Large Neutral Amino Acid Therapy Increases Tyrosine Levels in Adult Patients with Phenylketonuria: A Long-Term Study

 ,
, 
 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. LNAA Supplementation
2.3. Study Design
2.4. Statistical Analysis
3. Results
4. Discussion
5. Limitations
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blau, N.; van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef]
- Van Spronsen, F.J.; van Wegberg, A.M.; Ahring, K.; Belanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Gizewska, M.; et al. Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol. 2017, 5, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Camp, K.M.; Parisi, M.A.; Acosta, P.B.; Berry, G.T.; Bilder, D.A.; Blau, N.; Bodamer, O.A.; Brosco, J.P.; Brown, C.S.; Burlina, A.B.; et al. Phenylketonuria Scientific Review Conference: State of the science and future research needs. Mol. Genet. Metab. 2014, 112, 87–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Belanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Gizewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A. Diet and compliance in phenylketonuria. Eur. J. Pediatr. 2000, 159 (Suppl. 2), S136–S141. [Google Scholar] [CrossRef]
- Cazzorla, C.; Del Rizzo, M.; Burgard, P.; Zanco, C.; Bordugo, A.; Burlina, A.B.; Burlina, A.P. Application of the WHOQOL-100 for the assessment of quality of life of adult patients with inherited metabolic diseases. Mol. Genet. Metab. 2012, 106, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Bilder, D.A.; Burton, B.K.; Coon, H.; Leviton, L.; Ashworth, J.; Lundy, B.D.; Vespa, H.; Bakian, A.V.; Longo, N. Psychiatric symptoms in adults with phenylketonuria. Mol. Genet. Metab. 2013, 108, 155–160. [Google Scholar] [CrossRef]
- MacDonald, A.; Gokmen-Ozel, H.; van Rijn, M.; Burgard, P. The reality of dietary compliance in the management of phenylketonuria. J. Inherit. Metab. Dis. 2010, 33, 665–670. [Google Scholar] [CrossRef]
