
Scientific Rationale for the Treatment of Cognitive Deficits from Long COVID


Abstract
:1. Introduction
2. Search Strategy and Selection Criteria
3. Long COVID Preferentially Afflicts the Cognitive Functioning of the Prefrontal Cortex


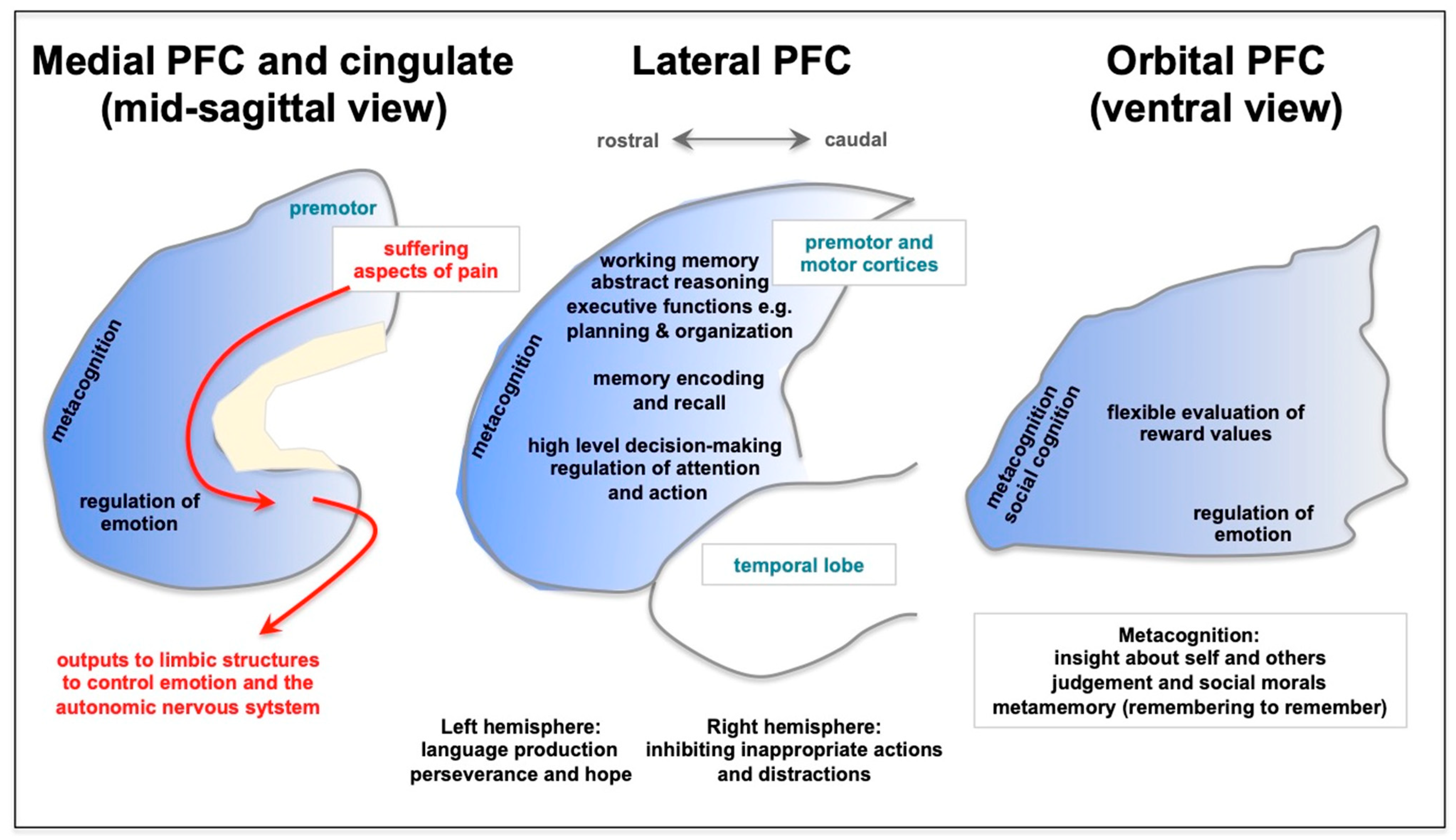{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Citation | Summary |
|---|---|
| Vanderlind et al. 2021 [23] | “Thirty-three studies met inclusion/exclusion criteria for review. Emerging findings link COVID-19 to cognitive deficits, particularly attention, executive function, and memory. Psychiatric symptoms occur at high rates in COVID-19 survivors, including anxiety, depression, fatigue, sleep disruption, and to a lesser extent posttraumatic stress.” |
| Perrottelli et al. 2022 [26] | “The available evidence revealed the presence of impairment in executive functions, speed of processing, attention and memory in subjects recovered from COVID-19.” |
| Zawilska and Kuczyńska. 2022 [27] | “Fatigue and cognitive dysfunction, such as concentration problems, short-term memory deficits, general memory loss, a specific decline in attention, language and praxis abilities, encoding and verbal fluency, impairment of executive functions, and psychomotor coordination, are amongst the most common and debilitating features of neuropsychatric symptoms of post COVID syndrome.” |
| Newhouse et al. 2022 [41] | “Many of these symptoms [42] are neuropsychiatric, such as inattention, impaired memory, and executive dysfunction; these are often colloquially termed “brain fog”.” |
| Bertuccelli et al. 2022 [28] | “Memory, attention, and executive functions appeared to be the most affected domains. Delayed recall and learning were the most impaired domains of memory. Among the executive functions, abstraction, inhibition, set shifting, and sustained and selective attention were most commonly impaired.” |
| Houben and Bonnechère. 2022 [29] | Meta-analysis showed impairments in attention, executive functioning, and verbal memory (recall) |
| Zeng et al. 2023 [30] | “Individuals with severe infection suffered more from PTSD, sleep disturbance, cognitive deficits, concentration impairment, and gustatory dysfunction. Survivors with mild infection had high burden of anxiety and memory impairment after recovery.” |
| Ceban et al. 2022 [31] | “The proportion of individuals exhibiting cognitive impairment was 0.22 (95% CI, 0.17, 0.28; p < 0.001; n = 13,232; I2 = 98.0). Moreover, narrative synthesis revealed elevations in proinflammatory markers and considerable functional impairment in a subset of individuals.” |
4. The Prefrontal Cortex Is Especially Vulnerable to Physiological and Psychological Stressors
5. Unique Neurotransmission and Neuromodulation Renders PFC Circuits Especially Vulnerable to Stress and Inflammation
6. Stress and Inflammatory Signaling in Brain—Similar Pathways Activated by Psychological and Physiological Stressors, Including Activation by COVID-19
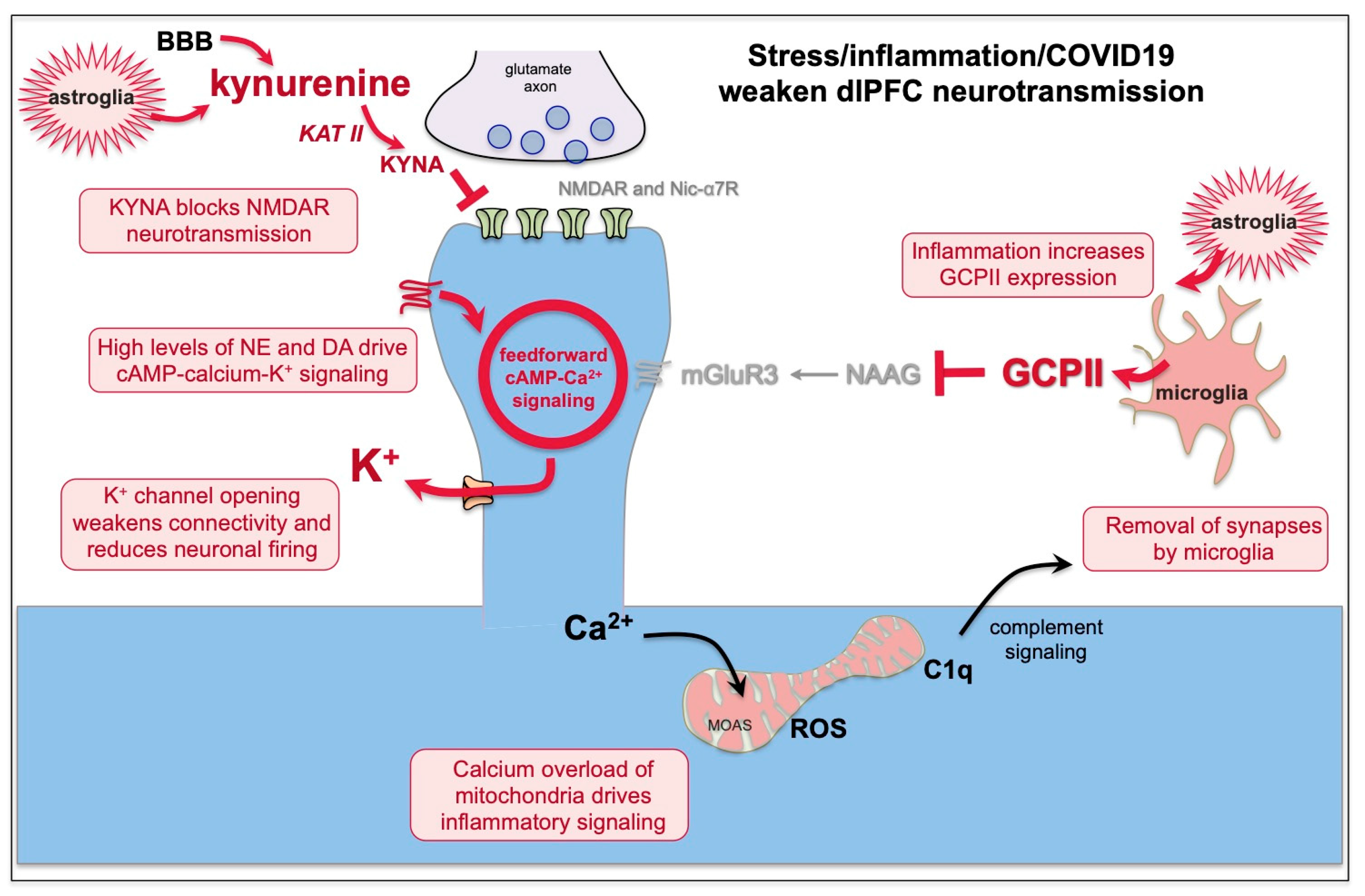
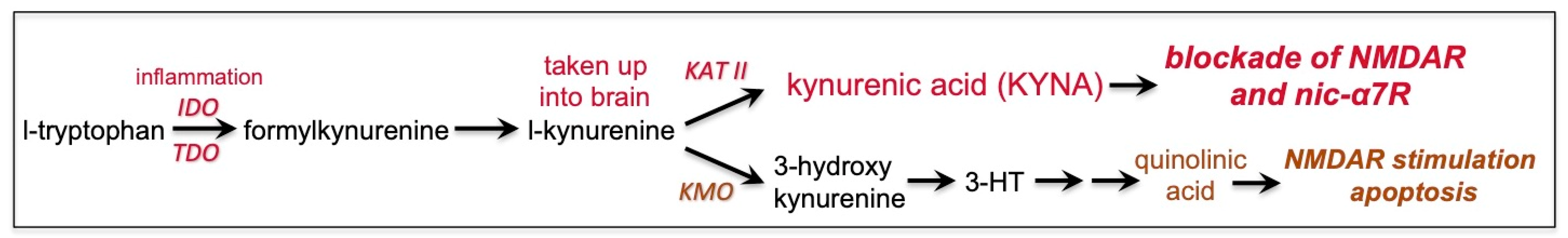
7. Kynurenic Acid Signaling Is Increased by Inflammation, Including by COVID-19, and Can Reduce NMDAR and nic-α7R Neurotransmission
8. GCPII Expression Is Increased by Inflammation, Including by COVID-19, and Reduces mGluR3 Regulation of cAMP-Calcium Signaling
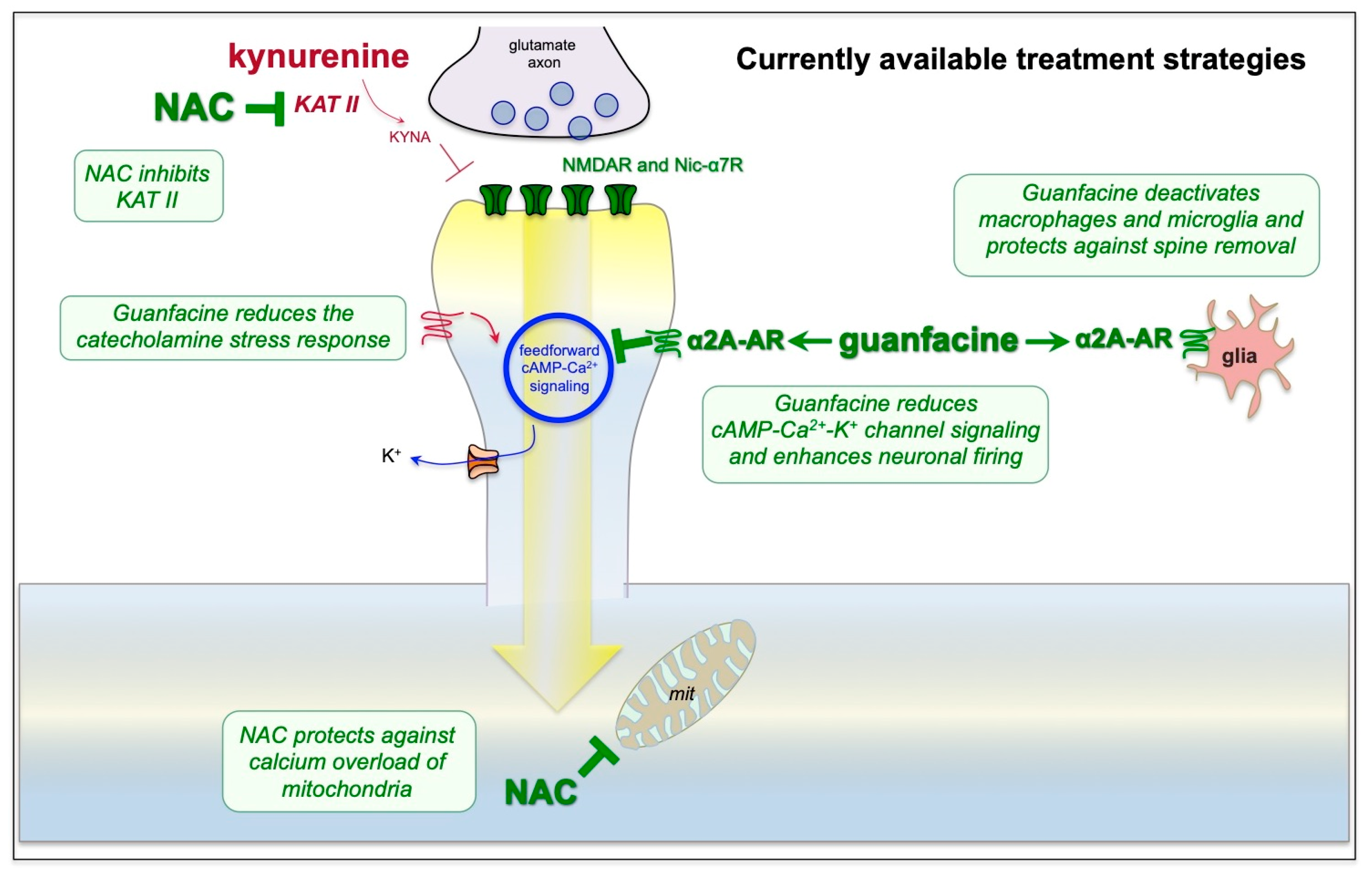
9. Strategies for Treatment Based on the Neuroscience
10. Outstanding Questions and Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Szczepanski, S.M.; Knight, R.T. Insights into human behavior from lesions to the prefrontal cortex. Neuron 2014, 83, 1002–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preuss, T.M.; Wise, S.P. Evolution of prefrontal cortex. Neuropsychopharmacology 2022, 47, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Alexander, L.; Wood, C.M.; Roberts, A.C. The ventromedial prefrontal cortex and emotion regulation: Lost in translation? J. Physiol. 2023, 601, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Elston, G.N.; Benavides-Piccione, R.; Elston, A.; Zietsch, B.; Defelipe, J.; Manger, P.; Casagrande, V.; Kaas, J.H. Specializations of the granular prefrontal cortex of primates: Implications for cognitive processing. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2006, 288, 26–35. [Google Scholar] [CrossRef]
