Slowing Down Ageing: The Role of Nutrients and Microbiota in Modulation of the Epigenome


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
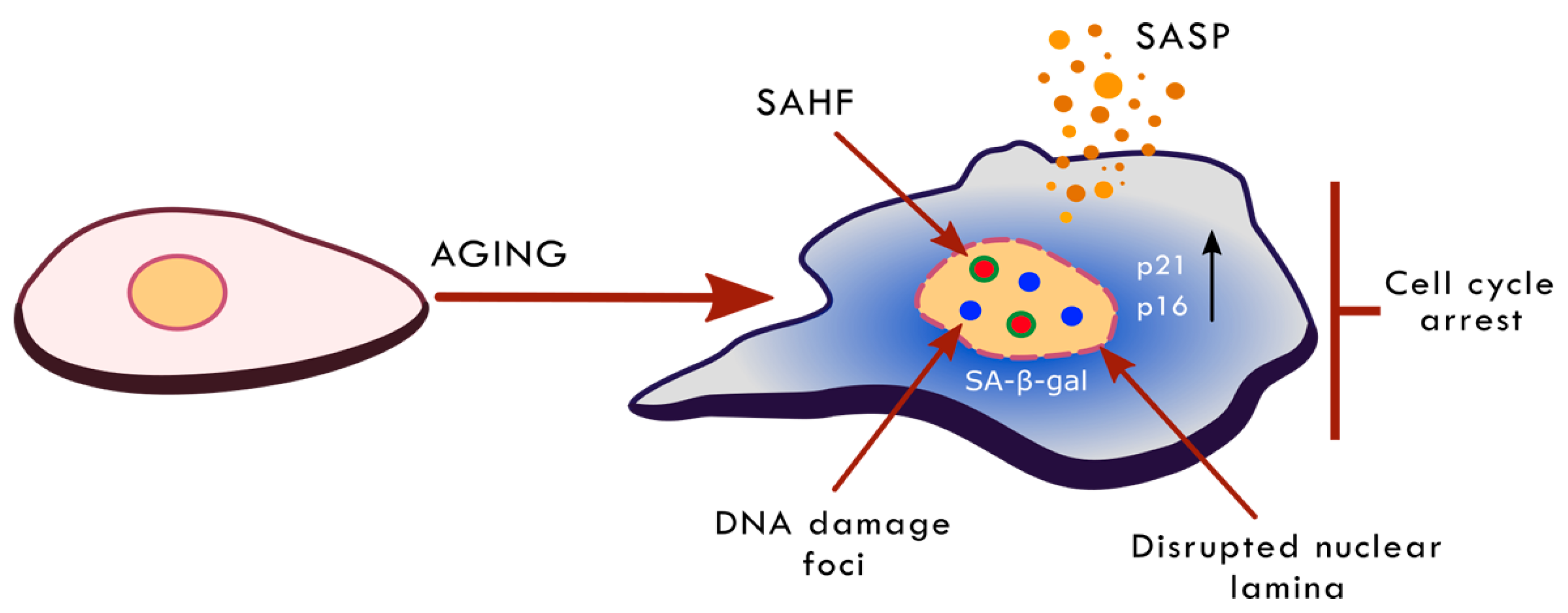
2. Ageing, Cellular Senescence and Anti-Ageing Interventions
3. Epigenetics and Nuclear Landscape; Ageing-Associated Changes of Chromatin Structure
3.1. DNA Methylation
3.2. Posttranslational Modification of Histones
3.3. Sirtuins
3.4. Non-Coding RNA
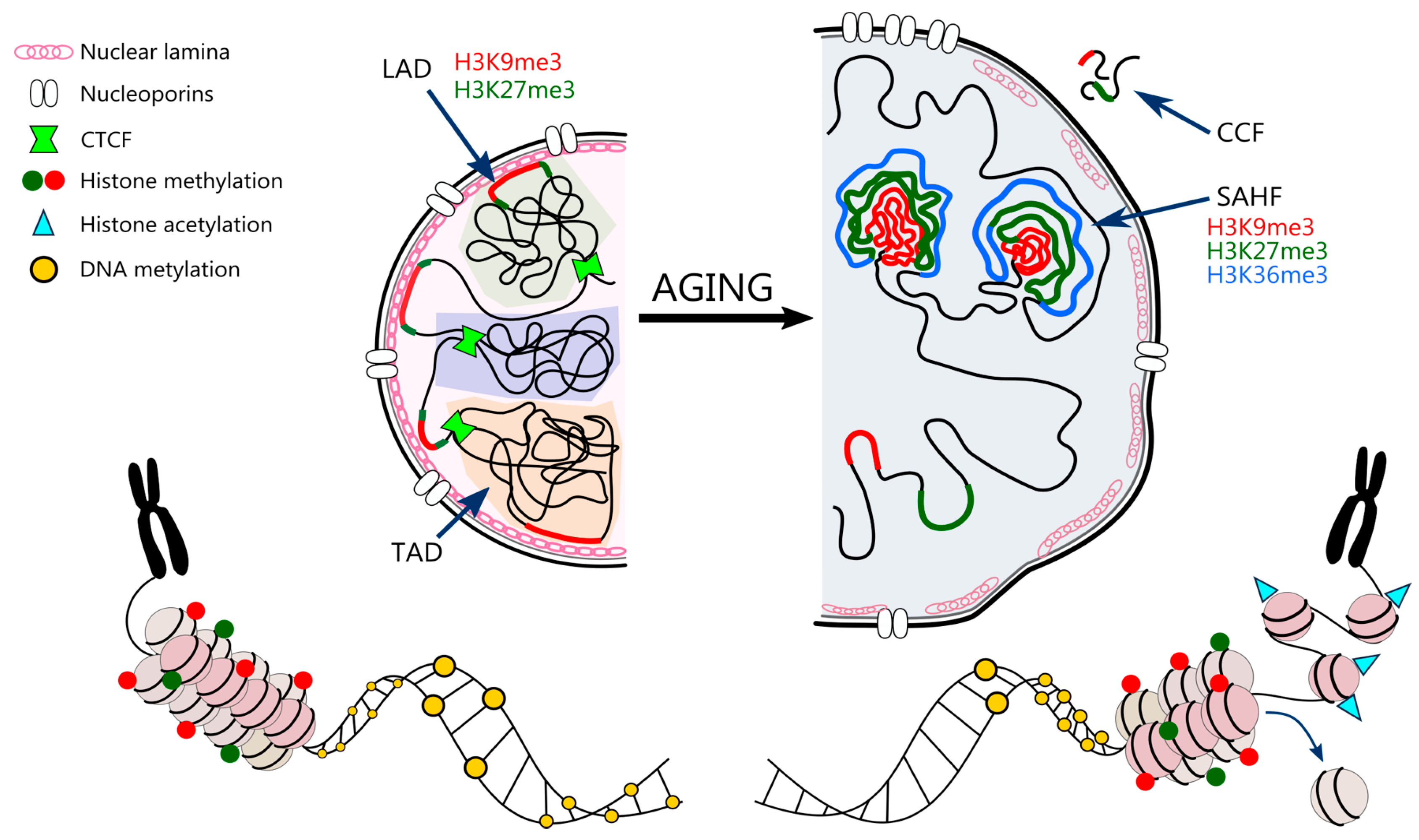
3.5. Nuclear Architecture
3.6. Nuclear Landscape in Cell Senescence and Ageing
3.7. Epigenetics in Age-Related Diseases
4. Impact of the Diet on Chromatin Structure and Gene Expression
4.1. Impact of Nutrients on DNA Methylation
4.2. Impact of Nutrients on Histones Modifications
4.3. Impact of Nutrients on miRNA Regulation
5. Impact of the Exercise on Gene Expression
6. Autophagy as a Target of Nutritionally-Altered Epigenetic Pattern
7. The Microbiota Changes in the Elderly
7.1. Gut Microbiota
7.2. Other than Gut Microbiota
8. Impact of the Diet on Microbiota
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Naylor, R.M.; Baker, D.J.; Van Deursen, J.M. Senescent cells: A novel therapeutic target for aging and age-related diseases. Clin. Pharmacol. Ther. 2013, 93, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.G.; Gibbs, M.E. Mechanisms and the clinical relevance of complex drug-drug interactions. Clin. Pharmacol. Adv. Appl. 2018, 10, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Bielak-Zmijewska, A.; Grabowska, W.; Ciolko, A.; Bojko, A.; Mosieniak, G.; Bijoch, Ł.; Sikora, E. The Role of Curcumin in the Modulation of Ageing. Int. J. Mol. Sci. 2019, 20, 1239. [Google Scholar] [CrossRef] [PubMed]
- Most, J.; Tosti, V.; Redman, L.M.; Fontana, L. Calorie restriction in humans: An update. Ageing Res. Rev. 2017, 39, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Fahy, G.M.; West, M.D.; Coles, L.S.; Harris, S.B. The Future of Aging: Pathways to Human Life Extension; Springer: New York, NY, USA, 2010. [Google Scholar]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [Green Version]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-ruiz, C.; Von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef]
