
Characterization and Hydrocarbon Degradation Potential of Variovorax sp. Strain N23 Isolated from the Antarctic Soil
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Enrichment of Phenanthrene-Degrading Consortia and Isolation of Cultivable Strains
2.2. DNA Extraction and 16S rRNA Gene Amplicon Sequencing of the Phenanthrene-Degrading Communities
2.3. Biodegradability of Various Hydrocarbons by Strain N23
2.4. Whole Genome Sequencing and Analysis of Strain N23
2.5. Phylogenetic Analysis of Strain N23
2.6. Statistical Analysis and Data Visualization
3. Results and Discussion
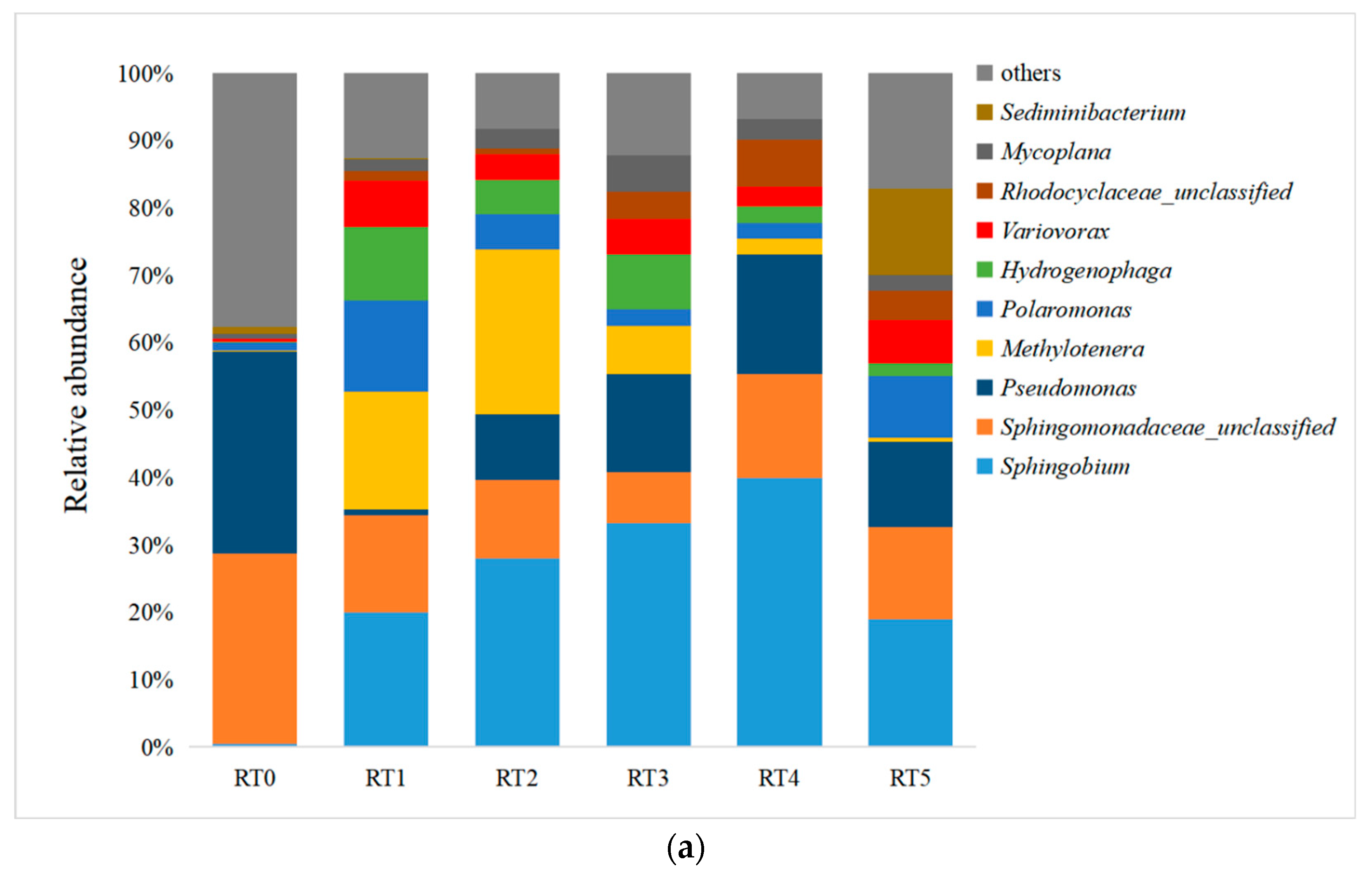
3.1. The Dynamics of Phenanthrene-Degrading Communities Derived from Antarctic Soil
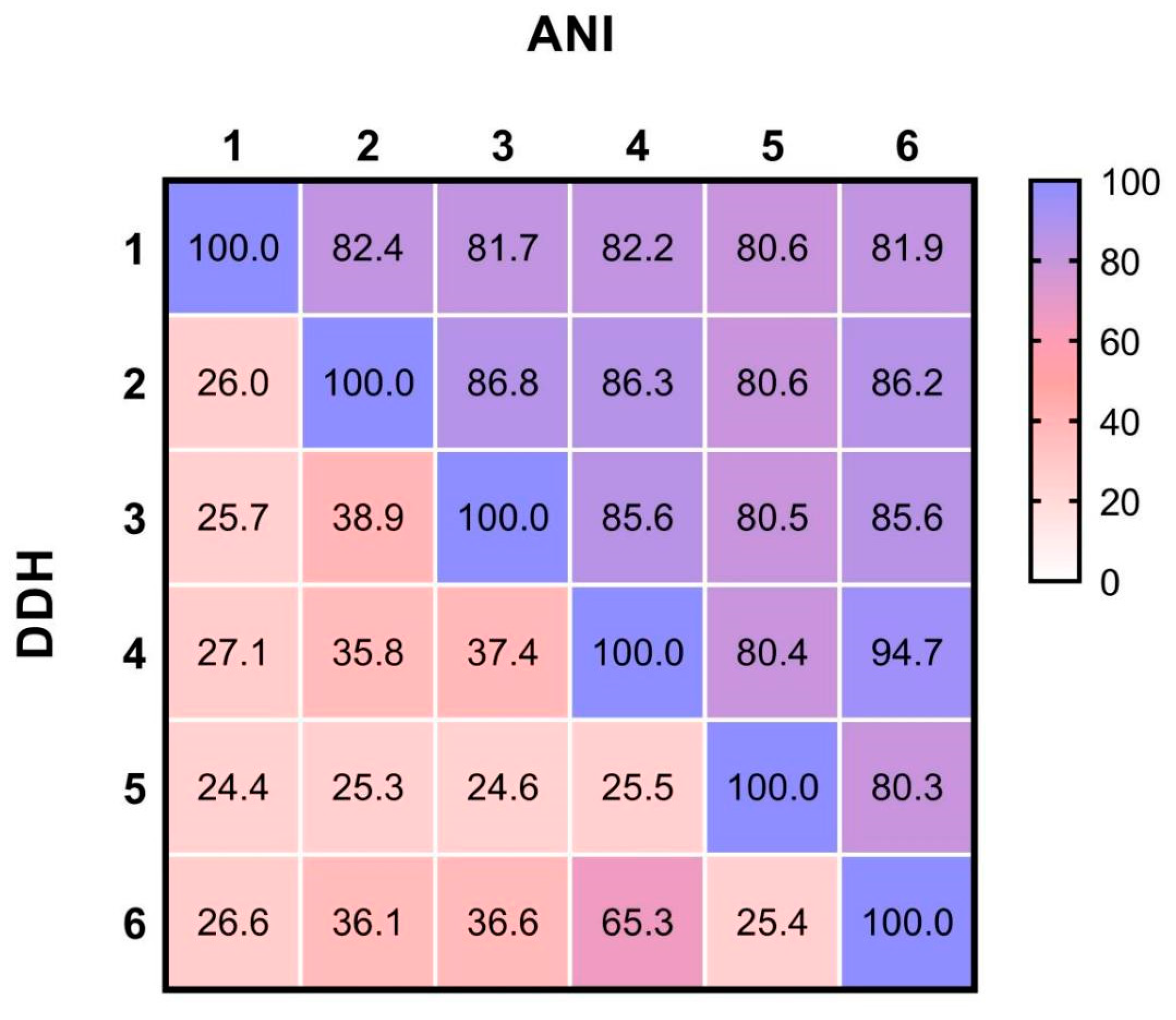
3.2. Phylogeny of Strain N23 and Proposal of a Novel Species
3.3. Hydrocarbon Utilization Profile of Strain N23
3.4. General Genomic Characteristics of Variovorax sp. Strain N23
3.5. Ecological Adaptability of Variovorax sp. Strain N23 to the Antarctic Environment
3.6. Identified Genes Encoding Hydrocarbon Metabolism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bargagli, R. Environmental contamination in Antarctic ecosystems. Sci. Total Environ. 2008, 400, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Mortelmans, K.; Haworth, S.; Lawlor, T.; Speck, W.; Tainer, B.; Zeiger, E. Salmonella mutagenicity tests: II. Results from the testing of 270 chemicals. Environ. Mutagen. 1986, 8 (Suppl. 7), 1–119. [Google Scholar] [CrossRef]
- Margesin, R.; Schinner, F. Biodegradation and bioremediation of hydrocarbons in extreme environments. Appl. Microbiol. Biotechnol. 2001, 56, 650–663. [Google Scholar] [CrossRef]
- McClintock, J.B.; Amsler, C.D.; Baker, B.J.; Moran, A.L.; Arthur Woods, H. Introduction to the Symposium: New Frontiers in Antarctic Marine Biology. Integr. Comp. Biol. 2020, 60, 1355–1357. [Google Scholar] [CrossRef] [PubMed]