- Cazzorla, C.; Bensi, G.; Biasucci, G.; Leuzzi, V.; Manti, F.; Musumeci, A.; Papadia, F.; Stoppioni, V.; Tummolo, A.; Vendemiale, M.; et al. Living with phenylketonuria in adulthood: The pku attitude study. Mol. Genet. Metab. Rep. 2018, 16, 39–45. [Google Scholar] [CrossRef]
- Ford, S.; O’Driscoll, M.; MacDonald, A. Living with Phenylketonuria: Lessons from the PKU community. Mol. Genet. Metab. Rep. 2018, 17, 57–63. [Google Scholar] [CrossRef]
- Jurecki, E.R.; Cederbaum, S.; Kopesky, J.; Perry, K.; Rohr, F.; Sanchez-Valle, A.; Viau, K.S.; Sheinin, M.Y.; Cohen-Pfeffer, J.L. Adherence to clinic recommendations among patients with phenylketonuria in the United States. Mol. Genet. Metab. 2017, 120, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Dimmock, D.; Levy, H.; Viau, K.; Bausell, H.; Bilder, D.A.; Burton, B.; Gross, C.; Northrup, H.; Rohr, F.; et al. Evidence-and consensus-based recommendations for the use of pegvaliase in adults with phenylketonuria. Genet. Med. 2019, 21, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Muntau, A.C.; Roschinger, W.; Habich, M.; Demmelmair, H.; Hoffmann, B.; Sommerhoff, C.P.; Roscher, A.A. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N. Engl. J. Med. 2002, 347, 2122–2132. [Google Scholar] [CrossRef] [PubMed]
- Pena, M.J.; Pinto, A.; Daly, A.; MacDonald, A.; Azevedo, L.; Rocha, J.C.; Borges, N. The use of glycomacropeptide in patients with phenylketonuria: A systematic review and meta-analysis. Nutrients 2018, 10, 1794. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, D.; Bruinenberg, V.M.; Mazzola, P.N.; van Faassen, M.H.; de Blaauw, P.; Kema, I.P.; Heiner-Fokkema, M.R.; van Anholt, R.D.; van der Zee, E.A.; van Spronsen, F.J. Large neutral amino acid supplementation exerts its effect through three synergistic mechanisms: Proof of principle in phenylketonuria mice. PLoS ONE 2015, 10, e0143833. [Google Scholar] [CrossRef] [PubMed]
