Nanotechnologies in Pancreatic Cancer Therapy




Abstract
:
1. Introduction
2. Nanotechnologies in Cancer
3. Physiology of Pancreatic Cancer
4. Nanotechnologies in Pancreatic Cancer
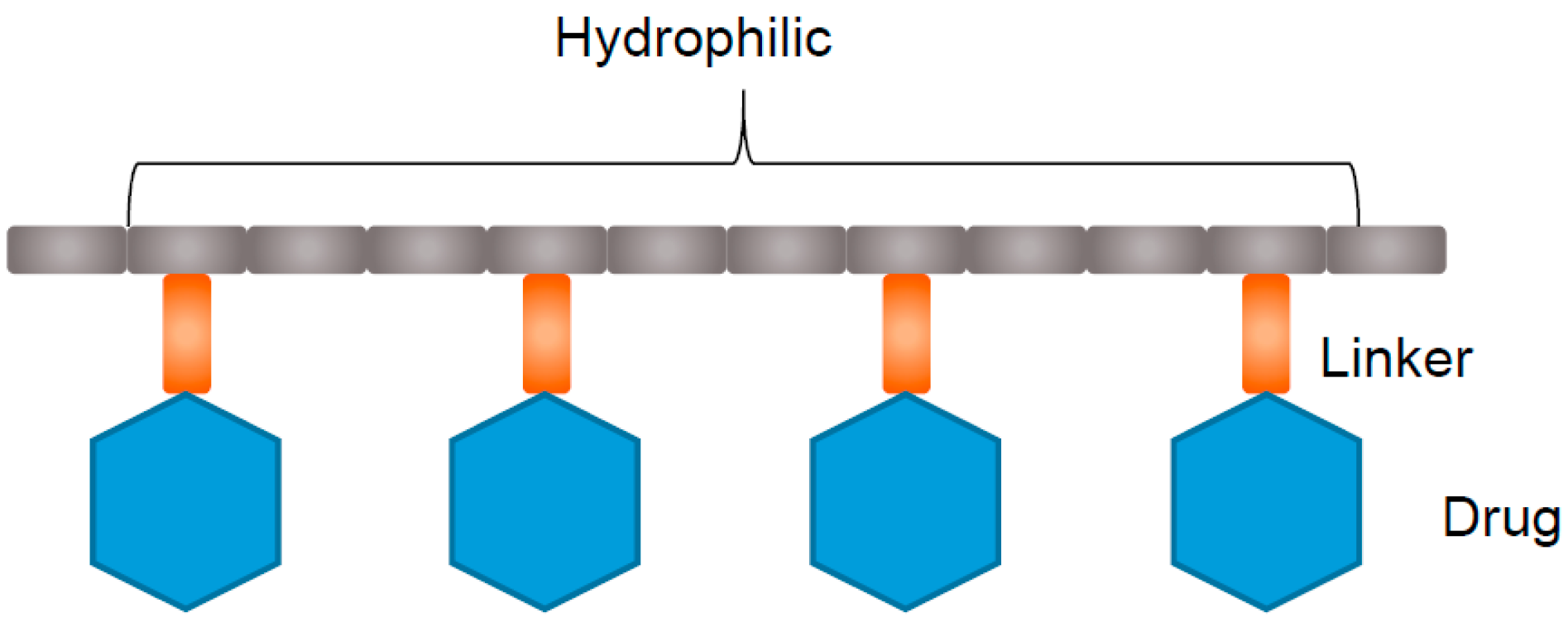
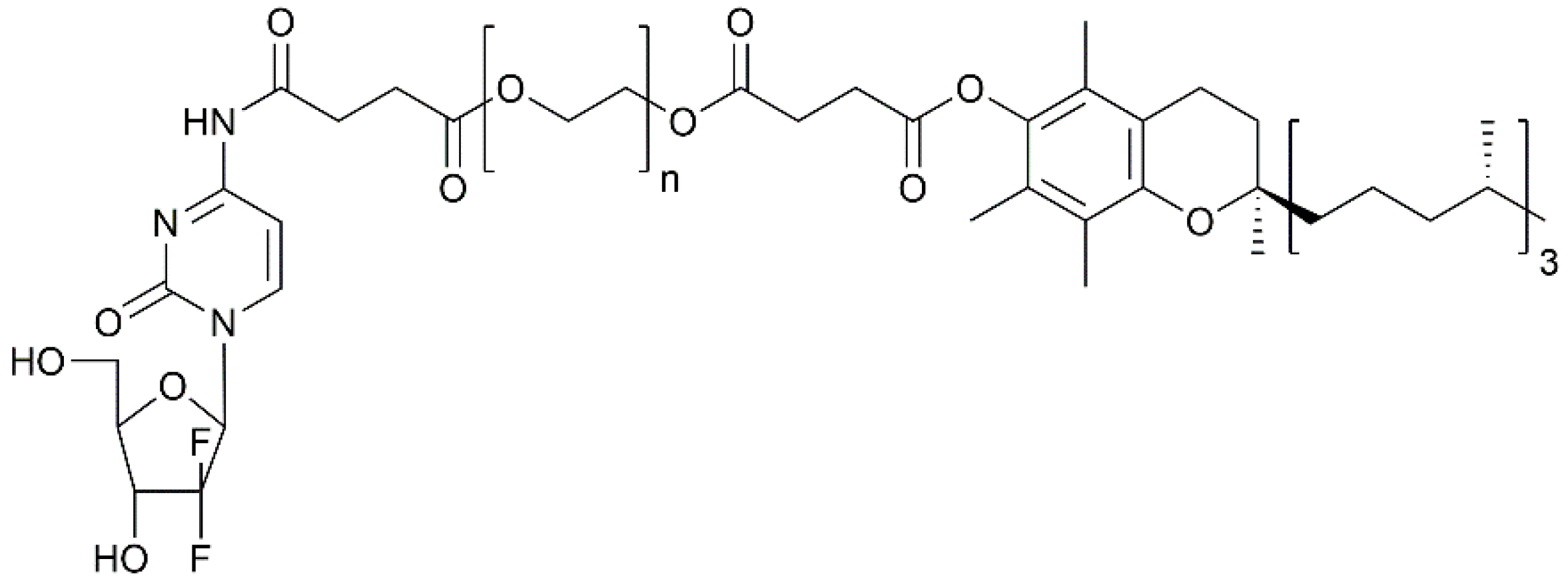
4.1. Polymer Drug Conjugates
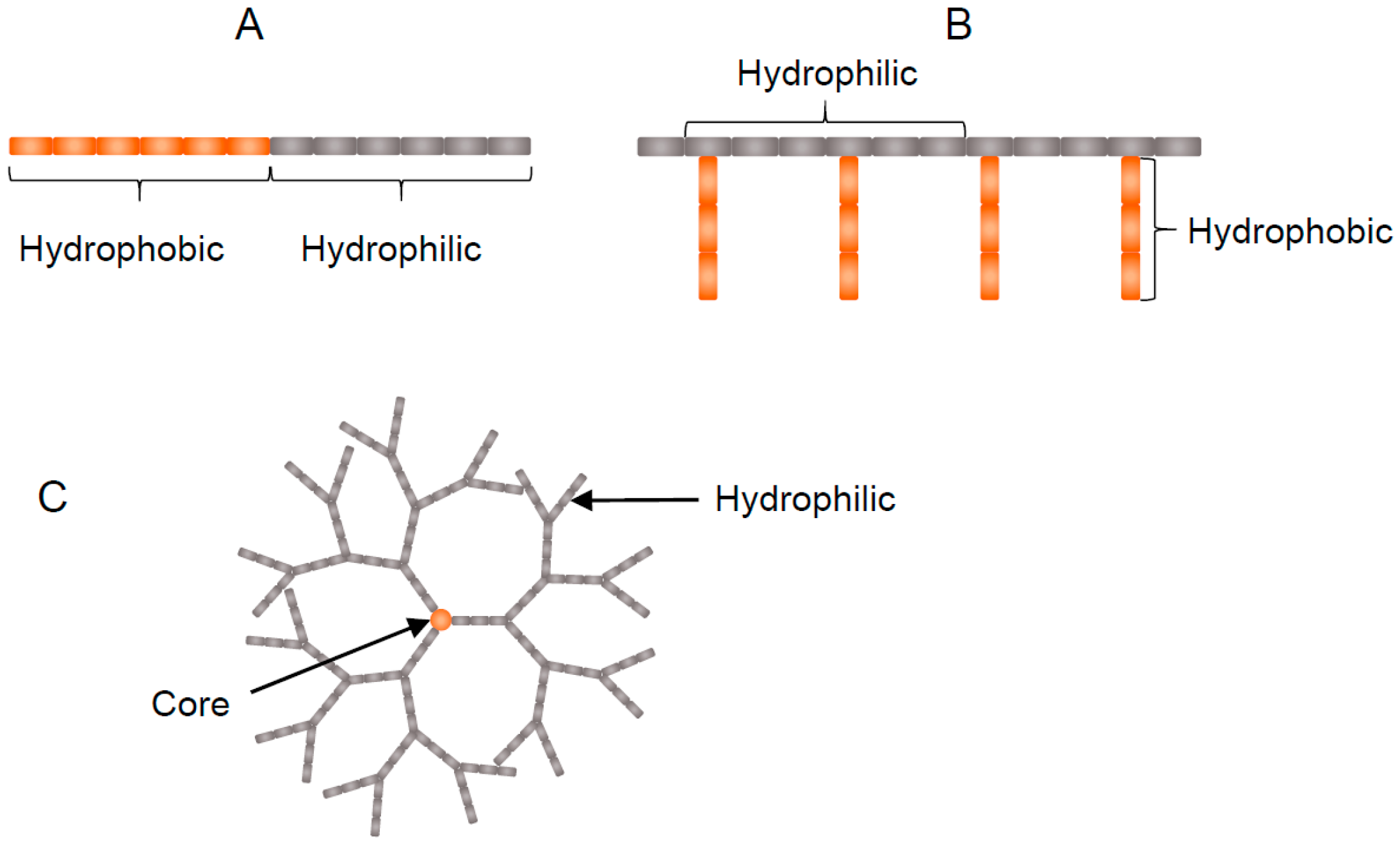
4.2. Amphiphilic Polymers
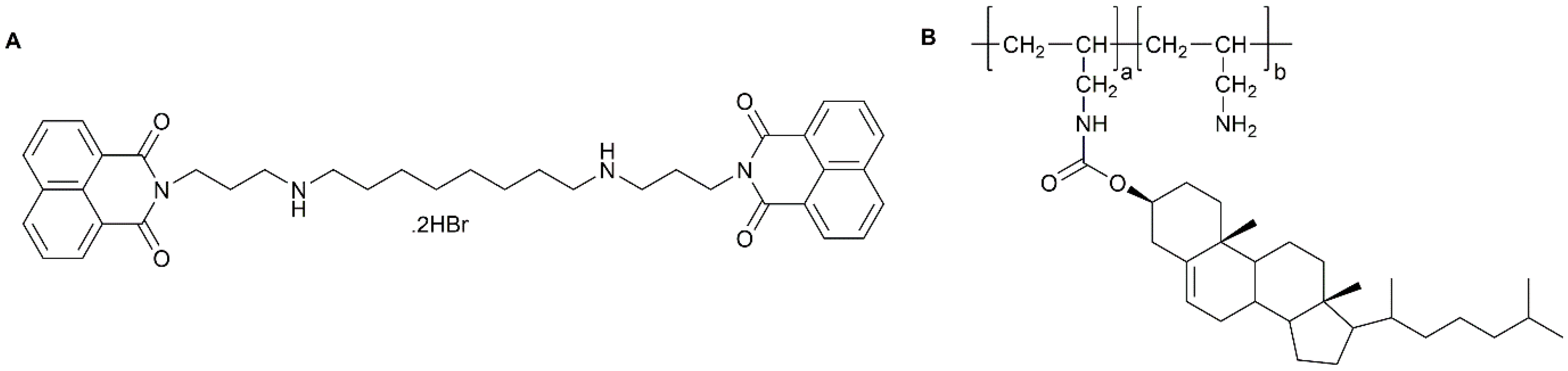
4.2.1. Block Copolymers
4.2.2. Graft Polymers
4.2.3. Dendrimers
4.2.4. Smart Polymers
4.3. Albumin
4.4. Inorganic Nanoparticles
4.4.1. Carbon Nanotubes
4.4.2. Quantum Dots
4.4.3. Iron Oxide Nanoparticles
4.4.4. Gold Nanoparticles
4.4.5. Hybrid Iron Oxide-Gold Nanoparticles
5. Conclusions and Future Perspectives
Author Contributions
Conflicts of Interest
References
- Lee, S.; Sissoko, M.; Hartshorn, K. Update on the Management of Pancreatic Cancer in Older Adults. Curr. Oncol. Rep. 2016, 18, 60. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. New Eng. J. Med. 2014, 371, 1039. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Löhr, J.-M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Garrido-laguna, I.; Hidalgo, M. Pancreatic cancer: From state-of-the-art treatments to promising novel therapies. Nat. Rev. Clin. Oncol. 2015, 12, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.-T.; Ye, Y.-P.; Deng, Y.-J.; Bian, X.-W.; Ding, Y.-Q. Metastatic cancer stem cells: From the concept to therapeutics. Am. J. Stem Cells 2014, 3, 46–62. [Google Scholar] [PubMed]
- Chadha, A.S.; Khoo, A.; Aliru, M.L.; Arora, H.K.; Gunther, J.R.; Krishnan, S. Recent Advances and Prospects for Multi-Modality Therapy in Pancreatic Cancer. Semin. Radiat. Oncol. 2016, 26, 320–337. [Google Scholar] [CrossRef] [PubMed]
- Government of Ontario. Metrolinx Five Year Strategy 2012–2017. In Canadian Research Index; Government of Ontario: Toronto, ON, Canada, 2011; pp. 1–33. [Google Scholar]
- Vaccaro, V.; Sperduti, I.; Vari, S.; Bria, E.; Melisi, D.; Garufi, C.; Nuzzo, C.; Scarpa, A.; Tortora, G.; Cognetti, F.; et al. Metastatic pancreatic cancer: Is there a light at the end of the tunnel? World J. Gastroenterol. 2015, 21, 4788. [Google Scholar] [CrossRef] [PubMed]
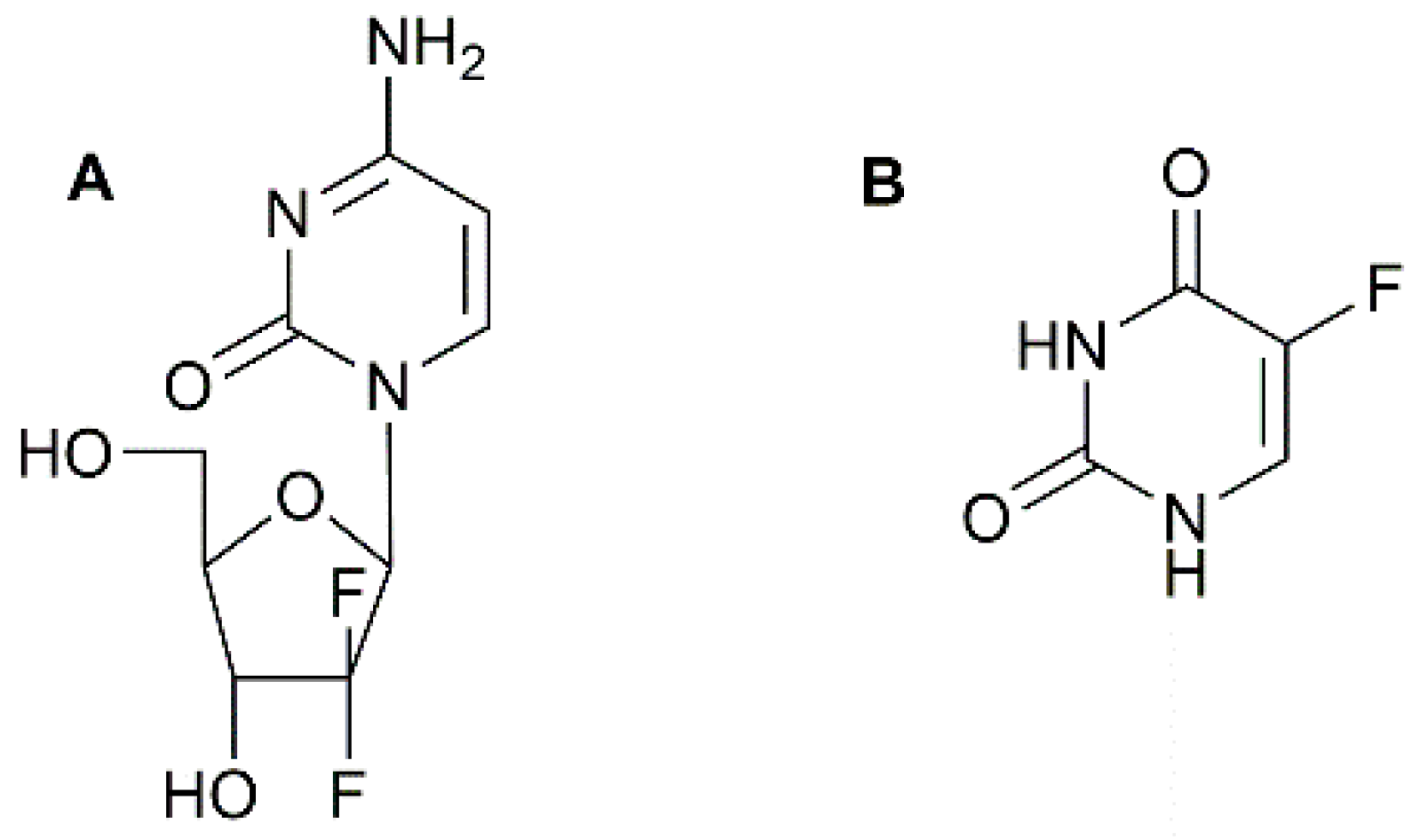
- Baraniak, J.; Pietkiewicz, A.; Kaczmarek, R.; Radzikowska, E.; Kulik, K.; Krolewska, K.; Cieslak, M.; Krakowiak, A.; Nawrot, B. N-Acyl-phosphoramidates as potential novel form of gemcitabine prodrugs. Bioorg. Med. Chem. 2014, 22, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kim, W.Y.; Huang, L. Systemic delivery of gemcitabine triphosphate via LCP nanoparticles for NSCLC and pancreatic cancer therapy. Biomaterials 2013, 34, 3447–3458. [Google Scholar] [CrossRef] [PubMed]
- Pliarchopoulou, K.; Pectasides, D. Pancreatic cancer: Current and future treatment strategies. Cancer Treat. Rev. 2009, 35, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Sotriolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Sa Cunha, A.; Rault, A.; Laurent, C.; Adhoute, X.; Vendrely, V.; Béllannée, G.; Brunet, R.; Collet, D.; Masson, B. Surgical Resection after Radiochemotherapy in Patients with Unresectable Adenocarcinoma of the Pancreas. J. Am. Coll. Surg. 2005, 201, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Malekigorji, M.; Curtis, A.D.M.; Hoskins, C. The Use of Iron Oxide Nanoparticles for Pancreatic Cancer Therapy. J. Nanomed. Res. 2014, 1, 1–12. [Google Scholar]
- Yuasa, T.; Inoshita, N.; Saiura, A.; Yamamoto, S.; Urakami, S.; Masuda, H.; Fujii, Y.; Fukui, I.; Ishikawa, Y.; Yonese, J. Clinical outcome of patients with pancreatic metastases from renal cell cancer. BMC Cancer 2015, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xue, X.; Liang, N.; Xu, D.; Liu, F.; Yu, X.; Zhang, J.D. Effect of chemoradiotherapy and neoadjuvant chemoradiotherapy in resectable pancreatic cancer: A systematic review and meta-analysis. J. Cancer Res. Clin. Oncol. 2014, 140, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Paulson, A.S.; Cao, H.S.T.; Tempero, M.A.; Lowy, A.M. Therapeutic advances in pancreatic cancer. Gastroenterology 2013, 144, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Demaria, S.; Formenti, S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J. Immunother. Cancer 2016, 4, 151. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, A.; Ahmed, A.M.; Mohiuddin, M.; Coleman, C.N. Exploiting sensitization windows of opportunity in hyper and hypo-fractionated radiation therapy. J. Thorac. Dis. 2014, 6, 287–302. [Google Scholar] [PubMed]
- Cid-Arregui, A.; Juarez, V. Perspectives in the treatment of pancreatic adenocarcinoma. World J. Gastroenterol. 2015, 21, 9297–9316. [Google Scholar] [CrossRef] [PubMed]
- Batmani, Y.; Khaloozadeh, H. Optimal drug regimens in cancer chemotherapy: A multi-objective approach. Comput. Biol. Med. 2013, 43, 2089–2095. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, E.T.; Safwat, G.M. Evaluation of cardioprotective activity of Lepidium sativum seed powder in albino rats treated with 5-fluorouracil. Beni-Suef Univ. J. Basic Appl. Sci. 2016, 5, 208–215. [Google Scholar] [CrossRef]
