Luteolin Shifts Oxaliplatin-Induced Cell Cycle Arrest at G0/G1 to Apoptosis in HCT116 Human Colorectal Carcinoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Reagents and Antibodies
2.3. Cell Cultures
2.4. Cell Viability Assay
2.5. Antioxidant Response Element (ARE)-Luciferase Reporter Gene Assay
2.6. Colony Formation Assay
2.7. Cell Cycle Analysis
2.8. Western Blot Analysis
2.9. Statistical Analysis
3. Results
3.1. Cytotoxicity of Oxaliplatin
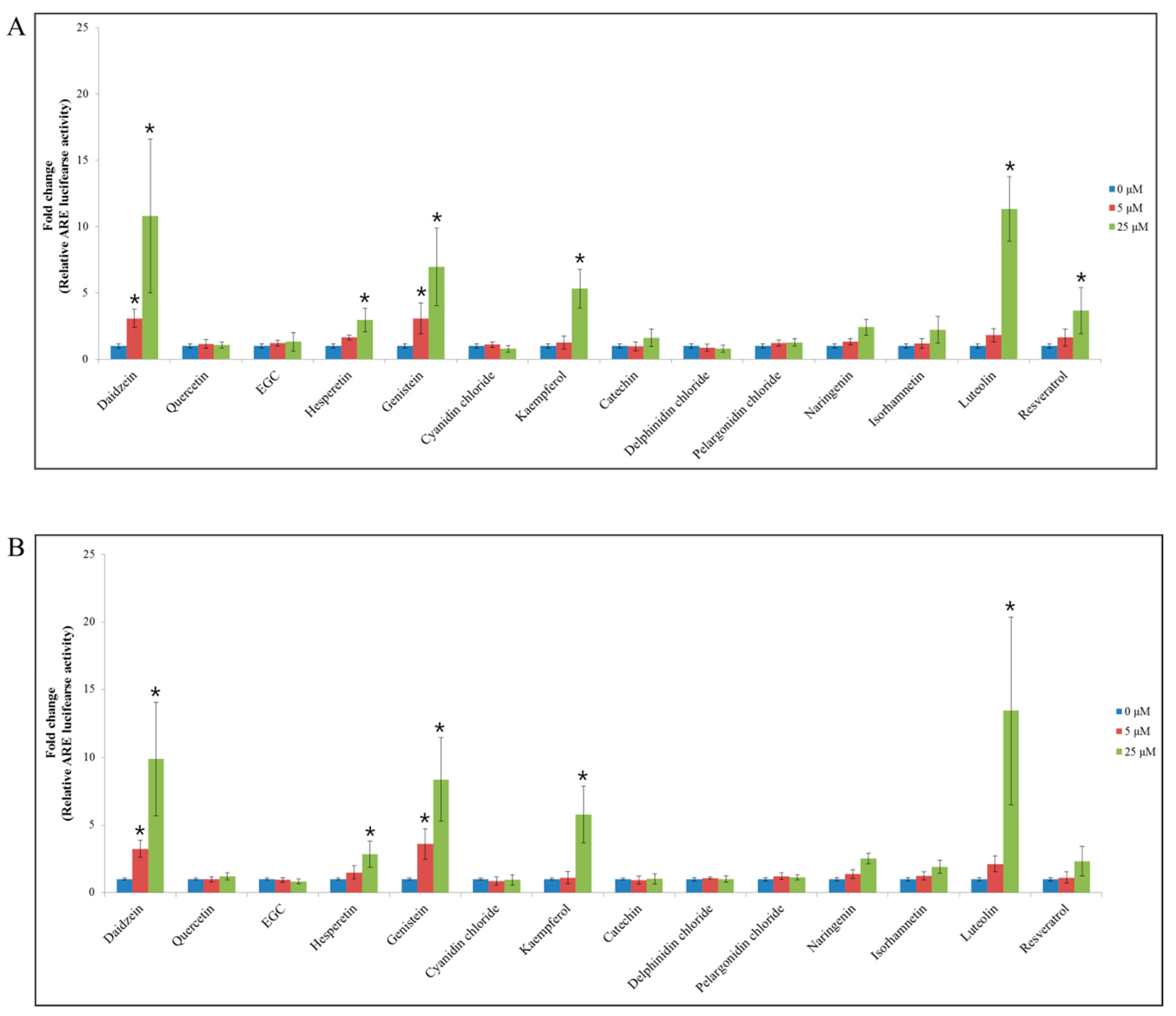
3.2. ARE-Luciferase Activity of Flavonoids
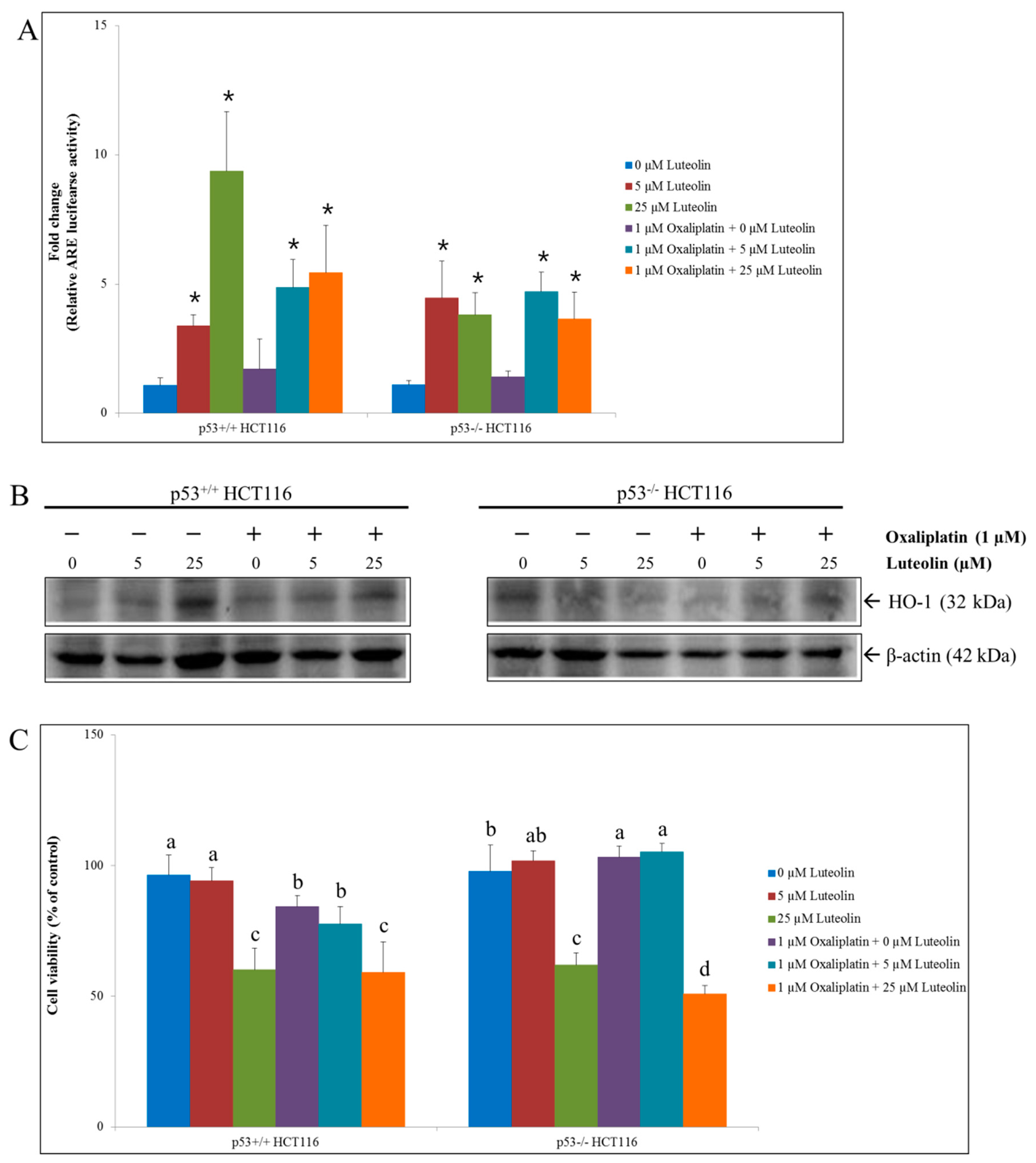
3.3. HCT116 Cell Viability by Luteolin and/or Oxaliplatin Treatment
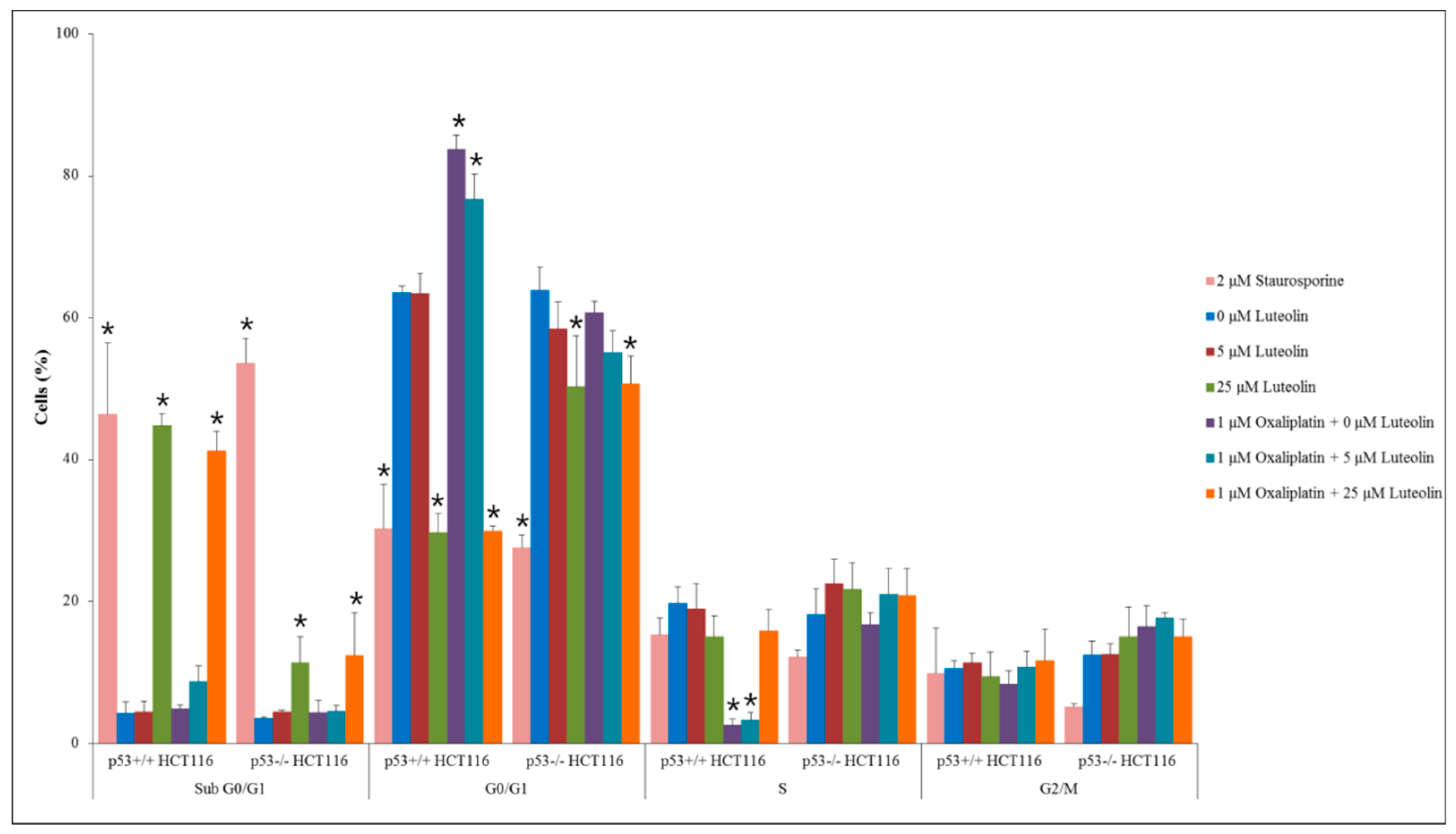
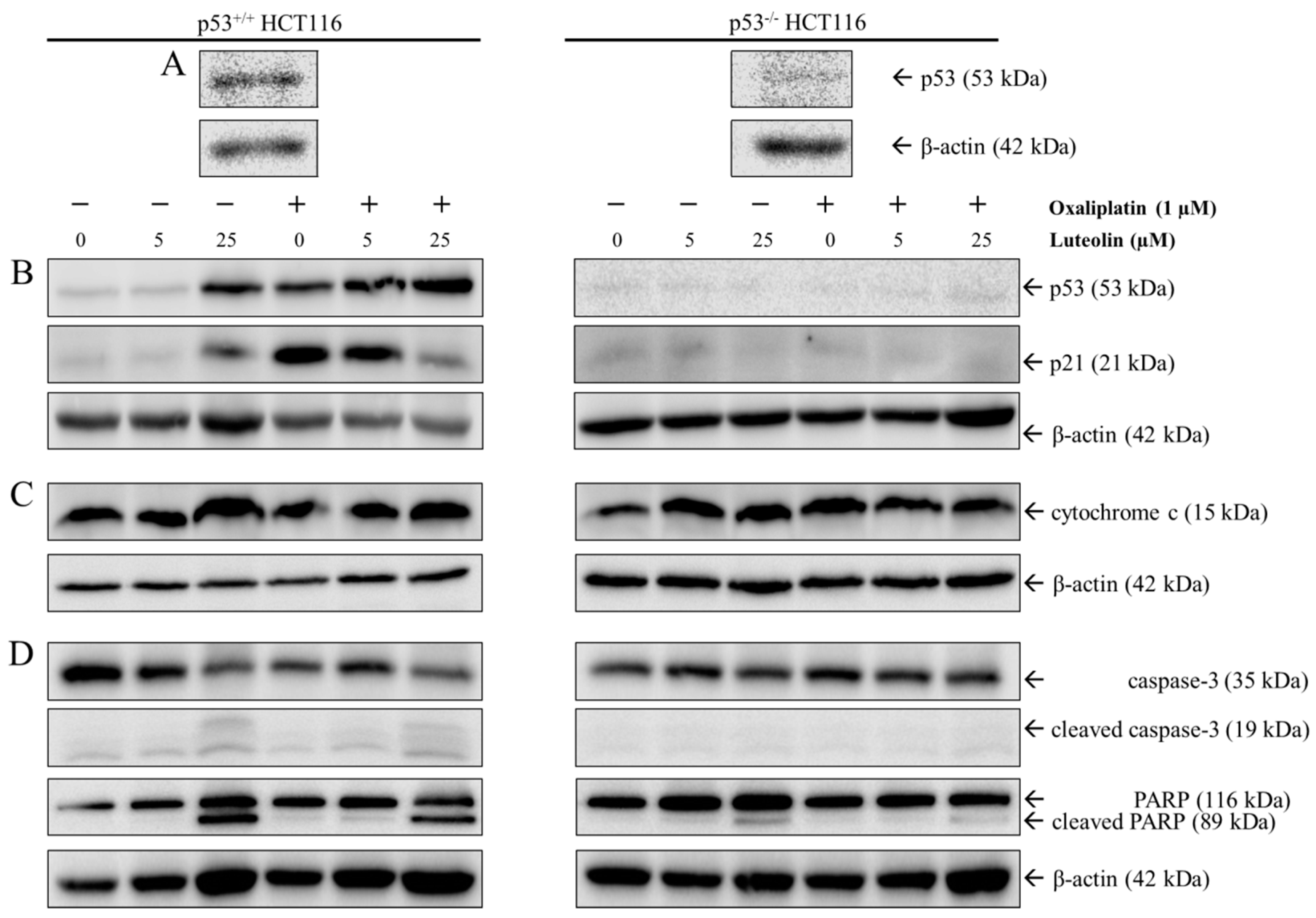
3.4. Mechanism of Cell Growth Inhibition by Luteolin and Oxaliplatin
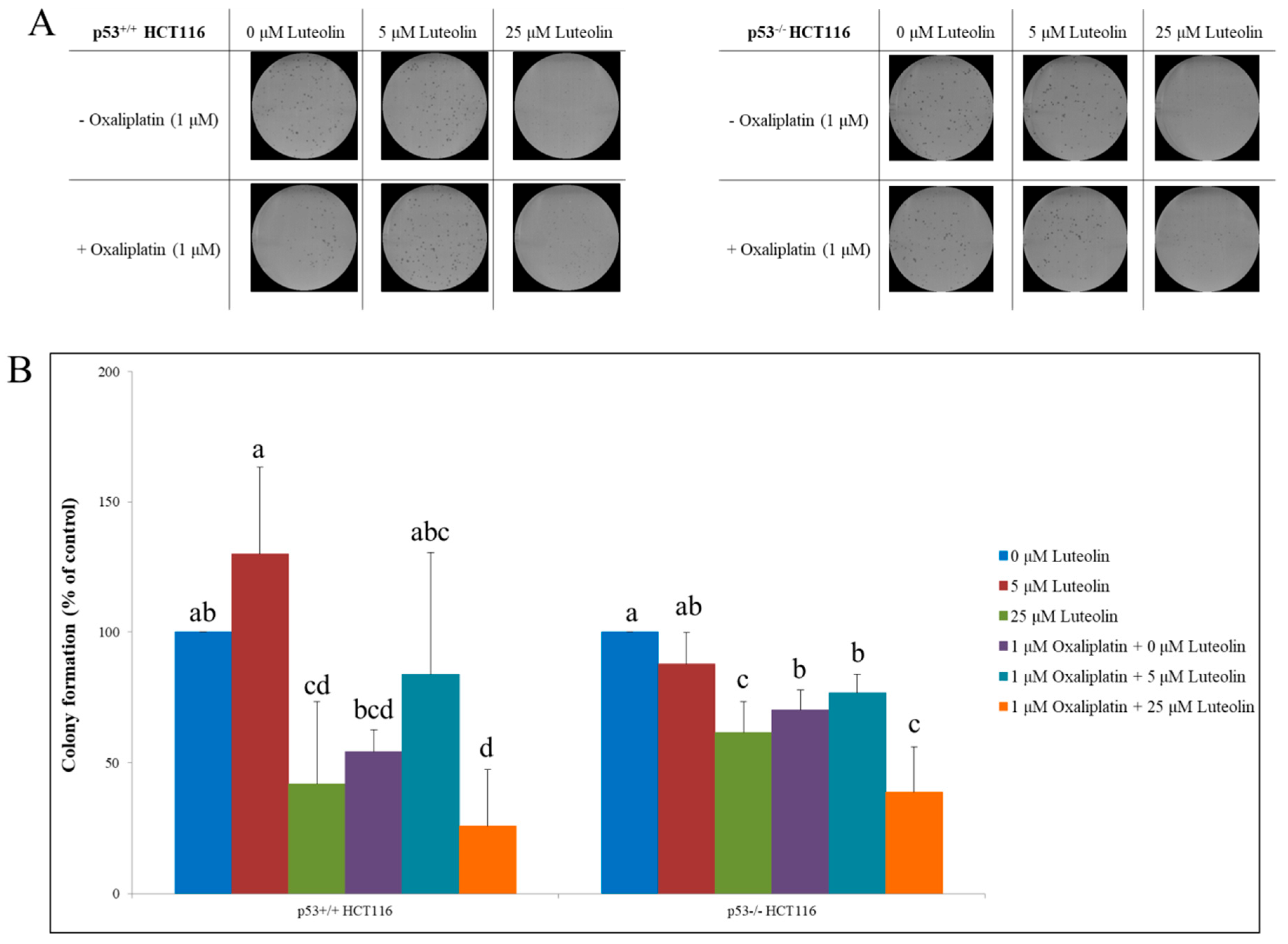
3.5. Colony-Forming Ability of HCT116 Cells Treated with Luteolin and/or Oxaliplatin
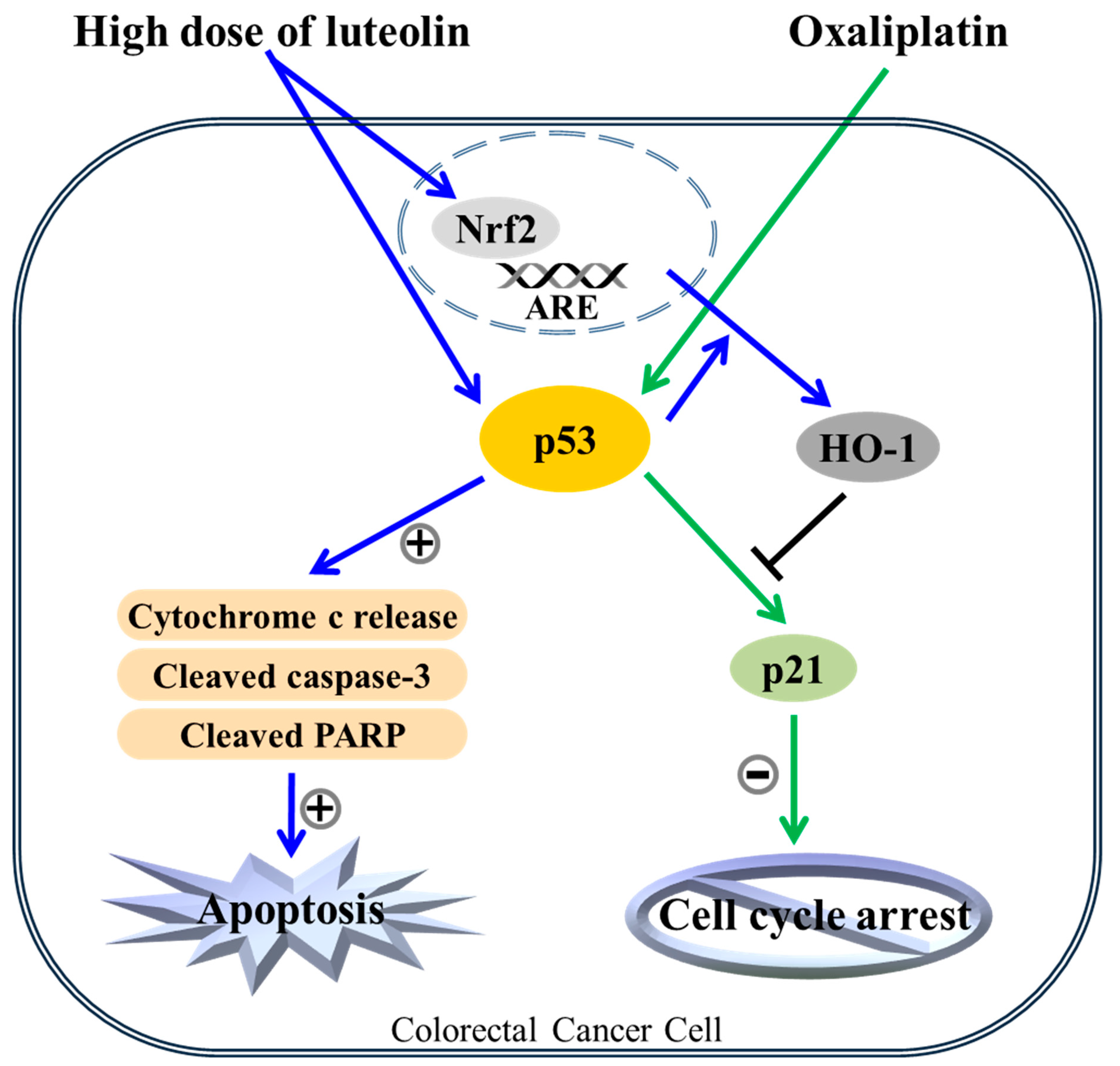
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Matés, J.M.; Segura, J.A.; Alonso, F.J.; Márquez, J. Intracellular redox status and oxidative stress: Implications for cell proliferation, apoptosis, and carcinogenesis. Arch. Toxicol. 2008, 82, 273–299. [Google Scholar] [CrossRef]
- Petrova, B.; Liu, K.; Tian, C.; Kitaoka, M.; Freinkman, E.; Yang, J.; Orr-Weaver, T.L. Dynamic redox balance directs the oocyte-to-embryo transition via developmentally controlled reactive cysteine changes. Proc. Natl. Acad. Sci. USA 2018, 115, E7978–E7986. [Google Scholar] [CrossRef] [PubMed]
