Omega-3 Fatty Acids and Insulin Resistance: Focus on the Regulation of Mitochondria and Endoplasmic Reticulum Stress

 , ,
, ,  ,
, {kind=link}
{kind=link}
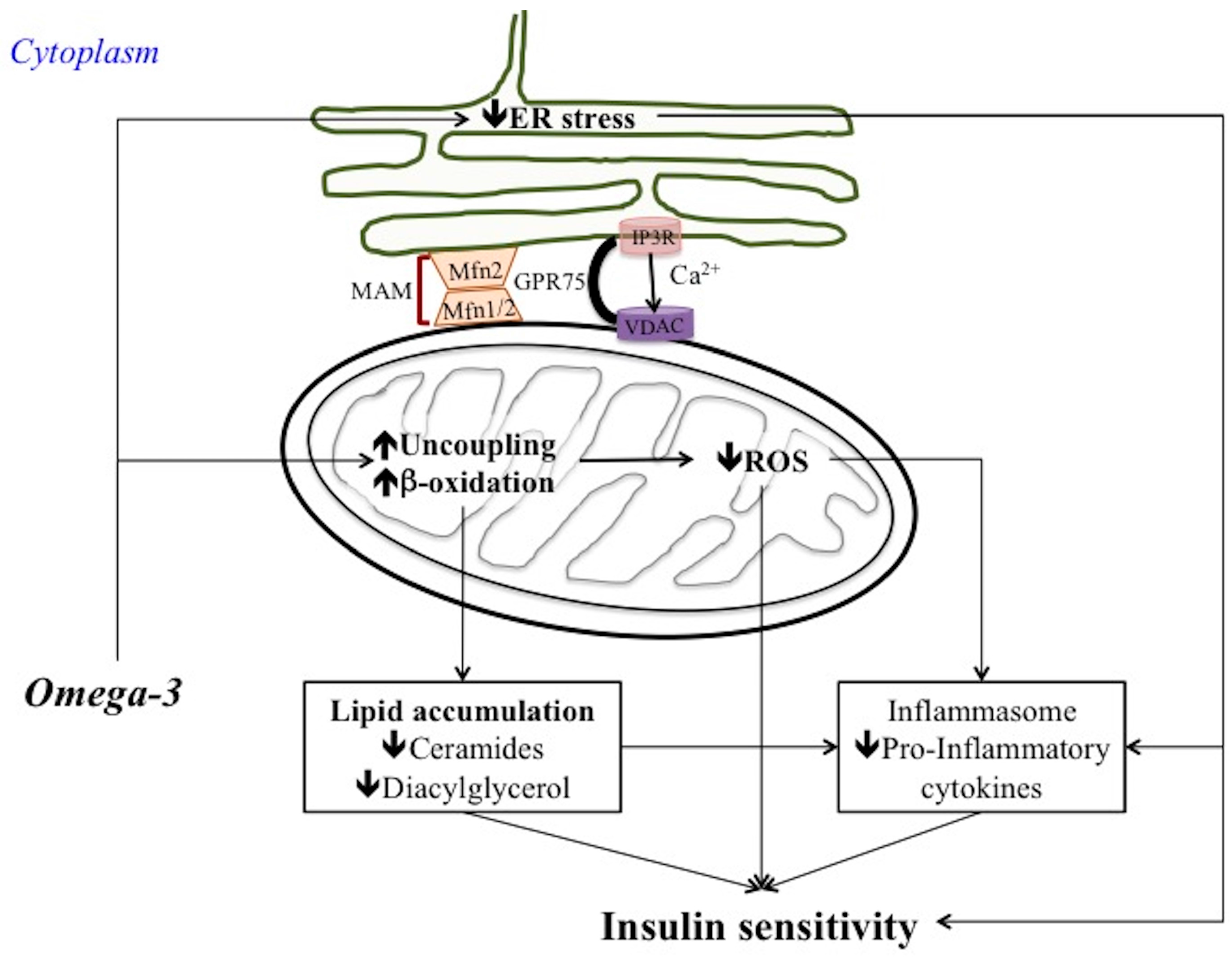
Abstract
:1. Introduction
2. Mechanisms Linking Mitochondria and Insulin Resistance
2.1. Mitochondrial Dysfunction or ROS Production?
2.2. Mitochondrial Dynamic Behaviour: Role of Mitofusin in Insulin Resistance
3. Mechanisms Linking ER Stress and Insulin Resistance
4. ER-Mitochondria Interaction in Inflammation and Insulin Resistance
5. Omega 3 PUFA as Important Bioactive Lipids
6. Omega 3 PUFA Regulation of Mitochondrial Bioenergetics and Dynamic Behaviour
7. Omega 3 PUFA Regulation of ER Stress, MAM and Inflammatory Process
7.1. Omega 3 PUFA Attenuate ER Stress
7.2. Omega 3 PUFA Inhibits Inflammasome Activation: Role of MAM
7.3. Omega 3 PUFA and Metaflammation: Few Considerations
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Martin, S.D.; McGee, S.L. The role of mitochondria in the aetiology of insulin resistance and type 2 diabetes. Biochim. Biophys. Acta 2014, 1840, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Lark, D.S.; Fisher-Wellman, K.H.; Neufer, P.D. High-fat load: Mechanism(s) of insulin resistance in skeletal muscle. Int. J. Obes. 2012, 2, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Hone, P.D.; Pacini, G. Hepatic dysfunction and insulin insensitivity in type 2 diabetes mellitus: A critical target for insulin-sensitizing agents. Diabetes Obes. Metab. 2008, 10, 699–718. [Google Scholar] [CrossRef]
- Boden, G. Obesity, insulin resistance and free fatty acids. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Suganami, T.; Tanaka, M.; Ogawa, Y. Adipose tissue inflammation and ectopic lipid accumulation. Endocr. J. 2012, 59, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.I.; Middelbeek, R.J.; Townsend, K.L.; An, D.; Nygaard, E.B.; Hitchcox, K.M.; Markan, K.R.; Nakano, K.; Hirshman, M.F.; Tseng, Y.H.; et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J. Clin. Investig. 2013, 123, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Bruns, O.T.; Reimer, R.; Hohenberg, H.; Ittrich, H.; Peldschus, K.; Kaul, M.G.; Tromsdorf, U.I.; Weller, H.; Waurisch, C.; et al. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 2011, 17, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Sidossis, L.; Kajimura, S. Brown and beige fat in humans: Thermogenic adipocytes that control energy and glucose homeostasis. J. Clin. Investig. 2015, 125, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Orava, J.; Nuutila, P.; Lidell, M.E.; Oikonen, V.; Noponen, T.; Viljanen, T.; Scheinin, M.; Taittonen, M.; Niemi, T.; Enerbäck, S.; et al. Different metabolic responses of human brown adipose tissue to activation by cold and insulin. Cell Metab. 2011, 14, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Orava, J.; Nuutila, P.; Noponen, T.; Parkkola, R.; Viljanen, T.; Enerbäck, S.; Rissanen, A.; Pietiläinen, K.H.; Virtanen, K.A. Blunted metabolic responses to cold and insulin stimulation in brown adipose tissue of obese humans. Obesity 2013, 21, 2279–2287. [Google Scholar] [CrossRef] [PubMed]
- Chondronikola, M.; Volpi, E.; Børsheim, E.; Porter, C.; Annamalai, P.; Enerback, S.; Lidell, M.E.; Saraf, M.K.; Labbe, S.M.; Hurren, N.M.; et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes 2014, 63, 4089–4099. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed]