- Myung, J.H.; Tam, K.A.; Park, S.; Cha, A.; Hong, S. Recent advances in nanotechnology-based detection and separation of circulating tumor cells. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Buzea, C.; Pacheco, I.; Robbie, K. Nanomaterials and nanoparticles: Sources and toxicity. Biointerphases 2007, 2, MR17–MR71. [Google Scholar] [CrossRef] [PubMed]
- Liang, X. Nanopharmaceutics: The Potential Application of Nanomaterials; World Scientific Publishing Co., Pte Ltd.: Singapore, 2013. [Google Scholar]
- Greish, K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol. Biol. 2010, 624, 25–37. [Google Scholar] [PubMed]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Goodman, T.T.; Chen, J.; Matveev, K.; Pun, S.H. Spatio-temporal modeling of nanoparticle delivery to multicellular tumor spheroids. Biotechnol. Bioeng. 2008, 101, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Popović, Z.; Chen, O.; Cui, J.; Fukumura, D.; Bawendi, M.G.; Jain, R.K. Fluorescent nanorods and nanospheres for real-time in vivo probing of nanoparticle shape-dependent tumor penetration. Angew. Chem. 2011, 50, 11417–11420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, F.; Leunig, M.; Huang, S.K.; Berk, D.A.; Papahadjopoulos, D.; Jain, R.K. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994, 54, 3352–3356. [Google Scholar] [PubMed]
- Xu, R.; Zhang, G.; Mai, J.; Deng, X.; Segura-Ibarra, V.; Wu, S.; Shen, J.; Liu, H.; Hu, Z.; Chen, L.; et al. An injectable nanoparticle generator enhances delivery of cancer therapeutics. Nat. Biotechnol. 2016, 34, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Chittasupho, C.; Anuchapreeda, S.; Sarisuta, N. CXCRA targeted dendrimer for anti-cancer drug delivery and breast cancer cell migration inhibition. Eur. J. Pharm. Biopharm. 2017, 119, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Luong, D.; Kesharwani, P.; Alsaab, H.O.; Sau, S.; Padhye, S.; Sarkar, F.H.; Iyer, A.K. Folic acid conjugated polymeric micelles loaded with a curcumin difluorinated analog for targeting cervical and ovarian cancers. Colloid Surf. B Biointerfaces 2017, 157, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Pantshwa, J.; Choonara, Y.E.; Kumar, P.; du Toit, L.C.; Penny, C.; Pillay, V. Synthesis of novel amphiphilic poly(-isopropylacrylamide)-poly(aspartic acid) nanomicelles for potential targeted chemotherapy in ovarian cancer. J. Drug Deliv. Sci. Technol. 2017, 39, 308–323. [Google Scholar] [CrossRef]
- Motawi, T.K.; El-Maraghy, S.A.; El Meshad, A.N.; Nady, O.M.; Hammam, O.A. Cromolyn chitosan nanoparticles as a novel protective approach for colorectal cancer. Chemic. Biol. Interact. 2017, 275, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.-H.; Luo, T.-Y.; Chiang, P.-F.; Yao, C.-J.; Lin, W.-J.; Peng, C.-L.; Shieh, M.-J. EGFR-targeted micelles containing near-infrared dye for enhanced photothermal therapy in colorectal cancer. J. Control. Release 2017, 258, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, G.; El Sadda, R.; Botchkina, G.; Ojima, I.; Egan, J.; Amiji, M. Nanoemulsion formulation of a novel taxoid DHA-SBT-1214 inhibits prostate cancer stem cell-induced tumor growth. Cancer Lett. 2017, 406, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.-Y.; Hsiao, J.-K.; Wang, Y.-P.; Lan, C.-H.; Wu, H.-C. Peptide-conjugated nanoparticles for targeted imaging and therapy of prostate cancer. Biomaterials 2016, 99, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, X.; Zhou, Q.; Sui, M.; Lu, Z.; Zhou, Z.; Tang, J.; Miao, Y.; Zheng, M.; Wang, W.; et al. Terminating the criminal collaboration in pancreatic cancer: Nanoparticle based synergistic therapy for overcoming fibroblast-induced drug resistance. Biomaterials 2017, 144, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Khare, V.; Al Shakarchi, W.; Gupta, P.N.; Curtis, A.D.M.; Hoskins, C. Synthesis and characterization of TPGS-gemcitabine prodrug micelles for pancreatic cancer therapy. RSC Adv. 2016, 6, 60126–60137. [Google Scholar] [CrossRef]
- Joubert, F.; Martin, L.; Perrier, S.; Pasparakis, G. Development of a gemcitabine-polymer conjugate with prolonged cytotoxicity against a pancreatic cancer cell line. ACS Macro Lett. 2017, 6, 535–540. [Google Scholar] [CrossRef]