- Defelipe, J. The evolution of the brain, the human nature of cortical circuits, and intellectual creativity. Front. Neuroanat. 2011, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Goldman-Rakic, P. Cellular Basis of Working Memory. Neuron 1995, 14, 477–485. [Google Scholar]
- Levy, R.; Dubois, B. Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cereb. Cortex 2006, 16, 916–928. [Google Scholar] [CrossRef] [Green Version]
- Friedman, N.P.; Robbins, T.W. The role of prefrontal cortex in cognitive control and executive function. Neuropsychopharmacology 2022, 47, 72–89. [Google Scholar] [CrossRef]
- Robbins, T.W. Dissociating executive functions of the prefrontal cortex. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1996, 351, 1463–1470. [Google Scholar] [CrossRef]
- Kimberg, D.Y.; Farah, M.J. A unified account of cognitive impairments following frontal lobe damage: The role of working memory in complex, organized behavior. J. Exp. Psychol. Gen. 1993, 122, 411–428. [Google Scholar] [CrossRef]
- Menon, V.; D’Esposito, M. The role of PFC networks in cognitive control and executive function. Neuropsychopharmacology 2022, 47, 90–103. [Google Scholar] [CrossRef]
- Dolcos, F.; McCarthy, G. Brain systems mediating cognitive interference by emotional distraction. J. Neurosci. 2006, 26, 2072–2079. [Google Scholar] [CrossRef] [Green Version]
- Dias, E.C.; McGinnis, T.; Smiley, J.F.; Foxe, J.J.; Schroeder, C.E.; Javitt, D.C. Changing plans: Neural correlates of executive control in monkey and human frontal cortex. Exp. Brain Res. 2006, 174, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.M. Prefrontal neurons in networks of executive memory. Brain Res. Bull. 2000, 52, 331–336. [Google Scholar] [CrossRef]
- Aron, A.R. The neural basis of inhibition in cognitive control. Neuroscientist 2007, 13, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Ungerleider, L.G.; Courtney, S.M.; Haxby, J.V. A neural system for human visual working memory. Proc. Natl. Acad. Sci. USA 1998, 95, 883–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujimoto, S.; Genovesio, A.; Wise, S.P. Frontal pole cortex: Encoding ends at the end of the endbrain. Trends Cogn. Sci. 2011, 15, 169–176. [Google Scholar] [CrossRef]
- Joyce, M.K.P.; Marshall, L.G.; Banik, S.L.; Wang, J.; Xiao, D.; Bunce, J.G.; Barbas, H. Pathways for Memory, Cognition and Emotional Context: Hippocampal, Subgenual Area 25, and Amygdalar Axons Show Unique Interactions in the Primate Thalamic Reuniens Nucleus. J. Neurosci. 2022, 42, 1068–1089. [Google Scholar] [CrossRef]
- Holmes, S.E.; Scheinost, D.; Finnema, S.J.; Naganawa, M.; Davis, M.T.; DellaGioia, N.; Nabulsi, N.; Matuskey, D.; Angarita, G.A.; Pietrzak, R.H.; et al. Lower synaptic density is associated with depression severity and network alterations. Nat. Commun. 2019, 10, 1529. [Google Scholar] [CrossRef] [Green Version]
- Helms, J.; Kremer, S.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Kummerlen, C.; Collange, O.; Boulay, C.; Fafi-Kremer, S.; Ohana, M.; et al. Neurologic Features in Severe SARS-CoV-2 Infection. N. Engl. J. Med. 2020, 382, 2268–2270. [Google Scholar] [CrossRef]
- Becker, J.H.; Lin, J.J.; Doernberg, M.; Stone, K.; Navis, A.; Festa, J.R.; Wisnivesky, J.P. Assessment of Cognitive Function in Patients After COVID-19 Infection. JAMA Netw. Open. 2021, 4, e2130645. [Google Scholar] [CrossRef]
- Hellmuth, J.; Barnett, T.A.; Asken, B.M.; Kelly, J.D.; Torres, L.; Stephens, M.L.; Greenhouse, B.; Martin, J.N.; Chow, F.C.; Deeks, S.G.; et al. Persistent COVID-19-associated neurocognitive symptoms in non-hospitalized patients. J. Neurovirol. 2021, 27, 191–195. [Google Scholar] [CrossRef]