- Shenk, S.; Saberi, M.; Olefsky, J.M. Insulin sensitivity: Modulation by nutrients and inflammation. J. Clin. Investig. 2008, 118, 2992–3002. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, L.; Mollica, M.P.; Lombardi, A.; Cavaliere, G.; Gifuni, G.; Barletta, A. From chronic overnutrition to insulin resistance: The role of fat-storing capacity and inflammation. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Mollica, M.P.; Lionetti, L.; Putti, R.; Cavaliere, G.; Gaita, M.; Barletta, A. From chronic overfeeding to hepaticinjury: Role of endoplasmic reticulum stress and inflammation. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Rieusset, J. Contribution of mitochondria and endoplasmic reticulum dysfunction in insulin resistance: Distinct or interrelated roles? Diabetes Metab. 2015, 41, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [PubMed]
- Rheinheimer, J.; de Souza, B.M.; Cardoso, N.S.; Bauer, A.C.; Crispim, D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: A systematic review. Metabolism 2017, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. Omega-3 fatty acids in inflammation and autoimmune disease. J. Am. Coll. Nutr. 2002, 21, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Aluko, R.E. Functional Foods and Nutriceuticals, 1st ed.; Springer: New York, NY, USA, 2012; p. 155. ISBN 978-1-4614-3479-5. [Google Scholar]
- Lalia, A.Z.; Lanza, I.R. Insulin-Sensitizing Effects of Omega-3 Fatty Acids: Lost in Translation? Nutrients 2016, 8, 329. [Google Scholar] [CrossRef] [PubMed]
- Civitarese, A.E.; Ravussin, E. Mitochondrial energetics and insulin resistance. Endocrinology 2008, 149, 950–995. [Google Scholar] [CrossRef] [PubMed]
- Holloszy, J.O. “Deficiency” of mitochondria in muscle does not cause insulin resistance. Diabetes 2013, 62, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Putti, R.; Migliaccio, V.; Sica, R.; Lionetti, L. Skeletal Muscle Mitochondrial Bioenergetics and Morphology in High Fat Diet Induced Obesity and Insulin Resistance: Focus on Dietary Fat Source. Front. Physiol. 2016, 6, 426. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [PubMed]
- Morino, K.; Petersen, K.F.; Dufour, S.; Befroy, D.; Frattini, J.; Shatzkes, N.; Neschen, S.; White, M.F.; Bilz, S.; Sono, S.; et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Investig. 2005, 115, 3587–3593. [Google Scholar] [CrossRef] [PubMed]
- Bruce, C.R.; Hoy, A.J.; Turner, N.; Watt, M.J.; Allen, T.L.; Carpenter, K.; Cooney, G.J.; Febbraio, M.A.; Kraegen, E.W. Kraegen Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high-fat diet-induced insulin resistance. Diabetes 2009, 58, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005, 54, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, M.; Sahlin, K.; Fernström, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Højlund, K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeks, J.; van Herpen, N.A.; Mensink, M.; Moonen-Kornips, E.; van Beurden, D.; Hesselink, M.K.; Schrauwen, P. Prolonged fasting identifies skeletal muscle mitochondrial dysfunction as consequence rather than cause of human insulin resistance. Diabetes 2010, 59, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muoio, D.M. Intramuscolar triacylglycerol and insulin resistance: Guilty as charged or wrongly accused? Biochim. Biophys. Acta 2010, 1801, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M.; Neufer, P.D. Lipid-induced mitochondrial stress and insulin resistance. Cell Metab. 