- Riera, P.T. Redox Status. Encyclopedia of Exercise Medicine in Health and Disease; Mooren, F.C., Ed.; Springer: Berlin, Germany, 2016; pp. 67–83. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Mahalingaiah, P.K.S.; Singh, K.P. Chronic oxidative stress increases growth and tumorigenic potential of MCF-7 breast cancer cells. PLoS ONE 2014, 9, e87371. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14–21. [Google Scholar] [CrossRef]
- Keum, Y.S. Regulation of Nrf2-Mediated Phase II Detoxification and Anti-oxidant Genes. Biomol. Ther. 2012, 20, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Daliu, P.; Santini, A.; Novellino, E. From pharmaceuticals to nutraceuticals: Bridging disease prevention and management. Expert Rev. Clin. Pharmacol. 2018, 28, 1–7. [Google Scholar] [CrossRef]
- Santini, A.; Tenore, G.C.; Novellino, E. Nutraceuticals: A paradigm of proactive medicine. Eur. J. Pharm. Sci. 2017, 96, 53–61. [Google Scholar] [CrossRef]
- Durazzo, A. Extractable and Non-extractable polyphenols: An overview. Non-extractable Polyphenols and Carotenoids: Importance in Human Nutrition and Health; Food Chemistry, Function Analysis No. 5; Saura-Calixto, F., Pérez-Jiménez, J., Eds.; Royal Society of Chemistry: London, UK, 2018; pp. 37–45. [Google Scholar] [CrossRef]
- Chikara, S.; Nagaprashantha, L.D.; Singhal, J.; Horne, D.; Awasthi, S.; Singhal, S.S. Oxidative stress and dietary phytochemicals: Role in cancer chemoprevention and treatment. Cancer Lett. 2018, 413, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Stepanić, V.; Kujundžić, R.N.; Trošelj, K.G. Epigenome, cancer prevention and flavonoids and curcumin. Epigentics and Epigenomics, 1st ed.; Payne, C.J., Ed.; IntechOpen: London, UK, 2014; pp. 173–209. [Google Scholar]
- Haminiuk, C.W.I.; Maciel, G.M.; Plata-Oviedo, M.S.V.; Peralta, R.M. Phenolic compounds in fruits—An overview. Int. J. Food Sci. Technol. 2012, 47, 2023–2044. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, 1–15. [Google Scholar] [CrossRef]