- Aislabie, J.; Saul, D.J.; Foght, J.M. Bioremediation of hydrocarbon-contaminated polar soils. Extrem. Life Under Extrem. Cond. 2006, 10, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Martinez Alvarez, L.; Bolhuis, H.; Mau, G.K.; Kok-Gan, C.; Sing, C.C.; Mac Cormack, W.; Ruberto, L. Identification of key bacterial players during successful full-scale soil field bioremediation in Antarctica. Int. Biodeterior. Biodegrad. 2022, 168, 105354. [Google Scholar] [CrossRef]
- De Sousa, S.T.P.; Cabral, L.; Lacerda Júnior, G.V.; Oliveira, V.M. Diversity of aromatic hydroxylating dioxygenase genes in mangrove microbiome and their biogeographic patterns across global sites. Microbiology 2017, 6, e00490. [Google Scholar] [CrossRef]
- Ciok, A.; Dziewit, L.; Grzesiak, J.; Budzik, K.; Gorniak, D.; Zdanowski, M.K.; Bartosik, D. Identification of miniature plasmids in psychrophilic Arctic bacteria of the genus Variovorax. FEMS Microbiol. Ecol. 2016, 92, fiw043. [Google Scholar] [CrossRef] [Green Version]
- Im, W.T.; Liu, Q.M.; Lee, K.J.; Kim, S.Y.; Lee, S.T.; Yi, T.H. Variovorax ginsengisoli sp. nov., a denitrifying bacterium isolated from soil of a ginseng field. Int. J. Syst. Evol. Microbiol. 2010, 60, 1565–1569. [Google Scholar] [CrossRef]
- Han, S.R.; Lee, J.H.; Kang, S.; Park, H.; Oh, T.J. Complete genome sequence of opine-utilizing Variovorax sp. strain PAMC28711 isolated from an Antarctic lichen. J. Biotechnol. 2016, 225, 46–47. [Google Scholar] [CrossRef]
- Jin, L.; Kim, K.K.; Ahn, C.Y.; Oh, H.M. Variovorax defluvii sp. nov., isolated from sewage. Int. J. Syst. Evol. Microbiol. 2012, 62, 1779–1783. [Google Scholar] [CrossRef] [Green Version]
- Miwa, H.; Ahmed, I.; Yoon, J.; Yokota, A.; Fujiwara, T. Variovorax boronicumulans sp. nov., a boron-accumulating bacterium isolated from soil. Int. J. Syst. Evol. Microbiol. 2008, 58, 286–289. [Google Scholar] [CrossRef] [Green Version]