- Jeyapalan, J.C.; Sedivy, J.M. Cellular senescence and organismal aging. Mech. Ageing Dev. 2008, 129, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16 Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Yu, R.; Dang, W. Chromatin architectural changes during cellular senescence and aging. Genes 2018, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.L.; Vastolo, V.; Ciccarelli, M.; Albano, L.; Macchia, P.E.; Ungaro, P. Dietary polyphenols and chromatin remodeling. Crit. Rev. Food Sci. Nutr. 2015, 57, 2589–2599. [Google Scholar] [CrossRef] [PubMed]
- Salazar, N.; Valdés-Varela, L.; González, S.; Gueimonde, M.; de los Reyes-Gavilán, C.G. Nutrition and the gut microbiome in the elderly. Gut. Microbes 2017, 8, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Hoile, S.P.; Grenfell, L.; Burdge, G.C. DNA methylation, ageing and the influence of early life nutrition. Proce. Nutr. Soc. 2014, 73, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, C.K.B. Functional foods, herbs and nutraceuticals: Towards biochemical mechanisms of healthy aging. Biogerontology 2004, 5, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S. Microbiota and aging. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 26–30. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, P.W.; Jeffery, I.B. Gut microbiota and aging. Science 2015, 350, 1214–1215. [Google Scholar] [CrossRef] [PubMed]
- Gorenne, I.; Kavurma, M.; Scott, S.; Bennett, M. Vascular smooth muscle cell senescence in atherosc. Cardiovasc. Res. 2006, 72, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Cell Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef] [PubMed]
- McShea, A.; Harris, P.L.R.; Webster, K.R.; Wahl, A.F.; Smith, M.A. Abnormal Expression Of the Cell Cycle Regulators P16 and Cdk4 In Alzheimers-Disease. Am. J. Pathol. 1997, 150, 1933–1939. [Google Scholar] [PubMed]