- Li, W.; Pung, D.; Su, Z.Y.; Guo, Y.; Zhang, C.; Yang, A.Y.; Zheng, X.; Du, Z.Y.; Zhang, K.; Kong, A.N.T. Epigenetic reactivation of Nrf2 in prostate TRAMP C1 cells by curcumin analogue FN1. Chem. Res. Toxicol. 2016, 29, 694–703. [Google Scholar] [CrossRef]
- Saw, C.L.; Cintrón, M.; Wu, T.Y.; Guo, Y.; Huang, Y.; Jeong, W.S.; Kong, A.N.T. Pharmacodynamics of dietary phytochemical indoles I3C and DIM: Induction of Nrf2-mediated phase II drug metabolizing and antioxidant genes and synergism with isothiocyanates. Biopharm. Drug Dispos. 2011, 32, 289–300. [Google Scholar] [CrossRef]
- Shu, L.; Cheung, K.L.; Khor, T.O.; Chen, C.; Kong, A.N.T. Phytochemicals: Cancer chemoprevention and suppression of tumor onset and metastasis. Cancer Metastasis Rev. 2010, 29, 483–502. [Google Scholar] [CrossRef]
- Tan, A.C.; Konczak, I.; Sze, D.M.; Ramzan, I. Molecular pathways for cancer chemoprevention by dietary phytochemicals. Nutr. Cancer 2011, 63, 495–505. [Google Scholar] [CrossRef]
- Chen, P.; Zhang, J.Y.; Sha, B.B.; Ma, Y.E.; Hu, T.; Ma, Y.C.; Sun, H.; Shi, J.X.; Dong, Z.M.; Li, P. Luteolin inhibits cell proliferation and induces cell apoptosis via down-regulation of mitochondrial membrane potential in esophageal carcinoma cells EC1 and KYSE450. Oncotarget 2017, 8, 27471–27480. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yin, J.; Rankin, G.O.; Chen, Y.C. Kaempferol induces G2/M cell cycle arrest via checkpoint kinase 2 and promotes apoptosis via death receptors in human ovarian carcinoma A2780/CP70 cells. Molecules 2018, 23, 1095. [Google Scholar] [CrossRef]