- Mittal, A.; Chitkara, D.; Behrman, S.W.; Mahato, R.I. Efficacy of gemcitabine conjugated and miRNA-405 complexed micelles for treatment of advanced pancreatic cancer. Biomaterials 2014, 35, 7007–7087. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, P.; Banerjee, S.; Padhye, S.; Sarkar, F.H.; Iyer, A.K. Parenterally administrable nano-micelles of 3,4-difluorobenzylidene curcumin for treating pancreatic cancer. Colloid Surf. B Biointerfaces 2015, 132, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Miura, Y.; Yamada, N.; Chida, T.; Liu, X.; Kim, A.; Sato, R.; Tsumura, R.; Koga, Y.; Yasunaga, M.; et al. Antibody fragment-conjugated polymeric micelles incorporating platinum drugs for targeted therapy of pancreatic cancer. Biomaterials 2015, 39, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Veeren, A.; Bhaw-Luximon, A.; Mukhopadhyay, D.; Jhurry, D. Mixed poly(vinyl pyrrolidone)-based drug-loaded nanomicelles shows enhanced efficacy against pancreatic cancer cell lines. Eur. J. Pharm. Sci. 2017, 102, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, C.; Ouaissi, M.; Lima, S.C.; Cheng, W.-P.; Loureiro, I.; Mas, E.; Lombardo, D.; Cordeiro-da-Silva, A.; Ouissi, A.; Kong Thoo Lin, P. In vitro and in vivo anticancer activity of a novel nano-sized formulation based on self-assembling polymers against pancreatic cancer. Pharm. Res. 2010, 27, 2694–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesharwani, P.; Xie, L.; Banerjee, S.; Mao, G.; Padhye, S.; Sarkar, F.H.; Iyer, A.K. Hyaluronic acide-conjugated polyamindoamine dendrimers for targeted delivery of 3,4-difluorobenzylidene curcumin to CD44 overexpressing pancreatic cancer cells. Colloids Surf. B Biointerfaces 2015, 136, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, K.; Esendagli, G.; Gürbüz, M.U.; Tülü, M.; Çalis, S. Effective targeting of gemcitabine to pancreatic cancer through PEG-cored Flt-1 antibody-conjugated dendrimers. Int. J. Pharm. 2017, 517, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Emamzadeh, M.; Desmaële, D.; Couvreur, P.; Pasparakis, G. Dual controlled delivery of squalenoyl-gemcitabine and paclitaxel using thermo-responsive polymeric micelles for pancreatic cancer. J. Control. Release 2017, 259, e90–e91. [Google Scholar] [CrossRef]
- Li, X.; Szewczuk, M.R.; Malardier-Jugroot, C. Folic acid-conjugated amphiphilic alternating copolymer as a new active tumor targeting drug delivery platform. Drug Des. Dev. Ther. 2016, 10, 4101–4110. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, N.Y.; Kennedy, A.M.; Shea, J.E.; Scaife, C.L.; Nam, K. Controlled and targeted tumor chemotherapy by ultrasound-activated nanoemulsions/microbubbles. J. Control. Release 2009, 138, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ramanathan, R.K.; Borad, M.J.; Laheru, D.A.; Smith, L.S.; Wood, T.E.; Korn, R.L.; Desai, N.; Trieu, V.; Iglesias, J.L.; et al. Gemcitabine Plus nab-Paclitaxel Is an Active Regimen in Patients With Advanced Pancreatic Cancer: A Phase I/II Trial. J. Clin. Oncol. 2011, 29, 4548–4554. [Google Scholar] [CrossRef] [PubMed]
- Frese, K.K.; Neesse, A.; Cook, N.; Bapiro, T.E.; Lolkema, M.P.; Jodrell, D.I.; Tuveson, D.A. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012, 2, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Arachchige, M.P.; Laha, S.S.; Naik, A.R.; Lewis, K.T.; Naik, R.; Jena, B.P. Functionalized nanoparticles enable tracking the rapid entry and release of doxorubicin in human pancreatic cancer cells. Micron 2017, 92, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Trabulo, S.; Aires, A.; Aicher, A.; Heeschen, C.; Cortajarena, A.L. Multifunctional iron oxide nanoparticles for selective targeting of pancreatic cancer cells. Biochim. Biophys. A 2017, 1861, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Malekigorji, M.; Alfahad, M.; Kong Thoo Lin, K.; Jones, S.; Curtis, A.; Hoskins, C. Thermally triggered theranostics for pancreatic cancer therapy. Nanoscale 2017, 9, 12735–12745. [Google Scholar] [CrossRef] [PubMed]
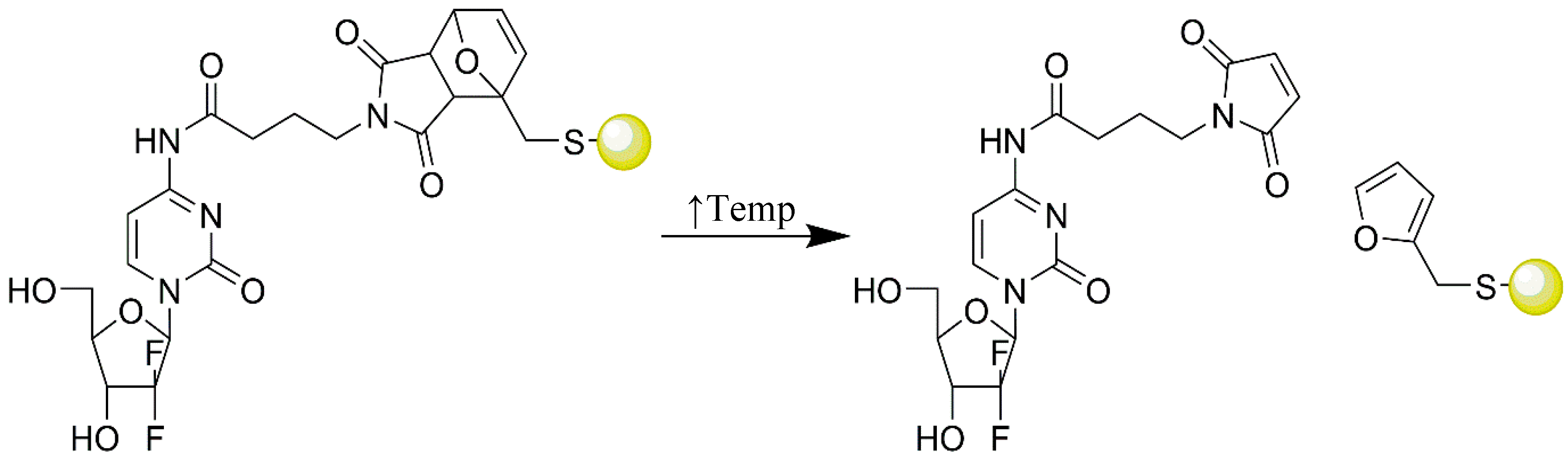
- Oluwasanmi, A.; Al-Shakarchi, W.; Manzur, A.; Aldebasi, M.H.; Elsini, R.S.; Albusair, M.K.; Haxton, K.J.; Curtis, A.D.M.; Hoskins, C. Diels Alder-mediated release of gemcitabine from hybrid nanoparticles for enhanced pancreatic cancer therapy. J. Control. Release 2017, in press. [Google Scholar] [CrossRef]
- Roshani, R.; McCarthy, F.; Hagemann, T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014, 345, 157–163. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, R.F.; Hruban, R.H.; Maitra, A.; Offerhaus, G.J.A. Reporting precursors to invasive pancreatic cancer: Pancreatic intraepithelial neoplasia, intraductal neoplasms and mucinous cystic neoplasm. Diagn. Histopathol. 2012, 18, 17–30. [Google Scholar] [CrossRef]
- Zhi, X.; Tao, J.; Xie, K.; Zhu, Y.; Li, Z.; Tang, J.; Wang, W.; Xu, H.; Zhang, J.; Xu, Z. MUC4-induced nuclear translocation of β-catenin: A novel mechanism for growth, metastasis and angiogenesis in pancreatic cancer. Cancer Lett. 2014, 346, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Z’graggen, K.; Centeno, B.A.; Fernandez-del Castillo, C.; Jimenez, R.E.; Werner, J.; Warshaw, A.L. Biological implications of tumor cells in blood and bone marrow of pancreatic cancer patients. Surgery 2001, 129, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Katiyar, S.K. Grape seed proanthocyanidins inhibit migration potential of pancreatic cancer cells by promoting mesenchymal-to-epithelial transition and targeting NF-κB. Cancer Lett. 2013, 334, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Hosoki, T. Dynamic CT of pancreatic tumors. Am. J. Roentgenol. 1983, 140, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Sofuni, A.; Iijima, H.; Moriyasu, F.; Nakayama, D.; Shimizu, M.; Nakamura, K.; Itokawa, F.; Itoi, T. Differential diagnosis of pancreatic tumors using ultrasound contrast imaging. J. Gastroenterol. 2005, 40, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-penetrating delivery of compounds and nanopartciles into tumours. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Grzesiak, J.J.; Ho, J.C.; Moossa, A.R.; Bouvet, M. The integrin-extracellular matrix axis in pancreatic cancer. Pancreas 2007, 35, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Dodson, L.F.; Hawkins, W.G.; Goedegebuure, P. Potential targets for pancreatic cancer immunotherapies. Immunotherapy 2011, 3, 517–537. [Google Scholar] [CrossRef] [PubMed]