2012, 15, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Wellman, K.H.; Neufer, P.D. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 2012, 23, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Choo, H.J.; Kim, J.H.; Kwon, O.B.; Lee, C.S.; Mun, J.Y.; Han, S.S.; Yoon, Y.S.; Yoon, G.; Choi, K.M.; Ko, Y.G. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 2006, 49, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Bouderba, S.; Sanz, M.N.; Sánchez-Martín, C.; El-Mir, M.Y.; Villanueva, G.R.; Detaille, D.; Koceïr, E.A. Hepatic mitochondrial alterations and increased oxidative stress in nutritional diabetes-prone Psammomys obesus model. Exp. Diabetes Res. 2012, 430176. [Google Scholar] [CrossRef]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Mantena, S.K.; Vaughn, D.P.; Andringa, K.K.; Eccleston, H.B.; King, A.L.; King, A.L.; Abrams, G.A.; Doeller, J.E.; Kraus, D.W.; Darley-Usmar, V.M.; et al. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem. J. 2009, 417, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.I.; Szendroedi, J.; Chmelik, M.; Krssák, M.; Moser, E.; Roden, M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 2011, 34, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Wong, E.D.; Meeusen, S.; Wagner, J.A. Studying the behaviour of mitochondria. Methods Enzymol. 2002, 351, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Holmström, M.H.; Iglesias-Gutierrez, E.; Zierath, J.R.; Garcia-Roves, P.M. Tissue-specific control of mitochondrial respiration in obesity-related insulin resistance and diabetes. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E731–E739. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, L.; Mollica, M.P.; Donizzetti, I.; Gifuni, G.; Sica, R.; Pignalosa, A.; Cavaliere, G.; Gaita, M.; De Filippo, C.; Zorzano, A.; et al. High-lard and high-fish-oil diets differ in their effects on function and dynamic behaviour of rat hepatic mitochondria. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Gan, K.X.; Wang, C.; Chen, J.H.; Zhu, C.J.; Song, G.Y. Mitofusin-2 ameliorates high-fat diet-induced insulin resistance in liver of rats. World J. Gastroenterol. 2013, 19, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, D.; Hernández-Alvarez, M.I.; Segalés, J.; Sorianello, E.; Muñoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signalling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [PubMed]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Zorzano, A.; Liesa, M.; Palacín, M. Mitochondrial dynamics as a bridge between mitochondrial dysfunction and insulin resistance. Arch. Physiol. Biochem. 2009, 115, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zorzano, A.; Liesa, M.; Palacin, M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int. J. Biochem. Cell Biol. 2009, 41, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Jin, P.; Yu, L.; Wang, Y.; Han, L.; Shi, T.; Li, X. Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS ONE 2014, 9, e92810. [Google Scholar] [CrossRef] [PubMed]