- Ullah, M.F.; Ahmad, A.; Zubair, H.; Khan, H.Y.; Wang, Z.; Sarkar, F.H.; Hadi, S.M. Soy isoflavone genistein induces cell death in breast cancer cells through mobilization of endogenous copper ions and generation of reactive oxygen species. Mol. Nutr. Food Res. 2011, 55, 553–559. [Google Scholar] [CrossRef]
- Wang, F.; Shan, Y. Sulforaphane retards the growth of UM-UC-3 xenographs, induces apoptosis, and reduces survivin in athymic mice. Nutr. Res. 2012, 32, 374–380. [Google Scholar] [CrossRef]
- Yao, H.; Xu, W.; Shi, X.; Zhang, Z. Dietary flavonoids as cancer prevention agents. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2011, 29, 1–31. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, C.Z.; Du, G.J.; Qi, L.W.; Calway, T.; He, T.C.; Du, W.; Yuan, C.S. Genistein induces G2/M cell cycle arrest and apoptosis via ATM/p53-dependent pathway in human colon cancer cells. Int. J. Oncol. 2013, 43, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Gutheil, W.G.; Reed, G.; Ray, A.; Anant, S.; Dhar, A. Crocetin: An agent derived from saffron for prevention and therapy for cancer. Curr. Pharm. Biotechnol. 2012, 13, 173–179. [Google Scholar] [CrossRef]
- Santini, A.; Novellino, E. Nutraceuticals: Shedding light on the grey area between pharmaceuticals and food. Expert Rev. Clin. Pharmacol. 2018, 11, 545–547. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The dual roles of Nrf2 in cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Graham, J.; Mushin, M.; Kirkpatrick, P. Oxaliplatin. Nat. Rev. Drug Discov. 2004, 3, 11–12. [Google Scholar] [CrossRef]
- Marchand, L.L. Cancer preventive effects of flavonoids—A review. Biomed. Pharmacother. 2002, 56, 296–301. [Google Scholar] [CrossRef]
- Choudhury, R.; Roy, S.G.; Tsai, Y.S.; Tripathy, A.; Graves, L.M.; Wang, Z. The splicing activator DAZAP1 ingegrates splicing control into MEK/Erk-regulated cell proliferation and migration. Nat. Commun. 2014, 5, 3078. [Google Scholar] [CrossRef]