- Felix, K.; Gaida, M.M. Neutrophil-derived proteases in the microenvironment of pancreatic cancer-active players in tumor progression. Int. J. Biol. Sci. 2016, 12, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Canal, F.; Sanchis, J.; Vicent, M.J. Polymer—Drug conjugates as nano-sized medicines. Curr. Opin. Biotechnol. 2011, 22, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, Q.; Liu, X.; Zhou, Z.; Tang, J.; Shen, Y. Enhanced anti-tumor efficacy by co-delivery of GDC-0449 with size-tunable polymeric SN38 nanoparticles in pancreatic cancer. J. Control. Release 2017, 259, e87–e88. [Google Scholar] [CrossRef]
- Ernsting, M.J.; Hoang, B.; Lohse, I.; Undzys, E.; Cao, P.; Do, T.; Gill, B.; Pintilie, M.; Hedley, D.; Li, S.-D. Targeting of metastasis-promoting tumor-associated fibroblasts and modulation of pancreatic tumor-associated stroma with a carboxymethylcellulose-docetaxel nanoparticle. J. Control. Release 2015, 206, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Bala, V.; Rao, S.S.; Boyd, B.J.; Prestidge, C.A. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J. Control. Release 2013, 172, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Khare, V.; Kour, S.; Alam, N.; Dubey, R.D.; Saneja, A.; Koul, M.; Gupta, A.P.; Singh, D.; Singh, S.K.; Saxena, A.K.; et al. Synthesis, characterization and mechanistic-insight into the anti-proliferative potential of PLGA-gemcitabine conjugate. Int. J. Pharm. 2014, 470, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Vandana, M.; Sahoo, S.K. Long circulation and cytotoxicity of PEGylated gemcitabine and its potential for the treatment of pancreatic cancer. Biomaterials 2010, 31, 9340–9356. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, G.; Mariano, L.; Salmaso, S.; Caliceti, P.; Gaetano, G. Folate-mediated targeting of polymeric conjugates of gemcitabine. Int. J. Pharm. 2006, 307, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Wang, H.; Chen, Y.; Li, Z.; Wang, Y.; Jun, Q.; Ji, J. Theranostic reduction-sensitive gemcitabine prodrug micelles for near infrared imaging and pancreatic cancer therapy. Nanoscale 2016, 8, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Maksimenko, A.; Caron, J.; Mougin, J.; Desmaële, D.; Couvreur, P. Gemcitabine-based therapy for pancreatic cancer using the squalenoyl nucleoside monophosphate nanoassemblies. Int. J. Pharm. 2015, 482, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal carcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Chiaravalli, M.; Reni, M.; O’Reilly, E.M. Pancreatic ductal adenocarcinoma: State of the Art 2017 and new therapeutic strategies. Cancer Treat. Rev. 2017, 60, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.I.; O’Reilly, R.K. To aggregate, or not to aggregate? Considerations in the design and application of polymeric thermally-responsive nanoparticles. Chem. Soc. Rev. 2013, 42, 7204–7213. [Google Scholar] [CrossRef] [PubMed]
- Letchford, K.; Burt, H. A Review of the formation and classification of amphiphilic block copolymer nanoparticulate structures: Micelles, nanospheres, nanocapsules and polymersomes. Eur. J. Pharm. Biopharm. 2007, 65, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Prompruk, K.; Govender, T.; Zhang, S.; Xiong, C.D.; Stolnik, S. Synthesis of a novel PEG-block-poly(aspartic acid-stat-phenylalanine) copolymer shows potential for formation of a micellar drug carrier. Int. J. Pharm. 2005, 297, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, L.; Zhao, C.; Zheng, J. Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer 2010, 51, 5283–5293. [Google Scholar] [CrossRef]
- Millili, P.G.; Selekman, J.A.; Blocker, K.M.; Johnson, D.A.; Naik, U.P.; Sullivan, M.O. Structural and Functional Consequences of Poly(ethylene glycol) Inclusion on DNA Condensation for Gene Delivery. Microsc. Res. Tech. 2010, 73, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.T.H.; Marcal, H.; Russell, R.A.; Holden, P.J.; Foster, L.J.R. Application of polyethylene glyson to promote cellular biocompatibility of polyhdroxybityrate films. Int. J. Polym. Sci. 2011. [Google Scholar] [CrossRef]
- Yoo, J.W.; Chambers, E.; Mitragotri, S. Factors that control the circulation time of nanoparticles in blood: Challenges, solutions and furture prospects. Curr. Pharm. Des. 2010, 16, 2298–2307. [Google Scholar] [CrossRef] [PubMed]