- Van Spronsen, F.J.; de Groot, M.J.; Hoeksma, M.; Reijngoud, D.J.; van Rijn, M. Large neutral amino acids in the treatment of PKU: From theory to practice. J. Inherit. Metab. Dis. 2010, 33, 671–676. [Google Scholar] [CrossRef]
- Kanai, Y.; Segawa, H.; Miyamoto, K.; Uchino, H.; Takeda, E.; Endou, H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem. 1998, 273, 23629–23632. [Google Scholar] [CrossRef]
- Christensen, H.N. Metabolism of amino acids and proteins. Annu. Rev. Biochem. 1953, 22, 233–260. [Google Scholar] [CrossRef]
- Pietz, J.; Kreis, R.; Rupp, A.; Mayatepek, E.; Rating, D.; Boesch, C.; Bremer, H.J. Large neutral amino acids block phenylalanine transport into brain tissue in patients with phenylketonuria. J. Clin. Investig. 1999, 103, 1169–1178. [Google Scholar] [CrossRef] [Green Version]
- Matalon, R.; Michals-Matalon, K.; Bhatia, G.; Burlina, A.B.; Burlina, A.P.; Braga, C.; Fiori, L.; Giovannini, M.; Grechanina, E.; Novikov, P.; et al. Double blind placebo control trial of large neutral amino acids in treatment of PKU: Effect on blood phenylalanine. J. Inherit. Metab. Dis. 2007, 30, 153–158. [Google Scholar] [CrossRef]
- Matalon, R.; Michals-Matalon, K.; Bhatia, G.; Grechanina, E.; Novikov, P.; McDonald, J.D.; Grady, J.; Tyring, S.K.; Guttler, F. Large neutral amino acids in the treatment of phenylketonuria (PKU). J. Inherit. Metab. Dis. 2006, 29, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Concolino, D.; Mascaro, I.; Moricca, M.T.; Bonapace, G.; Matalon, K.; Trapasso, J.; Radhakrishnan, G.; Ferrara, C.; Matalon, R.; Strisciuglio, P. Long-term treatment of phenylketonuria with a new medical food containing large neutral amino acids. Eur. J. Clin. Nutr. 2017, 71, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.; Moseley, K.D.; Yano, S.; Nelson, M., Jr.; Moats, R.A. Large neutral amino acid therapy and phenylketonuria: A promising approach to treatment. Mol. Genet. Metab. 2003, 79, 110–113. [Google Scholar] [CrossRef]