- Putti, R.; Sica, R.; Migliaccio, V.; Lionetti, L. Diet impact on mitochondrial bioenergetics and dynamics. Front. Physiol. 2015, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Hernández, A.; Leon-Aparicio, D.; Chavez-Reyes, J.; Olivares-Reyes, J.A.; DeJesus, S. Endoplasmic reticulum stress in insulin resistance and diabetes. Cell Calcium 2014, 56, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.K.; Jun, W.; Lee, J. Mechanism of ER Stress and Inflammation for Hepatic Insulin Resistance in Obesity. Ann. Nutr. Metab. 2015, 67, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes 2005, 54, S73–S78. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, M.; Ciani, L.; Kuriakose, G.; Westover, E.J.; Dura, M.; Covey, D.F.; Freed, J.H.; Maxfield, F.R.; Lytton, J.; et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in paral-lel with increased order of membrane lipids: Implications for depletion ofendoplasmic reticulum calcium stores and apoptosis in cholesterol-loadedmacrophages. J. Biol. Chem. 2004, 279, 37030–37039. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic retic-ulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartelet, A.; Widenmaier, S.B.; Schlein, C.; Johann, K.; Goncalves, R.L.S.; Eguchi, K.; Fischer, A.W.; Parlakgül, G.; Snyder, N.A.; Nguyen, T.B.; et al. Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Deldicque, L.; Van Proeyen, K.; Francaux, M.; Hespel, P. The unfolded protein response in human skeletal muscle is not involved in the onset of glucose tolerance impairment induced by a fat-rich diet. Eur. J. Appl. Physiol. 2011, 111, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.; Weigert, C.; Staiger, H.; Machicao, F.; Schick, F.; Machann, J.; Stefan, N.; Thamer, C.; Haring, H.-U.; Schleicher, E. Individual stearoyl-coa desaturase 1 expression modulates endoplasmic reticulum stress and inflammation in human myotubes and is associated with skeletal muscle lipid storage and insulin sensitivity in vivo. Diabetes 2009, 58, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Rieusset, J.; Chauvin, M.A.; Durand, A.; Bravard, A.; Laugerette, F.; Michalski, M.C.; Vidal, H. Reduction of endoplasmic reticulum stress using chemicalchaperones or Grp78 overexpression does not protect muscle cells frompalmitate-induced insulin resistance. Biochem. Biophys. Res. Commun. 2012, 417, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Hirabara, S.M.; Curi, R.; Maechler, P. Saturated fatty acid-induced insulin resistance is associated with mitochondrial dysfunction in skeletal muscle cells. J. Cell Physiol. 2010, 222, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Yuzefovych, L.; Wilson, G.; Rachek, L. Different effects of oleate versus palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: Role of oxidative stress. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E1096–E1105. [Google Scholar] [CrossRef] [PubMed]
- Rieusset, J. Role of Endoplasmic Reticulum-Mitochondria Communication in Type 2 Diabetes. Adv. Exp. Med. Biol. 2017, 997, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Thoudam, T.; Jeon, J.H.; Ha, C.M.; Lee, I.K. Role of Mitochondria-Associated Endoplasmic Reticulum Membrane in Inflammation-Mediated Metabolic Diseases. Mediat. Inflamm. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal 2011, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Tsai, T.F.; Wei, Y.H. Role of mitochondrial dysfunction and dysregulation of Ca2+ homeostasis in insulin insensitivity of mammalian cells. Ann. N. Y. Acad. Sci. 2015, 1350, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Wei, Y.H. Role of mitochondrial dysfunction and dysregulation of Ca2+ homeostasis in the pathophysiology of insulin resistance and type 2 diabetes. J. Biomed. Sci. 2017, 24, 70. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Hurst, S.; Hoek, J.; Sheu, S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerget. Biomembr. 2016, 49, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Shinjo, S.; Jiang, S.; Nameta, M.; Suzuki, T.; Kanai, M.; Nomura, Y.; Goda, N. Disruption of the mitochondria-associated ER membrane (MAM) plays a central role in palmitic acid-induced insulin resistance. Exp. Cell Res. 2017, 359, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Choi, H.; Min, J.S.; Park, S.J.; Kim, J.H.; Park, H.J.; Kim, B.; Chae, J.I.; Yim, M.; Lee, D.S. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem. 2013, 127, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Lane, T.; Flam, B.; Lockey, R.; Kolliputi, N.T. XNIP shuttling: The missing link bet6ween oxidative stress and inflammasome activation. Front. Physiol. 2013, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Vallerie, S.N.; Hotamisligil, G.S. The role of JNK proteins in metabolism. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Xu, R. Important bioactive properties of omega-3 fatty acids. Ital. J. Food Sci. 2015, 27, 129–135. [Google Scholar] [CrossRef]
- Meyer, B.J.; Mann, N.J.; Lewis, J.L.; Milligan, G.C.; Sinclair, A.J.; Howe, P.R.C. Dietary intakes and food sources of omega-6 and omega-3 polyunsaturated fatty acids. Lipids 2003, 38, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Lalia, A.Z.; Dasari, S.; Pallauf, M.; Fitch, M.; Hellerstein, M.K.; Lanza, I.R. Eicosapentaenoic acid but not docosahexaenoic acid restores skeletal muscle mitochondrial oxidative capacity in old mice. Aging Cell 2015, 14, 734–743. [Google Scholar] [CrossRef] [PubMed]
- The British Nutrition Foundation. Unsaturated Fatty Acids: Nutritional and Physiological Significance: The Report of the British Nutrition Foundation’s Task Force, 1st ed.; Chapman & Hall: London, UK, 1992; p. 211. ISBN 978-1-4899-4429-0. [Google Scholar]
- Buckley, J.D.; Howe, P.R. Long-chain omega-3 polyunsaturated fatty acids may be beneficial for reducing obesity-a review. Nutrients 2010, 2, 1212–1230. [Google Scholar] [CrossRef] [PubMed]
- Turinsky, J.; O’Sullivan, D.M.; Bayly, B.P. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J. Biol. Chem. 1990, 265, 16880–16885. [Google Scholar] [PubMed]
- Straczkowski, M.; Kowalska, I.; Baranowski, M.; Nikolajuk, A.; Otziomek, E.; Zabielski, P.; Adamska, A.; Blachnio, A.; Gorski, J.; Gorska, M. Increased skeletal muscle ceramide level in men at risk of developing type 2 diabetes. Diabetologia 2007, 50, 2366–2373. [Google Scholar] [CrossRef] [PubMed]
- Lanza, I.R.; Blachnio-Zabielska, A.; Johnson, M.L.; Schimke, J.M.; Jakaitis, D.R.; Lebrasseur, N.K.; Jensen, M.D.; SreekumaranNair, K.; Zabielski, P. Influence of fish oil on skeletal muscle mitochondrial energetics and lipid metabolites during high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1391–E1403. [Google Scholar] [CrossRef] [PubMed]
- Power, G.; Newsholme, E. Dietary fatty acids influence the activity and metabolic control of mitochondrial carnitine palmitoyl transferase I in ratheart and skeletal muscle. J. Nutr. 1997, 127, 2142–2150. [Google Scholar] [CrossRef] [PubMed]