- Sutton, D.; Nasongkla, N.; Blanco, E.; Gao, J. Functionalized Micellar Systems for Cancer Targeted Drug Delivery. Pharm. Res. 2007, 24, 1029–1046. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, G.; Dufresne, M.; Sant, V.P.; Kang, N.; Maysinger, D.; Leroux, J. Block copolymer micelles: Preparation, characterization and application in drug delivery. J. Control. Release 2005, 109, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Daman, Z.; Montazen, H.; Azizi, M.; Rezaie, F.; Ostad, S.N.; Amini, M.; Gilani, K. Polymeric micelles of PEG-PLA copolymer as a carrier for salinomycin against gemcitabine-resistant pancreatic cancer. Pharm. Res. 2015, 32, 3756–3767. [Google Scholar] [CrossRef] [PubMed]
- Jaidev, L.R.; Krishnan, U.M.; Swaminathan, S. Gemcitabine loaded biodegradable PLGA nanospheres for in vitro pancreatic cancer therapy. Mater. Sci. Eng. C 2014, 47, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Padhye, S.; Yang, H.; Jamadar, A.; Cui, Q.C.; Chavan, D.; Dominiak, K.; McKinney, J.; Banerjee, S.; Dou, Q.P.; Sarkar, F.H. New difluoro Knoevenagel condensates of curcumin, their Schiff bases and copper complexes as proteasome inhibitors and apoptosis inducers in cancer cells. Pharm. Res. 2009, 26, 1874–1880. [Google Scholar] [CrossRef] [PubMed]
- Dandawate, P.R.; Vyas, A.; Ahmad, A.; Banerjee, S.; Deshpande, J.; Swamy, K.V.; Jamadar, A.; Dumhe-Klaire, A.C.; Padhye, S.; Sarkar, F.H. Inclusion complex of novel curcumin analogue CDF and β-cyclodextrin (1:2) and its enhanced in vivo anticancer activity against pancreatic cancer. Pharm. Res. 2012, 29, 1775–1786. [Google Scholar] [CrossRef] [PubMed]
- Daruwalla, J.; Greish, K.; Malcontenti-Wilson, C.; Muralidharan, V.; Iyer, A.; Maeda, H.; Christophi, C. Styrene Maleic Acid-Pirarubicin Disrupts Tumor Microcirculation and Enhances the Permeability of Colorectal Liver Metastases. J. Vasc. Res. 2009, 46, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, C.; Kong Thoo Lin, P.; Cheng, W.-P. A review on comb shaped amphiphilic polymers for hydrophobic drug solubilization. Ther. Deliv. 2011, 3, 59–79. [Google Scholar] [CrossRef]
- Cheng, W.-P.; Gray, A.I.; Tetley, L.; Hang, T.L.B.; Schätzlein, A.G.; Uchegbu, I.F. Polyelectrolyte Nanoparticles with High Drug Loading Enhance the Oral Uptake of Hydrophobic Compounds. Biomacromolecules 2006, 7, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, C.; Kong Thoo Lin, P.; Tetley, L.; Cheng, W.-P. Novel fluorescent amphiphilic poly(allylamine) and their supramolecular self-assemblies in aqueous media. Polym. Adv. Technol. 2011, 20, 710–719. [Google Scholar]
- Uchegbu, I.F.; Schältzen, A.G.; Tetley, L. Polymeric chitosan based vesicles for drug delivery. J. Pharm. Pharmacol. 1998, 50, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Wang, W.; Wang, C.; Wang, Y.; Zhou, J.; Ding, Y.; Wang, X.; Jin, Y. A chitosan-graft-PEI-candesartan conjugate for targeted co-delivery of drug and gene in anti-angiogenesis cancer therapy. Biomaterials 2014, 35, 8450–8466. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Yeung, L.K.; Crooks, R.M. Size-Selective Hydrogenation of Olefins by Dendrimer-Encapsulated Palladium Nanoparticles. JACS 2001, 123, 6840–6846. [Google Scholar] [CrossRef]
- Topp, A.; Bauer, B.J.; Klimash, J.W.; Spindler, R.; Tomalia, D.A.; Amis, E.J. Probing the location of the terminal groups of dendrimers in dilute solution. Macromolecules 1999, 32, 7226–7231. [Google Scholar] [CrossRef]
- Ladd, E.; Sheikhi, A.; Li, N.; van de Ven, T.G.M.; Kakkar, A. Design and synthesis of dendrimers with facile surface group functionalisation and an evaluation of their bactericidal efficacy. Molecules 2017, 22, 868. [Google Scholar] [CrossRef] [PubMed]
- Chiba, F.; Twyman, J.L. Effect of Terminal-Group Functionality on the Ability of Dendrimers to Bind Proteins. Bioconjug. Chem. 2017, 28, 2046–2050. [Google Scholar] [CrossRef] [PubMed]
- Al-Jamal, K.; Ramaswamy, C.; Florence, A.F. Supramolecular structures from dendrons and dendrimers. Adv. Drug Deliv. Rev. 2005, 57, 2238–2270. [Google Scholar] [CrossRef] [PubMed]
- Svenson, S. Dendrimers as versatile platform in drug delivery applications. Eur. J. Pharm. Biopharm. 2009, 71, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Boas, U.; Heegaard, P.M.H. Dendrimers in drug research. Chem. Soc. Rev. 2004, 33, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Xu, T. The effect of dendrimers on the pharmacodynamic and pharmacokinetic behaviours of non-covalently or covalently attached drugs. Eur. J. Med. Chem. 2008, 43, 2291–2297. [Google Scholar] [CrossRef] [PubMed]