- Yano, S.; Moseley, K.; Azen, C. Large neutral amino acid supplementation increases melatonin synthesis in phenylketonuria: A new biomarker. J. Pediatr. 2013, 162, 999–1003. [Google Scholar] [CrossRef]
- Rocha, J.C.; MacDonald, A. Dietary intervention in the management of phenylketonuria: Current perspectives. Pediatric Health Med. Ther. 2016, 7, 155–163. [Google Scholar] [CrossRef]
- Dotremont, H.; Francois, B.; Diels, M.; Gillis, P. Nutritional value of essential amino acids in the treatment of adults with phenylketonuria. J. Inherit. Metab. Dis. 1995, 18, 127–130. [Google Scholar] [CrossRef]
- Lou, H.C.; Toft, P.B.; Andresen, J.; Mikkelsen, I.; Olsen, B.; Guldberg, P.; Guttler, F. Unchanged MRI of myelin in adolescents with PKU supplied with non-phe essential amino acids after dietary relaxation. Acta Paediatr. 1994, 83, 1312–1314. [Google Scholar] [CrossRef]
- Schindeler, S.; Ghosh-Jerath, S.; Thompson, S.; Rocca, A.; Joy, P.; Kemp, A.; Rae, C.; Green, K.; Wilcken, B.; Christodoulou, J. The effects of large neutral amino acid supplements in PKU: An MRS and neuropsychological study. Mol. Genet. Metab. 2007, 91, 48–54. [Google Scholar] [CrossRef]
- Van Vliet, D.; van der Goot, E.; Bruinenberg, V.M.; van Faassen, M.; de Blaauw, P.; Kema, I.P.; Heiner-Fokkema, M.R.; van der Zee, E.A.; van Spronsen, F.J. Large neutral amino acid supplementation as an alternative to the phenylalanine-restricted diet in adults with phenylketonuria: Evidence from adult Pah-enu2 mice. J. Nutr. Biochem. 2018, 53, 20–27. [Google Scholar] [CrossRef]