- Vanderlind, W.M.; Rabinovitz, B.B.; Miao, I.Y.; Oberlin, L.E.; Bueno-Castellano, C.; Fridman, C.; Jaywant, A.; Kanellopoulos, D. A systematic review of neuropsychological and psychiatric sequalae of COVID-19: Implications for treatment. Curr. Opin. Psychiatry 2021, 34, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Hampshire, A.; Trender, W.; Chamberlain, S.R.; Jolly, A.E.; Grant, J.E.; Patrick, F.; Mazibuko, N.; Williams, S.C.; Barnby, J.M.; Hellyer, P.; et al. Cognitive deficits in people who have recovered from COVID-19. EClinicalMedicine 2021, 39, 101044. [Google Scholar] [CrossRef]
- Spudich, S.; Nath, A. Nervous system consequences of COVID-19. Science 2022, 375, 267–269. [Google Scholar] [CrossRef]
- Perrottelli, A.; Sansone, N.; Giordano, G.M.; Caporusso, E.; Giuliani, L.; Melillo, A.; Pezzella, P.; Bucci, P.; Mucci, A.; Galderisi, S. Cognitive Impairment after Post-Acute COVID-19 Infection: A Systematic Review of the Literature. J. Pers. Med. 2022, 12, 2070. [Google Scholar] [CrossRef]
- Zawilska, J.B.; Kuczyńska, K. Psychiatric and neurological complications of long COVID. J. Psychiatr. Res. 2022, 156, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Bertuccelli, M.; Ciringione, L.; Rubega, M.; Bisiacchi, P.; Masiero, S.; Del Felice, A. Cognitive impairment in people with previous COVID-19 infection: A scoping review. Cortex 2022, 154, 212–230. [Google Scholar] [CrossRef] [PubMed]
- Houben, S.; Bonnechère, B. The Impact of COVID-19 Infection on Cognitive Function and the Implication for Rehabilitation: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2022, 19, 7748. [Google Scholar] [CrossRef] [PubMed]
- Zeng, N.; Zhao, Y.-M.; Yan, W.; Li, C.; Lu, Q.-D.; Liu, L.; Ni, S.-Y.; Mei, H.; Yuan, K.; Shi, L.; et al. A systematic review and meta-analysis of long term physical and mental sequelae of COVID-19 pandemic: Call for research priority and action. Mol. Psychiatry 2023, 28, 423–433. [Google Scholar] [CrossRef]
- Ceban, F.; Ling, S.; Lui, L.M.W.; Lee, Y.; Gill, H.; Teopiz, K.M.; Rodrigues, N.B.; Subramaniapillai, M.; Di Vincenzo, J.D.; Cao, B.; et al. Fatigue and cognitive impairment in Post-COVID-19 Syndrome: A systematic review and meta-analysis. Brain Behav. Immun. 2022, 101, 93–135. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Joyce, M.K.; Roberts, A.C. The Aversive Lens: Stress effects on the prefrontal-cingulate cortical pathways that regulate emotion. Neurosci. Biobehav. Rev. 2022, 145, 105000. [Google Scholar] [CrossRef]
- Herrera, E.; Pérez-Sánchez, M.D.C.; San Miguel-Abella, R.; Barrenechea, A.; Blanco, C.; Solares, L.; González, L.; Iza, C.; Castro, I.; Nicolás, E.; et al. Cognitive impairment in young adults with post COVID-19 syndrome. Sci. Rep. 2023, 13, 6378. [Google Scholar] [CrossRef] [PubMed]
- Gamberini, G.; Masuccio, F.G.; Cerrato, M.; Strazzacappa, M.; Ferraro, D.; Solaro, C. Previously independent patients with mild-symptomatic COVID-19 are at high risk of developing cognitive impairment but not depression or anxiety. J. Affect. Disord. 2023, 324, 645–651. [Google Scholar] [CrossRef]
- Taruffi, L.; Muccioli, L.; Mitolo, M.; Ferri, L.; Descovich, C.; Mazzoni, S.; Michelucci, R.; Lodi, R.; Liguori, R.; Cortelli, P.; et al. Neurological Manifestations of Long COVID: A Single-Center One-Year Experience. Neuropsychiatr. Dis. Treat. 2023, 19, 311–319. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Yuan, M.; Dang, W.; Bai, L.; Yang, R.; Wang, J.; Ma, Y.; Liu, B.; Liu, S.; Zhang, S.; et al. Long term neuropsychiatric consequences in COVID-19 survivors: Cognitive impairment and inflammatory underpinnings fifteen months after discharge. Asian J. Psychiatr. 2023, 80, 103409. [Google Scholar] [CrossRef] [PubMed]
- Cysique, L.A.; Jakabek, D.; Bracken, S.G.; Allen-Davidian, Y.; Heng, B.; Chow, S.; Dehhaghi, M.; Staats Pires, A.; Darley, D.R.; Byrne, A.; et al. Post-acute COVID-19 cognitive impairment and decline uniquely associate with kynurenine pathway activation: A longitudinal observational study. medRxiv 2022. [Google Scholar] [CrossRef]
- Reiken, S.; Sittenfeld, L.; Dridi, H.; Liu, Y.; Liu, X.; Marks, A.R. Alzheimer’s-like signaling in brains of COVID-19 patients. Alzheimer’s Dement. 2022, 18, 955–965. [Google Scholar] [CrossRef]