- Li, Y.H.; Niu, Y.B.; Sun, Y.; Zhang, F.; Liu, C.X.; Fan, L.; Mei, Q.B. Role of phytochemicals in colorectal cancer prevention. World J. Gastroenterol. 2015, 21, 9262–9272. [Google Scholar] [CrossRef]
- Birt, D.F.; Hendrich, S.; Wang, W. Dietary agents in cancer prevention: Flavonoids and isoflavonoids. Pharmacol. Ther. 2001, 90, 157–177. [Google Scholar] [CrossRef]
- Galati, G.; O’Brien, P.J. Potential toxicity of flavonoids and other dietary phenolics: Significance for their chemopreventive and anticancer properties. Free Radic. Biol. Med. 2004, 37, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Galati, G.; Teng, S.; Moridani, M.Y.; Chan, T.S.; O’Brien, P.J. Cancer chemoprevention and apoptosis mechanisms induced by dietary polyphenolics. Drug Metab. Drug Interact. 2000, 17, 311–349. [Google Scholar] [CrossRef]
- Liu, C.M.; Ma, J.Q.; Xie, W.R.; Liu, S.S.; Feng, Z.J.; Zheng, G.H.; Wang, A.M. Quercetin protects mouse liver against nickel-induced DNA methylation and inflammation associated with the Nrf2/HO-1 and p38/STAT1/NF-kB pathway. Food Chem. Toxical. 2015, 82, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Qiao, Z.; Wang, H.; Zhu, L.; Zhang, L. Flavonoids: Promising anticancer agents. Med. Res. Rev. 2003, 23, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Son, T.G.; Camandola, S.; Mattson, M.P. Hormetic dietary phytochemicals. Neuromol. Med. 2008, 10, 236–246. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar]
- Vargas, A.J.; Burd, R. Hormesis and synergy: Pathways and mechanisms of quercetin in cancer prevention and management. Nutr. Rev. 2010, 68, 418–428. [Google Scholar] [CrossRef]