- Burlina, A.B.; Bonafe, L.; Ferrari, V.; Suppiej, A.; Zacchello, F.; Burlina, A.P. Measurement of neurotransmitter metabolites in the cerebrospinal fluid of phenylketonuric patients under dietary treatment. J. Inherit. Metab. Dis. 2000, 23, 313–316. [Google Scholar] [CrossRef]
- Pilotto, A.; Blau, N.; Leks, E.; Schulte, C.; Deuschl, C.; Zipser, C.; Piel, D.; Freisinger, P.; Gramer, G.; Kolker, S.; et al. Cerebrospinal fluid biogenic amines depletion and brain atrophy in adult patients with phenylketonuria. J. Inherit. Metab. Dis. 2019, 42, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, R.; Kirkbride, B.; Newbould, E.; Durkalski, V.; Jaskiw, G.E. Relationships between large neutral amino acid levels in plasma, cerebrospinal fluid, brain microdialysate and brain tissue in the rat. Brain Res. 2010, 1334, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Oldendorf, W.H. Kinetic analysis of blood-brain barrier transport of amino acids. Biochim. Biophys. Acta 1975, 401, 128–136. [Google Scholar] [CrossRef]
- Taslimifar, M.; Buoso, S.; Verrey, F.; Kurtcuoglu, V. Functional polarity of microvascular brain endothelial cells supported by neurovascular unit computational model of large neutral amino acid homeostasis. Front. Physiol. 2018, 9, 171. [Google Scholar] [CrossRef]
- Cleary, M.; Trefz, F.; Muntau, A.C.; Feillet, F.; van Spronsen, F.J.; Burlina, A.; Belanger-Quintana, A.; Gizewska, M.; Gasteyger, C.; Bettiol, E.; et al. Fluctuations in phenylalanine concentrations in phenylketonuria: A review of possible relationships with outcomes. Mol. Genet. Metab. 2013, 110, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Taslimifar, M.; Buoso, S.; Verrey, F.; Kurtcuoglu, V. Propagation of plasma L-phenylalanine concentration fluctuations to the neurovascular unit in phenylketonuria: An in silico study. Front. Physiol. 2019, 10, 360. [Google Scholar] [CrossRef] [PubMed]
- Surtees, R.; Blau, N. The neurochemistry of phenylketonuria. Eur. J. Pediatr. 2000, 159 (Suppl. 2), S109–S113. [Google Scholar] [CrossRef]