- Anzai, Y.; Kim, H.; Park, J.Y.; Wakabayashi, H.; Oyaizu, H. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int. J. Syst. Evol. Microbiol. 2000, 50, 1563–1589. [Google Scholar] [CrossRef] [Green Version]
- Ghimire, N.; Kim, B.; Lee, C.M.; Oh, T.J. Comparative genome analysis among Variovorax species and genome guided aromatic compound degradation analysis emphasizing 4-hydroxybenzoate degradation in Variovorax sp. PAMC26660. BMC Genom. 2022, 23, 375. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, W.; Wang, E.; Wang, J.; Liu, Z.; Li, Y.; Wei, G. Microbial succession in response to pollutants in batch-enrichment culture. Sci. Rep. 2016, 6, 21791. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.; Xu, G.; Gao, W.; Li, Q.; Yang, B.; Yang, G.; Zheng, L. Isolation and characterization of Cycloclasticus strains from Yellow Sea sediments and biodegradation of pyrene and fluoranthene by their syntrophic association with Marinobacter strains. Int. Biodeterior. Biodegrad. 2014, 91, 45–51. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldenberger, D.; Perschil, I.; Ritzler, M.; Altwegg, M. A simple “universal” DNA extraction procedure using SDS and proteinase K is compatible with direct PCR amplification. PCR Methods Appl. 1995, 4, 368–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. A J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR recognition tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Wayne, L.G. International Committee on Systematic Bacteriology: Announcement of the Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Syst. Appl. Microbiol. 1988, 10, 99–100. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Vasileva-Tonkova, E.; Gesheva, V. Potential for biodegradation of hydrocarbons by microorganisms isolated from Antarctic soils. Z. Fur Naturforschung. C J. Biosci. 2004, 59, 140–145. [Google Scholar] [CrossRef]
- Zhou, M.; Liu, Z.; Wang, J.; Zhao, Y.; Hu, B. Sphingomonas Relies on Chemotaxis to Degrade Polycyclic Aromatic Hydrocarbons and Maintain Dominance in Coking Sites. Microorganisms 2022, 10, 1109. [Google Scholar] [CrossRef]