- Choi, B.H.; Kwak, M.K. Shadows of Nrf2 in cancer: Resistance to chemotherapy. Curr. Opin. Toxicol. 2016, 1, 20–28. [Google Scholar] [CrossRef]
- Ryoo, I.G.; Lee, S.H.; Kwak, M.K. Redox modulating Nrf2: A potential mediator of cancer stem cell resistance. Oxid. Med. Cell Longev. 2016, 2016, 2428153. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.J.; Jaramillo, M.C.; Zhang, Z.B.; Zheng, Y.X.; Yao, M.; Zhang, D.D.; Yi, X.F. Nrf2 induces cisplatin resistance through activation of autophage in ovarian carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 1502–1513. [Google Scholar] [PubMed]
- Wang, X.J.; Li, Y.; Luo, L.; Wnag, H.; Chi, Z.; Xin, A.; Li, X.; Tang, X. Oxaliplatin activates the Keap1/Nrf2 antioxidant system conferring protection against the cytotoxicity of anticancer drugs. Free Radic. Biol. Med. 2014, 70, 68–77. [Google Scholar] [CrossRef]
- Tang, W.; Stearns, R.A. Heterotropic cooperativity of cytochrome P450 3A4 and potential—Drug interactions. Curr. Drug Metab. 2001, 2, 185–198. [Google Scholar] [CrossRef]
- William-Faltaos, S.; Rouillard, D.; Lechat, P.; Bastian, G. Cell cycle arrest by oxaliplatin on cancer cells. Fundam. Clin. Pharmarcol. 2007, 21, 165–172. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Hyun, Y.J.; Park, J.E.; Shilnikova, K.; Zhen, A.X.; Kang, H.K.; Koh, Y.S.; Jeong, Y.J.; et al. Luteolin induces apoptotic cell death via antioxidant activity in human colon cancer cells. Int. J. Oncol. 2017, 51, 1169–1178. [Google Scholar] [CrossRef]
- Zuo, Q.; Wu, R.; Xiao, X.; Yang, C.; Yang, Y.; Wang, C.; Lin, L.; Kong, A.N.T. The dietary flavone luteolin epigenetically activates the Nrf2 pathway and blocks cell transformation in human colorectal cancer HCT116 cells. J. Cell Biochem. 2018, 119, 9573–9582. [Google Scholar] [CrossRef]