- Green, B.; Rahman, Y.; Firman, S.; Adam, S.; Jenkinson, F.; Nicol, C.; Adams, S.; Dawson, C.; Robertson, L.; Dunlop, C.; et al. Improved eating behaviour and nutrient intake in noncompliant patients with phenylketonuria after reintroducing a protein substitute: Observations from a multicentre study. Nutrients 2019, 11, 2035. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy | 1686 kJ |
|---|---|
| 399 kcal | |
| Fat | 5.3 g |
| of which saturated fat | 5.3 g |
| Carbohydrates | 12 g |
| of which sugars | 0 g |
| Fiber | 5.8 g |
| Equivalent protein | 70.79 g |
| Salt | 1.6 g |
| L-Arginine | 1.92 |
| Aspartate | 4.95 |
| L-phenylalanine | 0 g |
| L-isoleucine | 10 g |
| L-histidine | 3.36 g |
| L-leucine | 12 g |
| L-lysine | 5.44 g |
| L-methionine | 2.72 g |
| L-tyrosine | 24 g |
| L-threonine | 2.56 g |
| L-tryptophan | 8 g |
| L-valine | 10 g |
| N (%) | ||
|---|---|---|
| Gender | Female | 5 (42) |
| Male | 7 (58) | |
| Age range | 19–38 | |
| Marital status | Married/with partner | 6 (50) |
| Unmarried | 6 (50) | |
| Education level | Middle | 1 (8) |
| High | 7 (58) | |
| University | 4 (33) | |
| Employment status | Employed | 9 (75) |
| Unemployed | 1 (8) | |
| Student | 2 (17) | |
| Leisure time | Sport | 3 (25) |
| Social activity | 2 (17) | |
| Art, museum, exhibitions | 2 (17) | |
| Not specified | 5 (42) |
| AAM–MF | LNAA–MF | |||
|---|---|---|---|---|
| Mean ± SD | Median | Mean ± SD | Median | |
| Total proteins (g/day) | 72 ± 11 | 70 | 63 ± 13.45 | 59 |
| Total proteins (g/kg/day) | 1 ± 0.23 | 1.13 | 0.8 ± 0.24 | 0.75 |
| MF (g/day) | 51 ± 7.36 | 52 | 42 ± 9.78 | 38 |
| MF (g/kg/day) | 0.7 ± 0.23 | 0.8 | 0.5 ± 0.21 | 0.4 |
| Phe intake (mg/day) | 834 ± 455.41 | 700 | 709 ± 199.91 | 700 |
| Phe intake (mg/kg) | 12 ± 4.81 | 10.3 | 10 ± 1.8 | 10.2 |
| Tyr intake (g/day) | 4.8 ± 0.72 | 4.6 | 6.6 ± 0.61 | 6.5 |