- Woo, E.; Sansing, L.H.; Arnsten, A.F.T.; Datta, D. Chronic Stress Weakens Connectivity in the Prefrontal Cortex: Architectural and Molecular Changes. Chronic Stress 2021, 5, 24705470211029254. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Woo, E.; Yang, S.; Wang, M.; Datta, D. Unusual Molecular Regulation of Dorsolateral Prefrontal Cortex Layer III Synapses Increases Vulnerability to Genetic and Environmental Insults in Schizophrenia. Biol. Psychiatry 2022. ahead of print. [Google Scholar] [CrossRef]
- Newhouse, A.; Kritzer, M.D.; Eryilmaz, H.; Praschan, N.; Camprodon, J.A.; Fricchione, G.; Chemali, Z. Neurocircuitry Hypothesis and Clinical Experience in Treating Neuropsychiatric Symptoms of Postacute Sequelae of Severe Acute Respiratory Syndrome Coronavirus 2. J. Acad. Consult. Liaison Psychiatry 2022, 63, 619–627. [Google Scholar] [CrossRef]
- Bizjak, D.A.; Stangl, M.; Börner, N.; Bösch, F.; Durner, J.; Drunin, G.; Buhl, J.L.; Abendroth, D. Kynurenine serves as useful biomarker in acute, Long- and Post-COVID-19 diagnostics. Front. Immunol. 2022, 13, 1004545. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Flashman, L.; Saykin, A.J. Executive dysfunction following traumatic brain injury: Neural substrates and treatment strategies. NeuroRehabilitation 2002, 17, 33–44. [Google Scholar] [CrossRef] [Green Version]
- McAllister, T.W.; Flashman, L.A.; Sparling, M.B.; Saykin, A.J. Working memory deficits after traumatic brain injury: Catecholaminergic mechanisms and prospects for treatment—A review. Brain Inj. 2004, 18, 331–350. [Google Scholar] [CrossRef] [PubMed]
- Graner, J.; Oakes, T.R.; French, L.M.; Riedy, G. Functional MRI in the investigation of blast-related traumatic brain injury. Front. Neurol. 2013, 4, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozga, J.E.; Povroznik, J.M.; Engler-Chiurazzi, E.B.; Vonder Haar, C. Executive (dys)function after traumatic brain injury: Special considerations for behavioral pharmacology. Behav. Pharmacol. 2018, 29, 617–637. [Google Scholar] [CrossRef]
- Hoskison, M.M.; Moore, A.N.; Hu, B.; Orsi, S.A.; Kobori, N.; Dash, P.K. Persistent working memory dysfunction following traumatic brain injury: Evidence for a time-dependent mechanism. Neuroscience 2009, 159, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Huynh, J.; Hylin, M.J.; O’Malley, J.J.; Perez, A.; Moore, A.N.; Dash, P.K. Mild Traumatic Brain Injury Reduces Spine Density of Projection Neurons in the Medial Prefrontal Cortex and Impairs Extinction of Contextual Fear Memory. J. Neurotrauma 2018, 35, 149–156. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, J.; Shi, J.; Gong, Q.; Weng, X. Cerebral and functional adaptation with chronic hypoxia exposure: A multi-modal MRI study. Brain Res. 2010, 1348, 21–29. [Google Scholar] [CrossRef]
- Kauser, H.; Sahu, S.; Kumar, S.; Panjwani, U. Guanfacine is an effective countermeasure for hypobaric hypoxia-induced cognitive decline. Neuroscience 2013, 254, 110–119. [Google Scholar] [CrossRef]
- Kauser, H.; Sahu, S.; Panjwani, U. Guanfacine promotes neuronal survival in medial prefrontal cortex under hypobaric hypoxia. Brain Res. 2016, 1636, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.F.; Amat, J.; Baratta, M.V.; Paul, E.; Watkins, L.R. Behavioral control, the medial prefrontal cortex, and resilience. Dialogues Clin. Neurosci. 2006, 8, 397–406. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Wang, M. The Genie in the Bottle-Magnified calcium signaling in dorsolateral prefrontal cortex. Mol. Psychiatry 2021, 26, 3684–3700. [Google Scholar] [CrossRef] [PubMed]
- Liston, C.; McEwen, B.S.; Casey, B.J. Psychosocial stress reversibly disrupts prefrontal processing and attentional control. Proc. Nat. Acad. Sci. USA 2009, 106, 912–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hains, A.B.; Yabe, Y.; Arnsten, A.F.T. Chronic stimulation of alpha-2A-adrenoceptors with guanfacine protects rodent prefrontal cortex dendritic spines and cognition from the effects of chronic stress. Neurobiol. Stress 2015, 2, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansell, E.B.; Rando, K.; Tuit, K.; Guarnaccia, J.; Sinha, R. Cumulative adversity and smaller gray matter volume in medial prefrontal, anterior cingulate, and insula regions. Biol. Psychiatry 2012, 72, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yang, Y.; Wang, C.J.; Gamo, N.J.; Jin, L.E.; Mazer, J.A.; Morrison, J.H.; Wang, X.