- Laurente-Cebrián, S.; Bustos, M.; Marti, A.; Martinez, J.; Moreno-Aliaga, M. Eicosapentaenoic acid stimulates AMP-activated protein kinase and increases vistati secretion in cultured murine adipocytes. Clin. Sci. 2009, 117, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Motawi, T.; Hashem, R.; Rashed, L.; El-Razek, S. Comparative study between the effect of the peroxisome proliferator activated receptor-alpha ligands fenofibrate and n-3 polyunsaturated fatty acids on activation of 5′-AMP-activated protein kinase-alpha1 in high-fatfedrats. J. Pharm. Pharmacol. 2009, 61, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Flachs, P.; Horakova, O.; Brauner, P.; Rossmeisl, M.; Pecina, P.; Franssen-van Hal, N.; Ruzickova, J.; Sponarova, J.; Drahota, Z.; Vlcek, C.; et al. Polyunsaturated fatty acids of marine origin upregulate mitochondrial biogenesis and induce β-oxidation in white fat. Diabetologia 2005, 48, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- Baillie, R.; Takada, R.; Nakamura, M.; Clarke, S. Coordinate induction of peroxisomal acyl-CoAoxidase and UCP-3 by dietary fish oil: A mechanism for decreased body fat deposition. Prostaglandins Leukot. Essent. Fat. Acids 1999, 60, 351–356. [Google Scholar] [CrossRef]
- Lionetti, L.; Sica, R.; Mollica, M.P.; Putti, R. High-lard and high-fish oil diets differ in their effects on insulin resistance development, mitochondrial morphology and dynamic behaviour in rat skeletal muscle. Food Nutr. Sci. 2013, 4, 105–112. [Google Scholar] [CrossRef]
- Casanova, E.; Baselga-Escudero, L.; Ribas-Latre, A.; Arola-Arnal, A.; Bladé, C.; Arola, L.; Salvadó, M.J. Epigallocatechin gallate counteracts oxidative stress in docosahexaenoxic acid-treated myocytes. Biochim. Biophys. Acta 2014, 1837, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.; Naon, D.; Pich, S.; Soriano, F.X.; Vega, N.; Rieusset, J.; Laville, M.; Guillet, C.; Boirie, V.; Wallberg-Henriksson, H.; et al. Expression of Mfn2, the Charcot-Marie-Tooth neuropathytype 2A gene, in human skeletal muscle: Effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes 2005, 54, 2685–2693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, L.; Hu, W.; Zheng, Q.; Xiang, W. Mitochondrial dysfunction during in vitro hepatocyte steatos is reversed by omega-3 fatty acid-induced up-regulation of mitofusin 2. Metabolism 2011, 60, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Topczewski, F.; Pagliassotti, M.J. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E275–E281. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wong, S.; Xie, W.; Lei, T.; Luo, Z. Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat primary preadipocytes. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E576–E586. [Google Scholar] [CrossRef] [PubMed]
- Boslem, E.; MacIntosh, G.; Preston, A.M.; Bartley, C.; Busch, A.K.; Fuller, M.; Laybutt, D.; Meikle, P.; Biden, T. A lipidomic screen of palmitate-treated MIN6 beta-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem. J. 2011, 435, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, L.; Yang, X.; Shi, H.; Zhang, L. Alpha-Linolenic acid prevents endoplasmic reticulum stress-mediated apoptosis of stearic acid lipotoxicity on primary rat hepatocytes. Lipids Health Dis. 2011, 10, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X.; Shi, H.; Dong, L.; Bai, J. Effect of a-linolenic acid on endoplasmic reticulum stress-mediated apoptosis of palmitic acid lipotoxicity in primary rat hepatocytes. Lipids Health Dis. 2011, 10, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chen, X.; Chen, M.; Li, Y.; Li, Q.; Jiang, X.; Yang, Y.; Ling, W. Fish oil supplementation inhibits endoplasmic reticulum stress and improves insulin resistance: Involvement of AMP-activated protein kinase. Food Funct. 