- Gregson, B.H.; Metodieva, G.; Metodiev, M.V.; Golyshin, P.N.; McKew, B.A. Protein expression in the obligate hydrocarbon-degrading psychrophile Oleispira antarctica RB-8 during alkane degradation and cold tolerance. Environ. Microbiol. 2020, 22, 1870–1883. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.D.; Russell, M.H.; Alexandre, G. Diversity in bacterial chemotactic responses and niche adaptation. Adv. Appl. Microbiol. 2009, 66, 53–75. [Google Scholar]
- Wang, W.; Shao, Z. The long-chain alkane metabolism network of Alcanivorax dieselolei. Nat. Commun. 2014, 5, 5755. [Google Scholar] [CrossRef] [Green Version]
- Porter, S.L.; Wadhams, G.H.; Armitage, J.P. Signal processing in complex chemotaxis pathways. Nat. Rev. Microbiol. 2011, 9, 153–165. [Google Scholar] [CrossRef]
- De los Ríos, A.; Wierzchos, J.; Sancho, L.G.; Ascaso, C. Exploring the physiological state of continental Antarctic endolithic microorganisms by microscopy. FEMS Microbiol. Ecol. 2004, 50, 143–152. [Google Scholar]
- Simon, M.J.; Osslund, T.D.; Saunders, R.; Ensley, B.D.; Suggs, S.; Harcourt, A.; Suen, W.C.; Cruden, D.L.; Gibson, D.T.; Zylstra, G.J. Sequences of genes encoding naphthalene dioxygenase in Pseudomonas putida strains G7 and NCIB 9816-4. Gene 1993, 127, 31–37. [Google Scholar] [CrossRef]
- Kauppi, B.; Lee, K.; Carredano, E.; Parales, R.E.; Gibson, D.T.; Eklund, H.; Ramaswamy, S. Structure of an aromatic-ring-hydroxylating dioxygenase-naphthalene 1,2-dioxygenase. Structure 1998, 6, 571–586. [Google Scholar] [CrossRef] [Green Version]
- Habe, H.; Omori, T. Genetics of polycyclic aromatic hydrocarbon metabolism in diverse aerobic bacteria. Biosci. Biotechnol. Biochem. 2003, 67, 225–243. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liu, X.; Yang, W.; Xu, F.; Wang, W.; Feng, L.; Bartlam, M.; Wang, L.; Rao, Z. Crystal structure of long-chain alkane monooxygenase (LadA) in complex with coenzyme FMN: Unveiling the long-chain alkane hydroxylase. J. Mol. Biol. 2008, 376, 453–465. [Google Scholar] [CrossRef]