-J.; Arnsten, A.F. NMDA receptors subserve working memory persistent neuronal firing In dorsolateral prefrontal cortex. Neuron 2013, 77, 736–749. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Paspalas, C.D.; Jin, L.E.; Picciotto, M.R.; Arnsten, A.F.T.; Wang, M. Nicotinic α7 receptors enhance NMDA cognitive circuits in dorsolateral prefrontal cortex. Proc. Nat. Acad. Sci. USA 2013, 110, 12078–12083. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Ramos, B.; Paspalas, C.; Shu, Y.; Simen, A.; Duque, A.; Vijayraghavan, S.; Brennan, A.; Dudley, A.G.; Nou, E.; et al. Alpha2A-adrenoceptor stimulation strengthens working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell 2007, 129, 397–410. [Google Scholar] [CrossRef] [Green Version]
- Vornov, J.J.; Hollinger, K.R.; Jackson, P.F.; Wozniak, K.M.; Farah, M.H.; Majer, P.; Rais, R.; Slusher, B.S. Still NAAG’ing After All These Years: The Continuing Pursuit of GCPII Inhibitors. Adv. Pharmacol. 2016, 76, 215–255. [Google Scholar]
- Jin, L.E.; Wang, M.; Galvin, V.C.; Lightbourne, T.C.; Conn, P.J.; Arnsten, A.F.T.; Paspalas, C.D. mGluR2 vs. mGluR3 in Primate Prefrontal Cortex: Postsynaptic mGluR3 Strengthen Cognitive Networks. Cereb. Cortex 2018, 28, 974–987. [Google Scholar] [CrossRef] [Green Version]
- Finlay, J.M.; Zigmond, M.J.; Abercrombie, E.D. Increased dopamine and norepinephrine release in medial prefrontal cortex induced by acute and chronic stress: Effects of diazepam. Neuroscience 1995, 64, 619–628. [Google Scholar] [CrossRef]
- Goldstein, L.E.; Rasmusson, A.M.; Bunney, S.B.; Roth, R.H. Role of the amygdala in the coordination of behavioral, neuroendocrine and prefrontal cortical monoamine responses to psychological stress in the rat. J. Neurosci. 1996, 16, 4787–4798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zink, C.; Barker, P.; Sawa, A.; Weinberger, D.; Wang, A.; Quillian, H.; Ulrich, W.; Chen, Q.; Jaffe, A.; Kleinman, J.; et al. Missense Mutation in FOLH1 is Associated with Decreased NAAG Levels and Impaired Working Memory Circuitry and Cognition. Am. J. Psychiatry 2020, 177, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Datta, D.; Woo, E.; Duque, A.; Morozov, Y.M.; Arellano, J.; Slusher, B.S.; Wang, M.; Arnsten, A.F.T. Inhibition of glutamate-carboxypeptidase-II in dorsolateral prefrontal cortex: Potential therapeutic target for neuroinflammatory cognitive disorders. Mol. Psychiatry 2022, 27, 4252–4263. [Google Scholar] [CrossRef]
- Liston, C.; Miller, M.M.; Goldwater, D.S.; Radley, J.J.; Rocher, A.B.; Hof, P.R.; Morrison, J.H.; McEwen, B.S. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J. Neurosci. 2006, 26, 7870–7874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobori, N.; Clifton, G.L.; Dash, P.K. Enhanced catecholamine synthesis in the prefrontal cortex after traumatic brain injury: Implications for prefrontal dysfunction. J. Neurotrauma 2006, 23, 1094–1102. [Google Scholar] [CrossRef]
- Kobori, N.; Hu, B.; Dash, P.K. Altered adrenergic receptor signaling following traumatic brain injury contributes to working memory dysfunction. Neuroscience 2011, 172, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Kobori, N.; Moore, A.N.; Dash, P.K. Altered regulation of protein kinase a activity in the medial prefrontal cortex of normal and brain-injured animals actively engaged in a working memory task. J. Neurotrauma 2015, 32, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Yan, E.B.; Frugier, T.; Lim, C.K.; Heng, B.; Sundaram, G.; Tan, M.; Rosenfeld, J.V.; Walker, D.W.; Guillemin, G.J.; Morganti-Kossmann, M.C. Activation of the kynurenine pathway and increased production of the excitotoxin quinolinic acid following traumatic brain injury in humans. J. Neuroinflamm. 2015, 12, 110. [Google Scholar] [CrossRef] [Green Version]
- Szalardy, L.; Zadori, D.; Toldi, J.; Fulop, F.; Klivenyi, P.; Vecsei, L. Manipulating kynurenic acid levels in the brain—On the edge between neuroprotection and cognitive dysfunction. Curr. Top. Med. Chem. 2012, 12, 1797–1806. [Google Scholar] [CrossRef]