- Opitz, A.W.; Czymmek, K.J.; Wickstrom, E.; Wagnet, N.J. Uptake, efflux and mass transfer coefficient of fluorescent PAMAM dendrimers into pancreatic cancer cells. Biochim. Biophys. A 2013, 1828, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Cho, H.-J.; Yoon, H.-Y.; Yoon, I.S.; Ko, S.-H.; Shime, J.S.; Cho, J.-H.; Park, J.H.; Kim, K.; Kwon, I.C.; et al. Hyaluronic acid derivative-coated nanohybrid liposomes for cancer imaging and drug delivery. J. Control. Release 2014, 174, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Geng, H.; Wang, Y.; Gao, Y.; Huang, J.; Wang, Y.; Zhang, J.; Wang, S. Hyaluronic acid oligosaccharide modified redox-responsive mesoporous silica nanoparticles for targeted drug delivery. ACS Mater. Interfaces 2014, 6, 20290–20299. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, N.; Kataoka, K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol. Ther. 2006, 112, 630–648. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, J.-H.; Jeon, O.; Kwon, I.C.; Park, K. Engineered polymers for advanced drug delivery. Eur. J. Pharm. Biopharm. 2009, 71, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Galaev, I.Y.; Mattiasson, B. ‘Smart’ polymers and what they could do in biotechnology and medicine. Trends Biotechnol. 1999, 17, 335–340. [Google Scholar] [CrossRef]
- Cabane, E.; Zhang, X.; Langowska, K.; Palivan, C.; Meier, W. Stimuli-Responsive Polymers and Their Applications in Nanomedicine. Biointerphases 2012, 7, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiradharma, N.; Zhang, Y.; Venkataraman, S.; Hedrick, J.L.; Yang, Y.Y. Self-assembled nanostructures for delivery of anticancer therapeutics. Nano Today 2009, 4, 302–317. [Google Scholar] [CrossRef]
- Rapoport, N. Physical stimuli-responsive polymeric micelles for anti-cancer drug delivery. Prog. Polym. Sci. 2007, 32, 962–990. [Google Scholar] [CrossRef]
- Gil, E.S.; Hudson, S.M. Stimuli-responsive polymers and their bioconjugates. Prog. Polym. Sci. 2004, 29, 1173–1222. [Google Scholar] [CrossRef]
- Teranishi, R.; Matsuki, R.; Yuba, E.; Harada, A.; Kono, K. Doxorubicin delivery using pH and redox dual-responsive hollow nanocapdules with a cationic electrostatic barrier. Pharmaceutics 2017, 9, 4. [Google Scholar] [CrossRef]
- Li, A.; Wang, Y.; Chen, T.; Zhao, W.; Zhang, A.; Feng, S.; Liu, J. NIR-laser switched ICG/DOX loaded thermo-responsive polymeric capsule for chemo-photothermal targeted therapy. Eur. Polym. J. 2017, 92, 51–60. [Google Scholar] [CrossRef]
- Xiao, M.; González, E.; Monterroza, A.M.; Frey, M. Fabrication of thermo-responsive cotton fabrics using poly(vinyl caprolactam-co-hydroxyethyl acrylamide) copolymer. Carbohydr. Polym. 2017, 174, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Cheng, S.-X.; Zhang, X.-Z.; Zhuto, R.-X. Thermo-sensitive polymeric micelles based on poly(N-isopropylacrylamide) as drug carriers. Prog. Polym. Sci. 2009, 34, 893–910. [Google Scholar] [CrossRef]
- Li, J.; Wang, B.; Liu, P. Possibility of active targeting tumor by local hyperthermia with temperature sensitive nanoparticles. Med. Hypotheses 2007, 71, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Ameri, S.K.; Singh, P.K.; Sonkusale, S.R. Three dimensional graphene transistor for ultra-sensitive pH sensing directly in biological media. Anal. Chim. Acta 2016, 934, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, N.; Payne, A.; Dillon, C.; Shea, J.; Scaife, C.; Gupta, R. Focused ultrasound-mediated drug delivery to pancreatic cancer in a mouse model. J. Ther. Ultrasound 2013, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Peters, T. All about Albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Kim, B.; Seo, B.; Park, S.; Lee, C.; Kim, J.O.; Oh, K.T.; Lee, E.S.; Choi, H.G.; Youn, Y.S. Albumin nanoparticles with synergistic antitumor efficiency against metastatic lung cancers. Colloids Surf. B Biointerfaces 2017, 158, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. New Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Lee, C.; Lee, E.S.; Shin, B.S.; Youn, Y.S. Paclitaxel and curcumin co-bound albumin nanoparticles having antitumor potential to pancreatic cancer. Asian J. Pharm. Sci. 2016, 11, 708–714. [Google Scholar] [CrossRef]
- Ciofani, G.; Menciassi, A. Piezoelectric Nanomaterials for Biomedical Applications; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Liu, Z.; Peng, R. Inorganic nanomaterials for tumor angiogenesis imaging. Eur. J. Nuclear Med. Mol. Imaging 2010, 37, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Hemraj-Benny, T.; Wong, S.S. Covalent surface chemistry of single walled carbon nanotubes. Adv. Mater. 2005, 17, 17–29. [Google Scholar] [CrossRef]
- Klumpp, C.; Kostarelos, K.; Prato, M.; Bianco, A. Functionalized carbon nanotubes as emerging nanovectors for the delivery of therapeutics. Biochim. Biophys. Acta Biomembr. 2006, 1758, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Colvin, V.L. The potential environmental impact of engineered nanomaterials. Nat. Biotechnol. 2003, 21, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Prato, M.; Kostarelos, K.; Bianco, A. Functionalized carbon nanotubes in drug design and discovery. Acc. Chem. Res. 2007, 41, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Mocan, T.; Matea, C.T.; Cojocaru, I.; Ilie, I.; Tabaran, F.A.; Zaharie, F.; Iancu, C.; Bartos, D.; Mocan, L. Photothermal treatment of human pancreatic cancer using PEGylated multi-walled carbon nanotubes induces apoptosis by triggering mitochondrial membrane depolarization mechanism. J. Cancer 2014, 5, 679. [Google Scholar] [CrossRef] [PubMed]