- Wang, W.; Shao, Z. Enzymes and genes involved in aerobic alkane degradation. Front. Microbiol. 2013, 4, 116. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Features | Values |
|---|---|
| Genome size (bp) | 5,292,575 |
| G + C content (%) | 67.89 |
| Number of contigs | 47 |
| Number of scaffolds | 45 |
| Scaffold N50 (bp) | 319,074 |
| Scaffold N50 number | 7 |
| Max sequence length (bp) | 587,210 |
| Min sequence length (bp) | 1077 |
| Coding sequences | 5010 |
| CDS/total genome (%) | 90.25 |
| CRISPR elements | 2 |
| Non-coding RNA (ncRNA) | 73 |
| ncRNA/total genome (%) | 0.24 |
| rRNA | 6 (5S, 16S, 23S) |
| tRNA | 48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Cui, Z.; Hao, T.; Li, Y.; Luan, X.; Feng, K.; Zheng, L. Characterization and Hydrocarbon Degradation Potential of Variovorax sp. Strain N23 Isolated from the Antarctic Soil. Microbiol. Res. 2023, 14, 91-103. https://doi.org/10.3390/microbiolres14010009
Liu J, Cui Z, Hao T, Li Y, Luan X, Feng K, Zheng L. Characterization and Hydrocarbon Degradation Potential of Variovorax sp. Strain N23 Isolated from the Antarctic Soil. Microbiology Research. 2023; 14(1):91-103. https://doi.org/10.3390/microbiolres14010009
Chicago/Turabian StyleLiu, Jinyan, Zhisong Cui, Tong Hao, Yingchao Li, Xiao Luan, Ke Feng, and Li Zheng. 2023. "Characterization and Hydrocarbon Degradation Potential of Variovorax sp. Strain N23 Isolated from the Antarctic Soil" Microbiology Research 14, no. 1: 91-103. https://doi.org/10.3390/microbiolres14010009
APA StyleLiu, J., Cui, Z., Hao, T., Li, Y., Luan, X., Feng, K., & Zheng, L. (2023). Characterization and Hydrocarbon Degradation Potential of Variovorax sp. Strain N23 Isolated from the Antarctic Soil. Microbiology Research, 14(1), 91-103. https://doi.org/10.3390/microbiolres14010009