- Godlewska, B.R.; Minichino, A.; Emir, U.; Angelescu, I.; Lennox, B.; Micunovic, M.; Howes, O.; Cowen, P.J. Brain glutamate concentration in men with early psychosis: A magnetic resonance spectroscopy case-control study at 7 T. Transl. Psychiatry 2021, 11, 367. [Google Scholar] [CrossRef] [PubMed]
- Cannon, T.D.; Chung, Y.; He, G.; Sun, D.; Jacobson, A.; van Erp, T.G.; McEwen, S.; Addington, J.; Bearden, C.E.; Cadenhead, K.; et al. Progressive reduction in cortical thickness as psychosis develops: A multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol. Psychiatry 2014, 77, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindler, J.; Lim, C.K.; Weickert, C.S.; Boerrigter, D.; Galletly, C.; Liu, D.; Jacobs, K.R.; Balzan, R.; Bruggemann, J.; O’Donnell, M.; et al. Dysregulation of kynurenine metabolism is related to proinflammatory cytokines, attention, and prefrontal cortex volume in schizophrenia. Mol. Psychiatry 2020, 25, 2860–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, T.; Stefanoni, D.; Reisz, J.A.; Nemkov, T.; Bertolone, L.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hansen, K.C.; Hod, E.A.; et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight 2020, 5, e140327. [Google Scholar] [CrossRef]
- Cai, Y.; Kim, D.J.; Takahashi, T.; Broadhurst, D.I.; Yan, H.; Ma, S.; Rattray, N.J.W.; Casanovas-Massana, A.; Israelow, B.; Klein, J.; et al. Kynurenic acid may underlie sex-specific immune responses to COVID-19. Sci. Signal. 2021, 14, eabf8483. [Google Scholar] [CrossRef]
- Lawler, N.G.; Gray, N.; Kimhofer, T.; Boughton, B.; Gay, M.; Yang, R.; Morillon, A.C.; Chin, S.T.; Ryan, M.; Begum, S.; et al. Systemic Perturbations in Amine and Kynurenine Metabolism Associated with Acute SARS-CoV-2 Infection and Inflammatory Cytokine Responses. J. Proteome Res. 2021, 20, 2796–2811. [Google Scholar] [CrossRef]
- Almulla, A.F.; Supasitthumrong, T.; Amrapala, A.; Tunvirachaisakul, C.; Jaleel, A.K.A.; Oxenkrug, G.; Al-Hakeim, H.K.; Maes, M. The Tryptophan Catabolite or Kynurenine Pathway in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2022, 88, 1325–1339. [Google Scholar] [CrossRef]
- Ma, Q.; Ma, W.; Song, T.Z.; Wu, Z.; Liu, Z.; Hu, Z.; Han, J.B.; Xu, L.; Zeng, B.; Wang, B.; et al. Single-nucleus transcriptomic profiling of multiple organs in a rhesus macaque model of SARS-CoV-2 infection. Zool. Res. 2022, 43, 1041–1062. [Google Scholar] [CrossRef]
- Holmes, E.; Wist, J.; Masuda, R.; Lodge, S.; Nitschke, P.; Kimhofer, T.; Loo, R.L.; Begum, S.; Boughton, B.; Yang, R.; et al. Incomplete Systemic Recovery and Metabolic Phenoreversion in Post-Acute-Phase Nonhospitalized COVID-19 Patients: Implications for Assessment of Post-Acute COVID-19 Syndrome. J. Proteome Res. 2021, 20, 3315–3329. [Google Scholar] [CrossRef] [PubMed]
- Kucukkarapinar, M.; Yay-Pence, A.; Yildiz, Y.; Buyukkoruk, M.; Yaz-Aydin, G.; Deveci-Bulut, T.S.; Gulbahar, O.; Senol, E.; Candansayar, S. Psychological outcomes of COVID-19 survivors at sixth months after diagnose: The role of kynurenine pathway metabolites in depression, anxiety, and stress. J. Neural Transm. 2022, 129, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Chess, A.C.; Simoni, M.K.; Alling, T.E.; Bucci, D.J. Elevations of endogenous kynurenic acid produce spatial working memory deficits. Schizophr. Bull. 2007, 33, 797–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, K.S.; Wu, H.Q.; Schwarcz, R.; Bruno, J.P. Acute elevations of brain kynurenic acid impair cognitive flexibility: Normalization by the alpha7 positive modulator galantamine. Psychopharmacology 2012, 220, 627–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, R.; Campbell, B.M.; Strick, C.A.; Horner, W.; Hoffmann, W.E.; Kiss, T.; Chapin, D.S.; McGinnis, D.; Abbott, A.L.; Roberts, B.M.; et al. Reduction of brain kynurenic acid improves cognitive function. J. Neurosci. 2014, 34, 10592–10602. [Google Scholar] [CrossRef] [Green Version]