2017, 8, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, Y.; Kim, H.; Izawa, T.; Sakurai, K.; Hirako, S.; Wada, M.; Matsumoto, A. Protective effects of fish oil and pioglitazone on pancreatic tissue in obese KK mice with type 2 diabetes. Prostaglandins Leukot. Essent. Fat. Acids 2016, 115, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Hwang, H.J.; Hong, H.C.; Choi, H.Y.; Yoo, H.J.; Baik, S.H.; Choi, K.M. Resolvin D1 reduces ER stress-induced apoptosis and trigliceride accumulation through JNK pathway in HepG2 cells. Mol. Cell Endocrinol. 2014, 391, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Jiang, W.; Spinetti, T.; Tardivel, A.; Castillo, R.; Bourquin, C.; Guarda, G.; Tian, Z.; Tschopp, J.; Zhou, R. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 2013, 38, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Legrand-Poels, S.; Esser, N.; L’homme, L.; Scheen, A.; Paquot, N.; Piette, J. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem Pharmacol. 2014, 92, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–441. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; O’Neill, L.A.; Coll, R.C. Modulatory mechanisms controlling the NLRP3 inflammasome in inflammation: Recent developments. Curr. Opin. Immunol. 2013, 25, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule- driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15. [Google Scholar] [CrossRef] [PubMed]
- Horng, T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014, 35, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Ralston, J.C.; Lyons, C.L.; Kennedy, E.B.; Kirwan, A.M.; Roche, H.M. Fatty Acids and NLRP3 Inflammasome-Mediated Inflammation in Metabolic Tissues. Annu. Rev. Nutr. 2017, 37, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Kuda, O.; Rossmeisl, M.; Kopecky, J. Omega-3 fatty acids and adipose tissue biology. Mol. Asp. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Weigelt, C.; Cherradi, M.L.; Niemeier, A.; Tödter, K.; Heeren, J.; Scheja, L. Effects of adipocyte lipoprotein lipase on de novo lipogenesis and white adipose tissue browning. Biochim. Biophys. Acta 2013, 1831, 934–942. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lepretti, M.; Martucciello, S.; Burgos Aceves, M.A.; Putti, R.; Lionetti, L. Omega-3 Fatty Acids and Insulin Resistance: Focus on the Regulation of Mitochondria and Endoplasmic Reticulum Stress. Nutrients 2018, 10, 350. https://doi.org/10.3390/nu10030350
Lepretti M, Martucciello S, Burgos Aceves MA, Putti R, Lionetti L. Omega-3 Fatty Acids and Insulin Resistance: Focus on the Regulation of Mitochondria and Endoplasmic Reticulum Stress. Nutrients. 2018; 10(3):350. https://doi.org/10.3390/nu10030350
Chicago/Turabian StyleLepretti, Marilena, Stefania Martucciello, Mario Alberto Burgos Aceves, Rosalba Putti, and Lillà Lionetti. 2018. "Omega-3 Fatty Acids and Insulin Resistance: Focus on the Regulation of Mitochondria and Endoplasmic Reticulum Stress" Nutrients 10, no. 3: 350. https://doi.org/10.3390/nu10030350
APA StyleLepretti, M., Martucciello, S., Burgos Aceves, M. A., Putti, R., & Lionetti, L. (2018). Omega-3 Fatty Acids and Insulin Resistance: Focus on the Regulation of Mitochondria and Endoplasmic Reticulum Stress. Nutrients, 10(3), 350. https://doi.org/10.3390/nu10030350