg/mL) | 0.09 ± 0.01 | 0.11 ± 0.01 + | 0.07 ± 0.001 |
| Clast (μg/mL) | 0.08 ± 0.01 | 0.10 ± 0.01 | 0.07 ± 0.02 |
| Tlast (h) 2 | 4 | 4 | 4 |
| AUClast (μg * h/mL) | 0.33 ± 0.04 | 0.41 ± 0.03 + | 0.27 ± 0.005 |
| Fm (%) | NA | 10.32 ± 0.42 + | 6.81 ± 0.14 |
| F (%) | 8.57 ± 0.71 | NA | NA |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alrushaid, S.; Sayre, C.L.; Yáñez, J.A.; Forrest, M.L.; Senadheera, S.N.; Burczynski, F.J.; Löbenberg, R.; Davies, N.M. Pharmacokinetic and Toxicodynamic Characterization of a Novel Doxorubicin Derivative. Pharmaceutics 2017, 9, 35. https://doi.org/10.3390/pharmaceutics9030035
Alrushaid S, Sayre CL, Yáñez JA, Forrest ML, Senadheera SN, Burczynski FJ, Löbenberg R, Davies NM. Pharmacokinetic and Toxicodynamic Characterization of a Novel Doxorubicin Derivative. Pharmaceutics. 2017; 9(3):35. https://doi.org/10.3390/pharmaceutics9030035
Chicago/Turabian StyleAlrushaid, Samaa, Casey L. Sayre, Jaime A. Yáñez, M. Laird Forrest, Sanjeewa N. Senadheera, Frank J. Burczynski, Raimar Löbenberg, and Neal M. Davies. 2017. "Pharmacokinetic and Toxicodynamic Characterization of a Novel Doxorubicin Derivative" Pharmaceutics 9, no. 3: 35. https://doi.org/10.3390/pharmaceutics9030035
APA StyleAlrushaid, S., Sayre, C. L., Yáñez, J. A., Forrest, M. L., Senadheera, S. N., Burczynski, F. J., Löbenberg, R., & Davies, N. M. (2017). Pharmacokinetic and Toxicodynamic Characterization of a Novel Doxorubicin Derivative. Pharmaceutics, 9(3), 35. https://doi.org/10.3390/pharmaceutics9030035