- Andreoli, E.; Suxuki, R.; Orbaek, A.W.; Bhutani, M.S.; Hauge, R.H.; Adams, W.; Fleming, J.B.; Barron, A.R. Preparation and evaluation of polyethylenimine single walled carbon nanotube conjugates as vectors for pancreatic cancer therapy. J. Mater. Chem. B 2014, 2, 4740–4747. [Google Scholar] [CrossRef]
- Wang, S.; Lin, Q.; Chen, J.; Gao, H.; Fu, D.; Shen, S. Biocompatible polydopamine-encapsulated galodinium-loaded carbon nanotubes for MRI and color mapping guided photothermal dissection of tumor metastasis. Carbon 2017, 122, 53–62. [Google Scholar] [CrossRef]
- Matea, C.T.; Mocan, T.; Tabaran, F.; Pop, T.; Mosteanu, O.; Puia, C.; Iancu, C.; Mocan, L. Quantum dots in imaging, drug delivery and sensor applications. Int. J. Nanomed. 2017, 12, 5421–5431. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.-T.; Ding, H.; Roy, I.; Law, W.-C.; Bergey, E.J.; Maitra, A.; Prasad, P.N. Imaging pancreatic cancer using bioconjugated InP quantum dots. ACS Nano 2009, 3, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Nichlos, L.S.; Ashfaq, R.; Icaobuzo-Donahue, C.A. Claudin 4 Protein expression in primary and metastatic pancreatic cancer support for use as a therapeutic target. Am. J. Clin. Pathol. 2004, 121, 226–230. [Google Scholar] [CrossRef]
- Lee, K.H.; Galloway, J.F.; Park, J.; Dvoracek, C.M.; Dallas, M.; Konstantopoulous, K.; Maitra, A.; Searson, P.C. Quantitative molecular profiling of biomarkers for pancreatic cancer with functionalized quantum dots. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-Q.; Dai, Y.-D.; Kang, B.; Han, W.; Mao, L.; Chen, D. UV enhanced cytotoxicity of thiol-capped CdTe quantum dots in human pancreatic carcinoma cells. Toxicol. Lett. 2009, 188, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Saritas, E.U.; Goodwill, P.W.; Croft, L.R.; Konkle, J.J.; Lu, K.; Zheng, B.; Conolly, S.M. Magnetic Particle Imaging (MPI) for NMR and MRI researchers. J. Magn. Res. 2013, 229, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Lee, S.; Chen, X. Nanoparticle-based theranostic agents. Adv. Drug Deliv. Rev. 2010, 62, 1064–1079. [Google Scholar] [CrossRef] [PubMed]
- Rosen, J.E.; Chan, L.; Shieh, D.B.; Gu, F.X. Iron oxide nanoparticles for targeted cancer imaging and diagnostics. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Santhosh, P.B.; Ulrih, N.P. Multifunctional superparamagnetic iron oxide nanoparticles: Promising tools in cancer theranostics. Cancer Lett. 2012, 336, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Dutz, S.; Häfeli, U.O.; Mahmoudi, M. Magnetic fluid hyperthermia: Focus on superparamagnetic iron oxide nanoparticles. Adv. Coll. Interface Sci. 2011, 166, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Rochani, A.K.; Balasubramanian, S.; Girija, A.R.; Raveendran, A.; Borah, A.; Nagaoka, Y.; Nakajima, Y.; Maekawa, T.; Kumar, D.S. Dual mode of cancer cell destruction for pancreatic cancer therapy using Hsp90 inhibitor loaded polymeric nano magnetic formulation. Int. J. Pharm. 2016, 511, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, B.; Vandooren, J.; Locatelli, E.; Fiten, P.; Opdenakker, G.; Proost, P.; Krüger, A.; Lellouche, J.P.; Israel, L.L.; Shenkman, L.; et al. Matrix metalloproteinase-9 (MMP-9) as an activator of nanosystems for targeted drug delivery in pancreatic cancer. J. Control. Release 2016, 239, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Dobiasch, S.; Szanyi, S.; Kjaev, A.; Werner, J.; Strauss, S.; Weis, C.; Grenacher, L.; Kapilov-Buchman, K.; Israel, L.L.; Lellouche, J.-P.; et al. Synthesis and fucntionalization of protease-actvated nanoparticles with tissue plasmonigen activator peptides as targeting moiety and diagnostic tool for pancreatic cancer. J. Nanobiotechnol. 2016, 14, 81. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wei, Q.Q.; Wang, B.C.; Zhang, S.H.; Yuan, Z. Role of thiol-containing polyethylene glycol (thiol-PEG) in the modification process of gold nanoparticles (AuNPs): Stabilizer or coagulant? J. Colloid Interface Sci. 2013, 404, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Berguiga, L.; Elezgaray, J.; Roland, T.; Faivre-Moskalenko, C.; Argoul, F. Surface plasmon resonance characterization of thermally evaporated thin gold films. Surf. Sci. 2007, 601, 5445–5458. [Google Scholar] [CrossRef]
- Verma, S.; Rao, B.T.; Rai, S.; Kukreja, L.M. Influence of process parameters on surface plasmon resonance characteristics of densely packed gold nanoparticle films grown by pulsed laser deposition. Appl. Surf. Sci. 2012, 258, 4898–4905. [Google Scholar] [CrossRef]