- Busserrolles, J.; Megías, J.; Terencio, M.C.; Alcaraz, M.J. Heme oxygenase-1 inhibits apoptosis in Caco-2 cells with activatin of Akt pathway. Int. J. Biochem. Cell Biol. 2006, 38, 1510–1517. [Google Scholar] [CrossRef]
- Chen, W.; Jiang, T.; Wang, H.; Tao, S.; Lau, A.; Fang, D.; Zhang, D.D. Does Nrf2 contribute to p53-mediated control of cell survival and death? Antioxid. Redox. Signal. 2012, 17, 1670–1675. [Google Scholar] [CrossRef]
- Ji, L.; Wei, Y.; Jiang, T.; Wang, S. Correlation of Nrf2, NQO1, MRP1, cmyc and p53 in colorectal cancer and their relationships to clinicopathologic features and survival. Int. J. Clin. Exp. Pathol. 2014, 7, 1124–1131. [Google Scholar]
- Rotblat, B.; Melino, G.; Knight, R.A. Nrf2 and p53: Januses in cancer? Oncotarget 2012, 3, 1272–1283. [Google Scholar] [CrossRef]
- Agarwal, C.; Singh, R.P.; Dhanalakshmi, S.; Tyagi, A.K.; Tecklenburg, M.; Sclafani, R.A.; Agarwal, R. Silibinin upregulates the expression of cyclin-dependent kinase inhibitors and causes cell cycle arrest and apoptosis in human colon carcinoma HT-29 cells. Oncogene 2003, 22, 8271–8282. [Google Scholar] [CrossRef] [Green Version]
- Pucci, B.; Kasten, M.; Giordano, A. Cell cycle and apoptosis. Neoplasia 2000, 2, 291–299. [Google Scholar] [CrossRef]
- Ahmad, N.; Adhami, V.M.; Afaq, F.; Feyes, D.K.; Mukhtar, H. Resveratrol causes WAF-1/p21-mediated G1-phase arrest of cell cycle and induction of apoptosis in human epidermoid carcinoma A431 cells. Clin. Cancer Res. 2001, 7, 1466–1473. [Google Scholar]
- Shen, G.; Xu, C.; Chen, C.; Hebbar, V.; Kong, A.N.T. p53-independent G1 cell cycle arrest of human colon carcinoma cells HT-29 by sulforaphane is associated with induction of p21CIP1 and inhibition of expression of cyclin D1. Cancer Chemother. Pharmacol. 2006, 57, 317–327. [Google Scholar] [CrossRef]