| Tyr intake (mg/kg) | 73 ± 21.54 | 74.7 | 100 ± 26.1 | 103.7 |
| Patient | Molecular Analysis | 12-month Period Prior to LNAA | 12-month LNAA Treatment Period | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean Phe Values a (SD) | Mean Tyr Values a (SD) | Mean Phe/Tyr Values a (SD) | DBS Frequency | Mean Phe Values a (SD) | Mean Tyr Values a (SD) | Mean Phe/Tyr Values a (SD) | DBS Frequency | ||
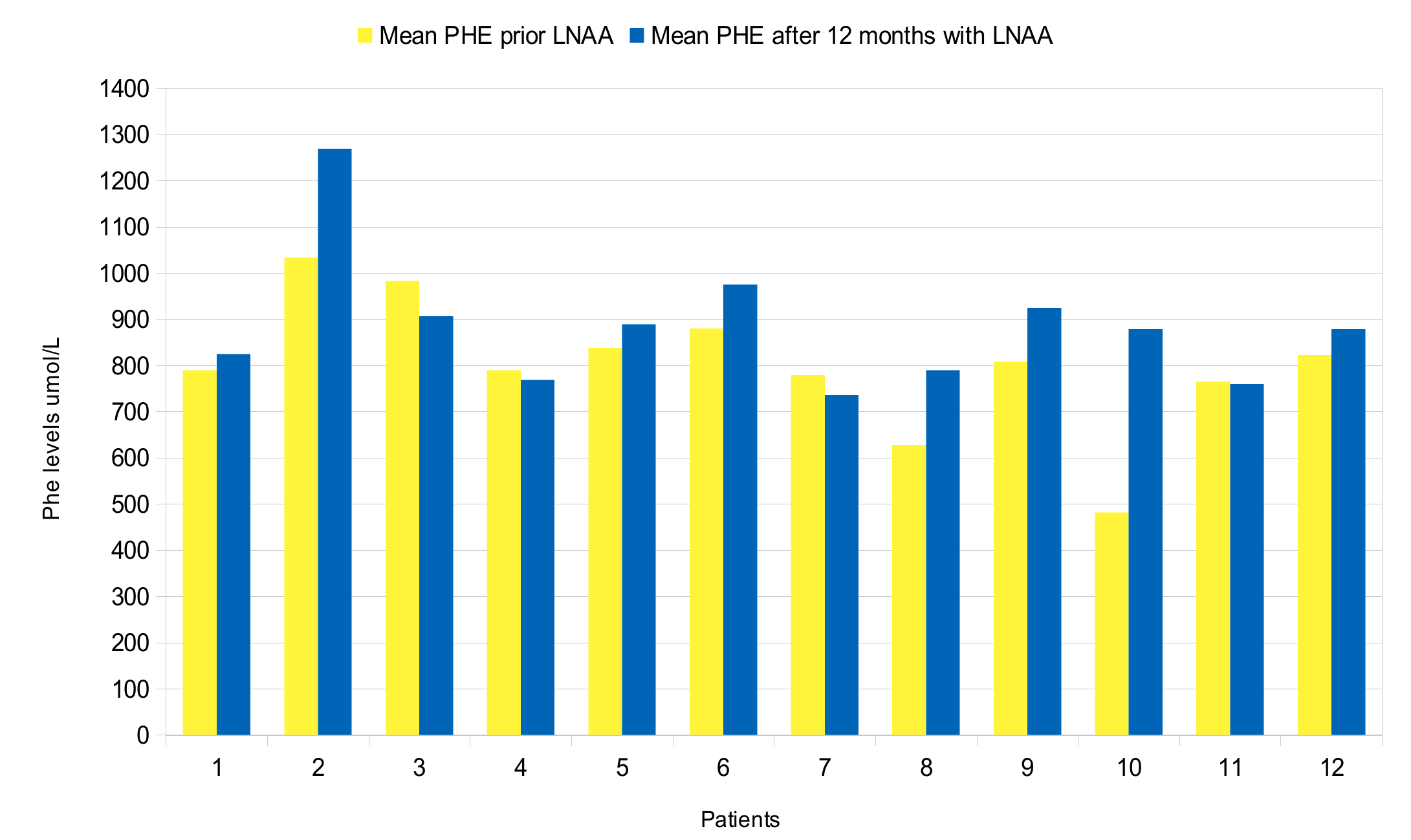
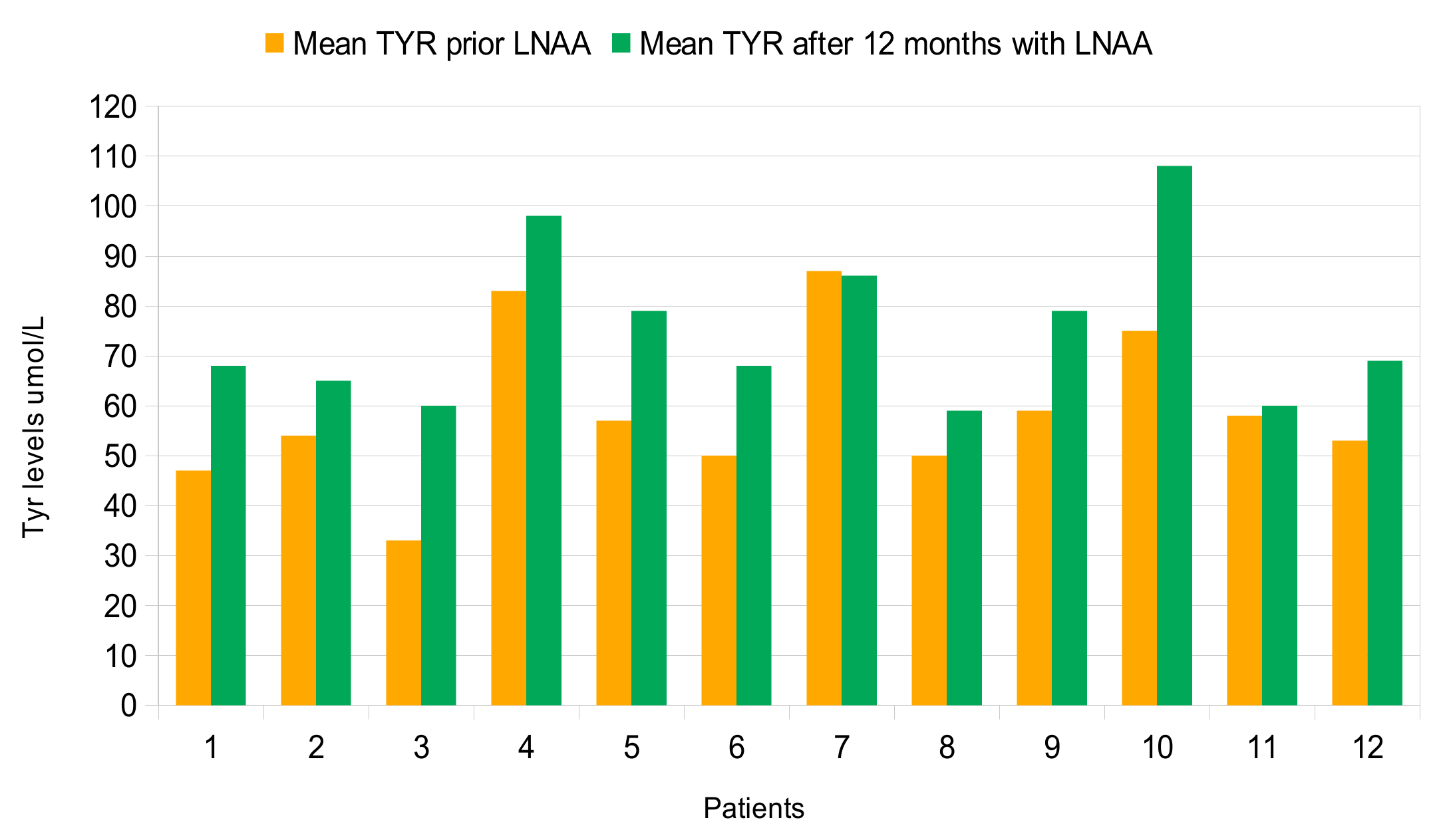
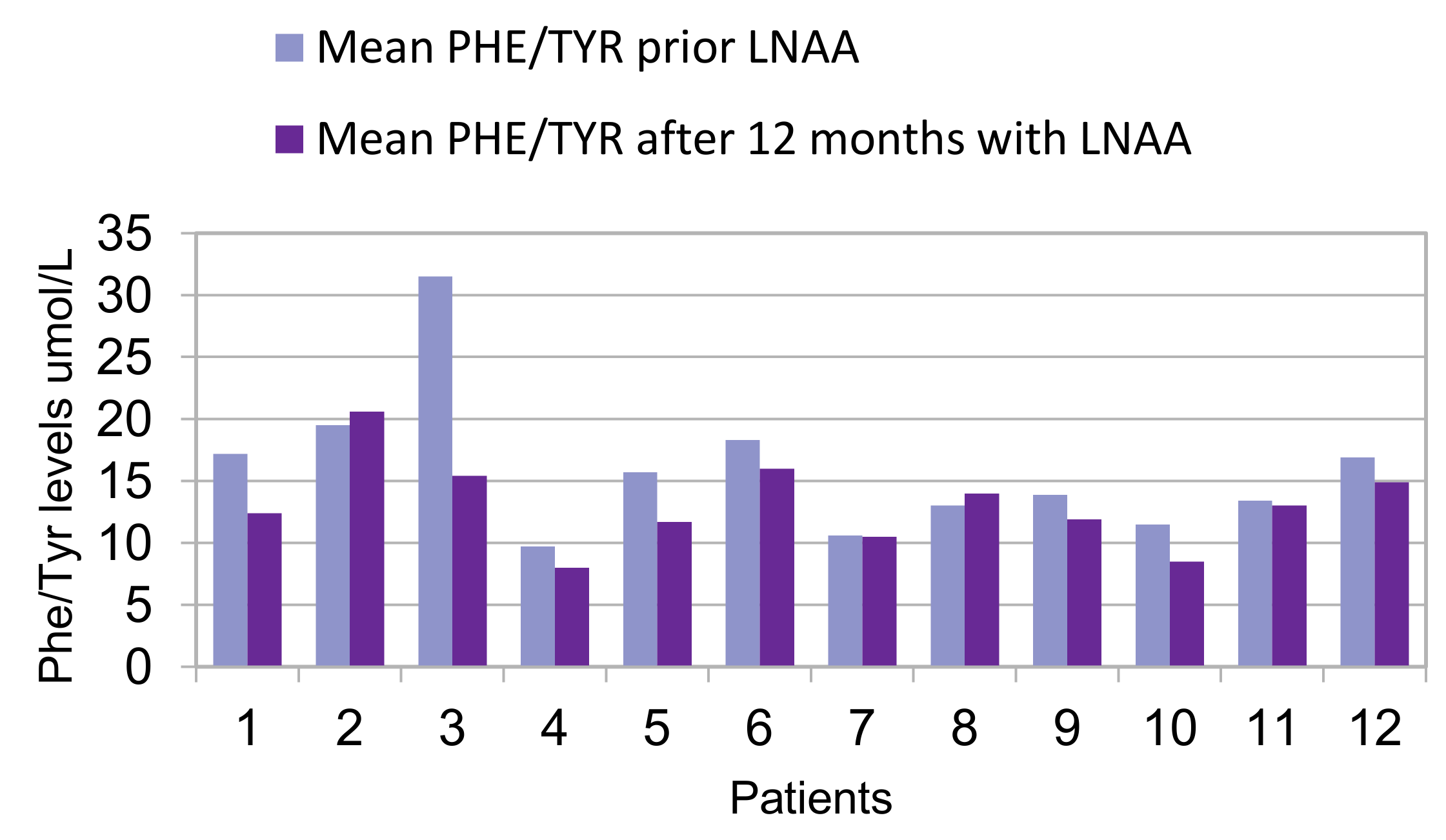
| 1 | c.473G > A/c.1315 + 1G > A | 790 (80) | 47 (8) | 17.2 (2.5) | 8 | 825 (114) | 68 (10) | 12.4 (2.4) | 17 |
| 2 | c.473G > A/c.526C > T | 1033 (198) | 54 (10) | 19.5 (4.4) | 5 | 1269 (265) | 65 (14) | 20.6 (7.6) | 14 |
| 3 | c.473G > A/c.473G > A | 983 (142) | 32 (7) | 31.5 (7.3) | 10 | 907 (166) | 60 (8) | 15.4 (3) | 20 |
| 4 | c.842 + 3G > C in heterozygosis | 790 (118) | 83 (14) | 9.7 (2.2) | 18 | 769 (144) | 98 (18) | 8 (1.4) | 34 |
| 5 | c.47_48delCT/c.1315 + 2T > C | 838 (179) | 57 (12) | 15.7 (6.6) | 20 | 889 (149) | 79 (19) | 11.7 (2.7) | 24 |
| 6 | c.842C > T/c.1315 + 1G > A | 880 (160) | 49 (11) | 18.3 (4) | 38 | 975 (148) | 68 (29) | 16 (5) | 39 |
| 7 | c.842C > T/c.1315 + 1G > A | 779 (108) | 87 (35) | 10.6 (5) | 25 | 736 (93) | 86 (41) | 10.5 (4.9) | 27 |
| 8 | c.842C > T/c.1315 + 1G > A | 628 (148) | 50 (13) | 13 (5.2) | 26 | 790 (147) | 59 (15) | 14 (4.5) | 29 |
| 9 | c.1222C > T/c.1315 + 1G > A | 808 (135) | 59 (7) | 19.9 (2.8) | 12 | 925 (139) | 79 (13) | 11.9 (1.8) | 18 |
| 10 | c.782G > A/c.782G > A | 842 (94) | 75 (12) | 11.5 (2.3) | 23 | 879 (91) | 108 (35) | 8.5 (1.9) | 36 |
| 11 | c.782G > A/c.1066 − 11G > A | 765 (234) | 58 (11) | 13.4 (3.9) | 23 | 760 (124) | 60 (11) | 13 (2.7) | 24 |
| 12 | c.1222C > T/macro deletion in exon 3 | 823 (117) | 53 (19) | 16.9 (5) | 18 | 1000 (163) | 68 (17) | 15.2 (3.7) | 15 |