- Blanco Ayala, T.B.; Ramírez Ortega, D.R.; Ovalle Rodríguez, P.O.; Pineda, B.; Pérez de la Cruz, G.P.; González Esquivel, D.G.; Schwarcz, R.; Sathyasaikumar, K.V.; Jiménez Anguiano, A.J.; Pérez de la Cruz, V.P. Subchronic N-acetylcysteine Treatment Decreases Brain Kynurenic Acid Levels and Improves Cognitive Performance in Mice. Antioxidants 2021, 10, 147. [Google Scholar] [CrossRef]
- Zhang, W.; Murakawa, Y.; Wozniak, K.M.; Slusher, B.; Sima, A.A. The preventive and therapeutic effects of GCPII (NAALADase) inhibition on painful and sensory diabetic neuropathy. J. Neurol. Sci. 2006, 247, 217–223. [Google Scholar] [CrossRef]
- Rahn, K.A.; Watkins, C.C.; Alt, J.; Rais, R.; Stathis, M.; Grishkan, I.; Crainiceau, C.M.; Pomper, M.G.; Rojas, C.; Pletnikov, M.V.; et al. Inhibition of glutamate carboxypeptidase II (GCPII) activity as a treatment for cognitive impairment in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 20101–20106. [Google Scholar] [CrossRef] [Green Version]
- Rais, R.; Jiang, W.; Zhai, H.; Wozniak, K.M.; Stathis, M.; Hollinger, K.R.; Thomas, A.G.; Rojas, C.; Vornov, J.J.; Marohn, M.; et al. FOLH1/GCPII is elevated in IBD patients, and its inhibition ameliorates murine IBD abnormalities. JCI Insight 2016, 1, e88634. [Google Scholar] [CrossRef] [Green Version]
- Arteaga Cabeza, O.; Zhang, Z.; Smith Khoury, E.; Sheldon, R.A.; Sharma, A.; Zhang, F.; Slusher, B.S.; Kannan, R.M.; Kannan, S.; Ferriero, D.M. Neuroprotective effects of a dendrimer-based glutamate carboxypeptidase inhibitor on superoxide dismutase transgenic mice after neonatal hypoxic-ischemic brain injury. Neurobiol. Dis. 2021, 148, 105201. [Google Scholar] [CrossRef]
- Neale, J.H.; Olszewski, R. A role for N-acetylaspartylglutamate (NAAG) and mGluR3 in cognition. Neurobiol. Learn. Mem. 2019, 158, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.; Leslie, S.N.; Woo, E.; Amancharla, N.; Elmansy, A.; Lepe, M.; Mecca, A.P.; Slusher, B.S.; Nairn, A.C.; Arnsten, A.F.T. Glutamate Carboxypeptidase II in aging rat prefrontal cortex impairs working memory performance. Front. Aging Neurosci. 2021, 13, 760270. [Google Scholar] [CrossRef] [PubMed]
- Hollinger, K.R.; Sharma, A.; Tallon, C.; Lovell, L.; Thomas, A.G.; Zhu, X.; Wiseman, R.; Wu, Y.; Kambhampati, S.P.; Liaw, K.; et al. Dendrimer-2PMPA selectively blocks upregulated microglial GCPII activity and improves cognition in a mouse model of multiple sclerosis. Nanotheranostics 2022, 6, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Dodd, S.; Dean, O.; Copolov, D.L.; Malhi, G.S.; Berk, M. N-acetylcysteine for antioxidant therapy: Pharmacology and clinical utility. Expert. Opin. Biol. Ther. 2008, 8, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Peterson, P.L.; Lee, C.P. Effect of N-acetylcysteine on mitochondrial function following traumatic brain injury in rats. J. Neurotrauma 1999, 16, 1067–1082. [Google Scholar] [CrossRef]
- Whitney, K.; Nikulina, E.; Rahman, S.N.; Alexis, A.; Bergold, P.J. Delayed dosing of minocycline plus N-acetylcysteine reduces neurodegeneration in distal brain regions and restores spatial memory after experimental traumatic brain injury. Exp. Neurol. 2021, 45, 113816. [Google Scholar] [CrossRef]
- Bhatti, J.; Nascimento, B.; Akhtar, U.; Rhind, S.G.; Tien, H.; Nathens, A.; da Luz, L.T. Systematic Review of Human and Animal Studies Examining the Efficacy and Safety of N-Acetylcysteine (NAC) and N-Acetylcysteine Amide (NACA) in Traumatic Brain Injury: Impact on Neurofunctional Outcome and Biomarkers of Oxidative Stress and Inflammation. Front. Neurol. 2018, 8, 744. [Google Scholar] [CrossRef] [Green Version]
- Mcpherson, R.; Mangram, A.J.; Barletta, J.F.; Dzandu, J.K. N-Acetylcysteine Is Associated with Reduction Of Post Concussive Symptoms In Elderly Patients: A Pilot Study. J. Trauma Acute Care Surg. 2022. ahead of print. [Google Scholar] [CrossRef]
- Dominari, A.; Hathaway, I.D.; Kapasi, A.; Paul, T.; Makkar, S.S.; Castaneda, V.; Gara, S.; Singh, B.M.; Agadi, K.; Butt, M.; et al. Bottom-up analysis of emergent properties of N-acetylcysteine as an adjuvant therapy for COVID-19. World J. Virol. 2021, 10, 34–52. [Google Scholar] [CrossRef]