- Waldman, T.; Kinzler, K.W.; Vogelstein, B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995, 55, 5187–5190. [Google Scholar]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell. 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Slattery, M.L.; Sweeney, C.; Herrick, J.; Wolff, R.K.; Albertsen, H. APC mutations and other genetic and epigenetic changes in colon cancer. Mol. Cancer Res. 2007, 5, 165–170. [Google Scholar] [CrossRef]
- Martinez-Balibrea, E.; Martínez-Cardús, A.; Ginés, A.; Ruiz de Porras, V.; Moutinho, C.; Layos, L.; Manzano, J.L.; Bugés, C.; Bystrup, S.; Esteller, M.; et al. Tumor-related molecular mechanisms of oxaliplatin resistance. Mol. Cancer Ther. 2015, 14, 1767–1776. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, C.H.; Moon, N.; Oh, J.; Kim, J.-S. Luteolin Shifts Oxaliplatin-Induced Cell Cycle Arrest at G0/G1 to Apoptosis in HCT116 Human Colorectal Carcinoma Cells. Nutrients 2019, 11, 770. https://doi.org/10.3390/nu11040770
Jang CH, Moon N, Oh J, Kim J-S. Luteolin Shifts Oxaliplatin-Induced Cell Cycle Arrest at G0/G1 to Apoptosis in HCT116 Human Colorectal Carcinoma Cells. Nutrients. 2019; 11(4):770. https://doi.org/10.3390/nu11040770
Chicago/Turabian StyleJang, Chan Ho, Nayoung Moon, Jisun Oh, and Jong-Sang Kim. 2019. "Luteolin Shifts Oxaliplatin-Induced Cell Cycle Arrest at G0/G1 to Apoptosis in HCT116 Human Colorectal Carcinoma Cells" Nutrients 11, no. 4: 770. https://doi.org/10.3390/nu11040770
APA StyleJang, C. H., Moon, N., Oh, J., & Kim, J. -S. (2019). Luteolin Shifts Oxaliplatin-Induced Cell Cycle Arrest at G0/G1 to Apoptosis in HCT116 Human Colorectal Carcinoma Cells. Nutrients, 11(4), 770. https://doi.org/10.3390/nu11040770