- Vigderman, L.; Zubarev, E.R. Therapeutic platforms based on gold nanoparticles and their covalent conjugates with drug molecules. Adv. Drug Deliv. Rev. 2013, 65, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Mocan, L.; Ilie, I.; Tabaran, F.A.; Dana, B.; Zaharie, F.; Zdrehus, C.; Puia, C.; Mocan, T.; Muntean, V.; Teodora, P.; et al. Surface plasmon resonance-induced photoactivation of gold nanoparticles as mitochondria-targeted therapeutic agents for pancreatic cancer. Expert Opin. Ther. Target 2013, 17, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Xiong, X.; Chakraborty, P.K.; Shameer, K.; Avrvizo, R.R.; Kudgus, R.A.; Dwivedi, S.K.D.; Hossen, M.N.; Gillies, E.M.; Robertson, J.D.; et al. Gold nanoparticle reprorgrammes pancreatic tumor microenvironment and inhibits tumor growth. ACS Nano 2016, 10, 10636–10651. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, Z.; Kim, D.H.; Li, W.; Nicolai, J.; Procissi, D.; Huan, Y.; Han, G.; Omary, R.A.; Larson, A.C. Photothermal ablation of pancreatic cancer cells with hybrid iron-oxide core gold-shell nanoparticles. Int. J. Nanomed. 2013, 8, 3437–3446. [Google Scholar] [CrossRef] [PubMed]
- Oluwasanmi, A.; Malekigorji, M.; Jones, S.; Curtis, A.; Hoskins, C. Potential of hybrid iron oxide-gold nanoparticles as thermal triggers for pancreatic cancer therapy. RSC Adv. 2016, 6, 95044–95054. [Google Scholar] [CrossRef]
- Sonuç Karaboğa, M.N.; Şimşek, Ç.S.; Sezgintürk, M.K. AuNPs modified, disposable, ITO based biosensor: Early diagnosis of heat shock protein 70. Biosens. Bioelectron. 2016, 84, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.C.; Glaus, C.; Chen, J.; Welch, M.J.; Xia, Y. Inorganic nanoparticle-based contrast agents for molecular imaging. Trends Mol. Med. 2010, 16, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zheng, J. Clearance pathways and tumor targeting of imaging nanoparticles. ACS Nano 2015, 9, 6655–6674. [Google Scholar] [CrossRef] [PubMed]
- Henriksen-Lacey, M.; Carregal-Romero, S.; Liz-Marzán, L.M. Current challenges toward in vitro cellular validation of inorganic nanoparticles. Bioconjug. Chem. 2017, 28, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yu, M.; Nig, X.; Zhou, C.; Yang, S.; Zheng, J. PEGylation and zwitterionization: Pros and cons in the renal clearance and tumour targeting of near-IR-emitting gold nanoparticles. Angew. Chem. 2013, 52, 12572–12576. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
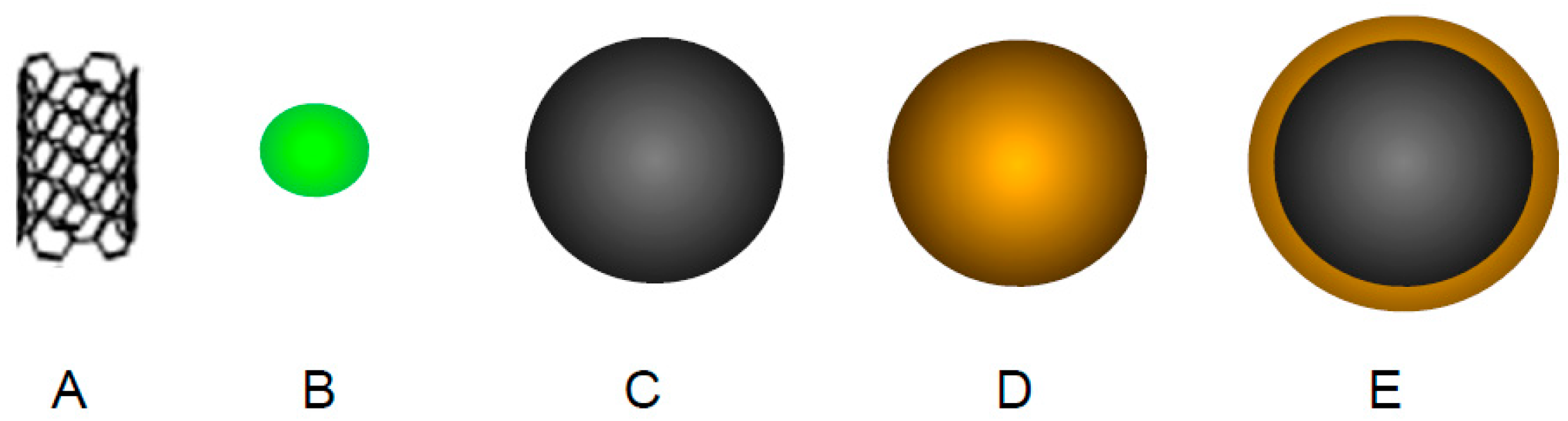{kind=link}
{kind=link}
| Type of Nano-System | Name of Nano-System | Drug Formulated | Testing Phase | |
|---|---|---|---|---|
| Polymer-drug conjugate | Poly(ethylene glycol)-P(HEMASN38) | SN38 | Preclinical: In vivo | [39] |
| Poly (TPGS)-PEG-GEM | Gemcitabine | Preclinical: In vitro | [40] | |
| Methacrylate-based GEM-monomer conjugate 3 | Gemcitabine | Preclinical: In vitro | [41] | |
| Poly(ethylene glycol)-block-poly(2-methyl-2-carboxyl-propylenecarbonate)-graft-dodecanol-graft-cationic ligand | Gemcitabine | Preclinical: In vivo | [42] | |
| Block copolymer | Styrene-maleic acid | CDF | Preclinical: In vitro | [43] |
| Poly(ethylene glycol)-b-poly(glutamic acid) | Oxaliplatin | Preclinical: In vivo | [44] | |
| Mixed micelles | Poly(vinyl pyrrolidone-b-polycaprolactone) (PVP-b-PCL) and poly(vinyl pyrrolidone-b-poly(dioxanone-co-methyl dioxanone)) (PVP-b-P(DX-co-MeDX) | Gemcitabine, doxorubicin, doxorubicin hydrochloride, 5-fluorouricil and paclitaxel | Preclinical: In vitro | [45] |
| Graft polymer | Poly(allylamine)-g-cholesterol | BNIPDAoct | Preclinical: In vivo | [46] |
| Dendrimer | PAMAM—hyaluronic acid | CDF | Preclinical: In vitro | [47] |
| Poly(ethylene glycol)—PAMAM—poly(ethylene glycol)-Flt-2 | Gemcitabine Hydrochloride | Preclinical: In vivo | [48] | |
| Thermo-responsive polymer | Poly(diEGMAco-OEGMA300)-b-PEHMA | Squalenoyl-gemcitabine | Preclinical: In vitro | [49] |
| pH-responsive polymer | Poly(styrene-alt-maleic anhydride) | Curcumin | Preclinical: In vitro | [50] |
| Ultrasound-responsive nano-emulsion | PEG-PLLA | Paclitaxel | Preclinical: In vivo | [51] |
| Albumin | Abraxane® | Paclitaxel | FDA approved 2013 | [52] |
| Abraxane®/Gemcitabine | Paclitaxel & gemcitabine | Phase III | [53] | |
| Inorganic nanoparticle | Iron oxide-dextran-DOX | Doxorubicin | Preclinical: In vitro | [54] |
| Iron oxide-antiCD47-GEM | Gemcitabine | Preclinical: In vitro | [55] | |
| Iron oxide-gold | BNIPDSpm | Preclinical: In vivo | [56] | |
| Iron oxide-gold-GEM | Gemcitabine | Preclinical: In vivo | [57] | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manzur, A.; Oluwasanmi, A.; Moss, D.; Curtis, A.; Hoskins, C. Nanotechnologies in Pancreatic Cancer Therapy. Pharmaceutics 2017, 9, 39. https://doi.org/10.3390/pharmaceutics9040039
Manzur A, Oluwasanmi A, Moss D, Curtis A, Hoskins C. Nanotechnologies in Pancreatic Cancer Therapy. Pharmaceutics. 2017; 9(4):39. https://doi.org/10.3390/pharmaceutics9040039
Chicago/Turabian StyleManzur, Ayesha, Adeolu Oluwasanmi, Darren Moss, Anthony Curtis, and Clare Hoskins. 2017. "Nanotechnologies in Pancreatic Cancer Therapy" Pharmaceutics 9, no. 4: 39. https://doi.org/10.3390/pharmaceutics9040039
APA StyleManzur, A., Oluwasanmi, A., Moss, D., Curtis, A., & Hoskins, C. (2017). Nanotechnologies in Pancreatic Cancer Therapy. Pharmaceutics, 9(4), 39. https://doi.org/10.3390/pharmaceutics9040039