- Blanco-Ayala, T.; Sathyasaikumar, K.V.; Uys, J.D.; Pérez-de-la-Cruz, V.; Pidugu, L.S.; Schwarcz, R. N-Acetylcysteine Inhibits Kynurenine Aminotransferase II. Neuroscience 2020, 444, 160–169. [Google Scholar] [CrossRef]
- Biederman, J.; Melmed, R.D.; Patel, A.; McBurnett, K.; Konow, J.; Lyne, A.; Scherer, N.; SPD503 Study Group. A randomized, double-blind, placebo-controlled study of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. Pediatrics 2008, 121, e73–e84. [Google Scholar] [CrossRef] [PubMed]
- Rubia, K.; Alegría, A.A.; Brinson, H. Brain abnormalities in attention-deficit hyperactivity disorder: A review. Rev. Neurol. 2014, 58 (Suppl. S1), S3–S16. [Google Scholar] [PubMed]
- Ota, T.; Yamamuro, K.; Okazaki, K.; Kishimoto, T. Evaluating Guanfacine Hydrochloride in the Treatment of Attention Deficit Hyperactivity Disorder (ADHD) in Adult Patients: Design, Development and Place in Therapy. Drug Des. Dev. Ther. 2021, 15, 1965–1969. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.T.; Wang, M. The Evolutionary Expansion of mGluR3-NAAG-GCPII Signaling: Relevance to Human Intelligence and Cognitive Disorders. Am. J. Psychiatry 2020, 177, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Gyoneva, S.; Traynelis, S.F. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J. Biol. Chem. 2013, 288, 15291–15302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tan, S.L.; Du, J.; Chen, Y.; Jia, J.; Feng, J.G.; Liu, K.X.; Zhou, J. Dexmedetomidine alleviates neuroinflammation, restores sleep disorders and neurobehavioral abnormalities in rats with minimal hepatic encephalopathy. Int. Immunopharmacol. 2021, 96, 107795. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Higuchi, H.; Honda-Wakasugi, Y.; Miyake, S.; Nishioka, Y.; Yabuki-Kawase, A.; Maeda, S.; Miyawaki, T. Dexmedetomidine inhibits LPS-induced inflammatory responses through peroxisome proliferator-activated receptor gamma (PPARγ) activation following binding to α2 adrenoceptors. Eur. J. Pharmacol. 2021, 892, 173733. [Google Scholar] [CrossRef]
- McAllister, T.W.; McDonald, B.C.; Flashman, L.A.; Ferrell, R.B.; Tosteson, T.D.; Yanofsky, N.N.; Grove, M.R.; Saykin, A.J. Alpha-2 adrenergic challenge with guanfacine one month after mild traumatic brain injury: Altered working memory and BOLD response. Int. J. Psychophysiol. 2011, 82, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, T.; Mizushima, C.; Noborio, Y.; Kimoto, Y.; Nakaharu, Y.; Shimamoto, S. Efficacy of combination therapy with gabapentin and guanfacine for paroxysmal sympathetic hyperactivity following hypoxic encephalopathy: A case report. J. Int. Med. Res. 2021, 49, 3000605211009721. [Google Scholar] [CrossRef]
- Connor, D.F.; Grasso, D.J.; Slivinsky, M.D.; Pearson, G.S.; Banga, A. An open-label study of guanfacine extended release for traumatic stress related symptoms in children and adolescents. J. Child Adolesc. Psychopharmacol. 2013, 23, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Fesharaki-Zadeh, A.; Lowe, N.; Arnsten, A.F. Clinical experience with the α2A-adrenoceptor agonist, guanfacine, and N-acetylcysteine for the treatment of cognitive deficits in “Long-COVID19”. Neuroimmunol. Rep. 2023, 3, 100154. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fesharaki Zadeh, A.; Arnsten, A.F.T.; Wang, M. Scientific Rationale for the Treatment of Cognitive Deficits from Long COVID. Neurol. Int. 2023, 15, 725-742. https://doi.org/10.3390/neurolint15020045
Fesharaki Zadeh A, Arnsten AFT, Wang M. Scientific Rationale for the Treatment of Cognitive Deficits from Long COVID. Neurology International. 2023; 15(2):725-742. https://doi.org/10.3390/neurolint15020045
Chicago/Turabian StyleFesharaki Zadeh, Arman, Amy F. T. Arnsten, and Min Wang. 2023. "Scientific Rationale for the Treatment of Cognitive Deficits from Long COVID" Neurology International 15, no. 2: 725-742. https://doi.org/10.3390/neurolint15020045
APA StyleFesharaki Zadeh, A., Arnsten, A. F. T., & Wang, M. (2023). Scientific Rationale for the Treatment of Cognitive Deficits from Long COVID. Neurology International, 15(2), 725-742. https://doi.org/10.3390/neurolint15020045