| Overall Patient Population | 12-Month Period Prior to LNAA | 12-Month LNAA Treatment Period | p-Value |
|---|---|---|---|
| Phe μmol/L, mean (SD) | 752 (143) | 894 (145) | 0.0522 |
| Tyr μmol/L, mean (SD) | 59 (13) | 75 (16) | 0.0195 |
| Phe/Tyr ratio μmol/L, mean (SD) | 16 (4) | 13 (3) | 0.049 |
| DBS frequency | 19 (9) | 25 (8) | 0.0088 |
| Overall Patient Population | 12-Month Period Prior to LNAA | 6-Month LNAA Treatment Period | 12-Month LNAA Treatment Period |
|---|---|---|---|
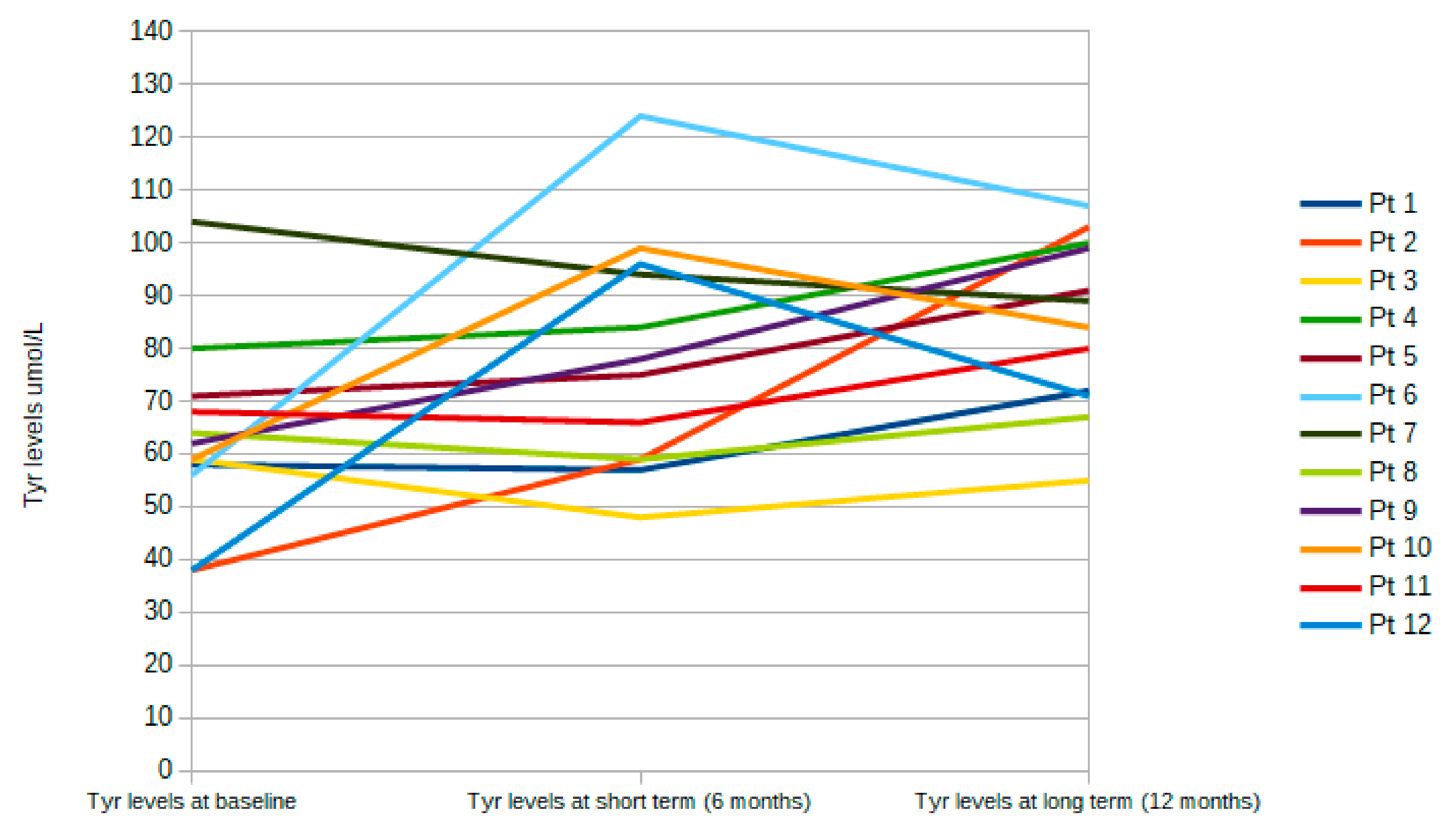
| Phe μmol/L, mean (SD) | 752 (143) | 645 (101)* | 894 (145) ** |
| Tyr μmol/L, mean (SD) | 59 (13) | 65 (16)* | 75 (16) ** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burlina, A.P.; Cazzorla, C.; Massa, P.; Polo, G.; Loro, C.; Gueraldi, D.; Burlina, A.B. Large Neutral Amino Acid Therapy Increases Tyrosine Levels in Adult Patients with Phenylketonuria: A Long-Term Study. Nutrients 2019, 11, 2541. https://doi.org/10.3390/nu11102541
Burlina AP, Cazzorla C, Massa P, Polo G, Loro C, Gueraldi D, Burlina AB. Large Neutral Amino Acid Therapy Increases Tyrosine Levels in Adult Patients with Phenylketonuria: A Long-Term Study. Nutrients. 2019; 11(10):2541. https://doi.org/10.3390/nu11102541
Chicago/Turabian StyleBurlina, Alessandro P., Chiara Cazzorla, Pamela Massa, Giulia Polo, Christian Loro, Daniela Gueraldi, and Alberto B. Burlina. 2019. "Large Neutral Amino Acid Therapy Increases Tyrosine Levels in Adult Patients with Phenylketonuria: A Long-Term Study" Nutrients 11, no. 10: 2541. https://doi.org/10.3390/nu11102541
APA StyleBurlina, A. P., Cazzorla, C., Massa, P., Polo, G., Loro, C., Gueraldi, D., & Burlina, A. B. (2019). Large Neutral Amino Acid Therapy Increases Tyrosine Levels in Adult Patients with Phenylketonuria: A Long-Term Study. Nutrients, 11(10), 2541. https://doi.org/10.3390/nu11102541