- Cohen, G. The pathobiology of Parkinson’s disease: Biochemical aspects of dopamine neuron senescence. J. Neural Transm. Suppl. 1983, 19, 89–103. [Google Scholar] [PubMed]
- Price, J.S.; Waters, J.G.; Darrah, C.; Pennington, C.; Edwards, D.R.; Donell, S.T.; Clark, I.M. The role of chondrocyte senescence in osteoarthritis. Aging Cell 2002, 1, 57–65. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Evans, G.; Tonne, J.M.; Verzosa, G.C.; Stout, M.B.; Mazula, D.L.; Palmer, A.K.; Baker, D.J.; Jensen, M.D.; et al. Exercise prevents diet-induced cellular senescence in adipose tissue. Diabetes 2016, 65, 1606–1615. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, O.; Dumont, P.; Dierick, J.F.; Pascal, T.; Frippiat, C.; Chainiaux, F.; Sluse, F.; Eliaers, F.; Remacle, J. Stress-Induced Premature Senescence: Essence of Life, Evolution, Stress, and Aging. Ann. N. Y. Acad. Sci. 2010, 908, 85–98. [Google Scholar] [CrossRef]
- Fridlyanskaya, I.; Alekseenko, L.; Nikolsky, N. Senescence as a general cellular response to stress: A mini-review. Exp. Gerontol. 2015, 72, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Bielak-Zmijewska, A.; Mosieniak, G.; Sikora, E. Is DNA damage indispensable for stress-induced senescence? Mech. Ageing Dev. 2018, 170, 13–21. [Google Scholar] [CrossRef]
- Rebelo-Marques, A.; Lages, A.D.S.; Andrade, R.; Ribeiro, C.F.; Mota-Pinto, A.; Carrilho, F.; Espregueira-Mendes, J. Aging hallmarks: The benefits of physical exercise. Front. Endocrinol. 2018, 9, 258. [Google Scholar] [CrossRef]
- Sikora, E.; Bielak Zmijewska, A.; Mosieniak, G. What is and what is not cell senescence. Postepy Biochem. 2018, 64, 110–118. [Google Scholar] [CrossRef]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [Green Version]
- D’Adda Di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.J.; Chiang, Y.J.; Hathcock, K.S.; Horikawa, I.; Sedelnikova, O.A.; Hodes, R.J.; Bonner, W.M. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenet. Chromatin 2008, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, W.; Kucharewicz, K.; Wnuk, M.; Lewinska, A.; Suszek, M.; Przybylska, D.; Mosieniak, G.; Sikora, E.; Bielak-Zmijewska, A. Curcumin induces senescence of primary human cells building the vasculature in a DNA damage and ATM-independent manner. Age 2015, 37, 9744. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, P.; Howell, P.R.; Anderson, R.M. Aging and Caloric Restriction Research: A Biological Perspective With Translational Potential. EBioMedicine 2017, 21, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingram, D.K.; de Cabo, R. Calorie restriction in rodents: Caveats to consider. Ageing Res. Rev. 2017, 39, 15–28. [Google Scholar] [CrossRef]
- Weindruch, R. The retardation of aging by caloric restriction: Studies in rodents and primates. Toxicol. Pathol. 1996, 24, 742–745. [Google Scholar] [CrossRef]
- Lane, M.A.; Baer, D.J.; Rumpler, W.V.; Weindruch, R.; Ingram, D.K.; Tilmont, E.M.; Cutler, R.G.; Roth, G.S. Calorie restriction lowers body temperature in rhesus monkeys, consistent with a postulated anti-aging mechanism in rodents. Proc. Natl. Acad. Sci. USA 1996, 93, 4159–4164. [Google Scholar] [CrossRef]
- Masoro, E.J. Overview of caloric restriction and ageing. Mech. Ageing Dev. 2005, 126, 913–922. [Google Scholar] [CrossRef]
- Hanjani, N.; Vafa, M. Protein restriction, epigenetic diet, intermittent fasting as new approaches for preventing age-associated diseases. Int. J. Prev. Med. 2018, 9, 58. [Google Scholar]
- Mattson, M.P.; Longo, V.D.; Harvie, M. Impact of intermittent fasting on health and disease processes. Ageing Res. Rev. 2017, 39, 46–58. [Google Scholar] [CrossRef]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Beasley, T.M.; Kemnitz, J.W.; Johnson, S.C.; Weindruch, R.; Anderson, R.M. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 2014, 5, 3557. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Antebi, A.; Bartke, A.; Barzilai, N.; Brown-Borg, H.M.; Caruso, C.; Curiel, T.J.; de Cabo, R.; Franceschi, C.; Gems, D.; et al. Interventions to slow aging in humans: Are we ready? Aging Cell 2015, 14, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Cava, E.; Fontana, L. Will calorie restriction work in humans? Aging 2013, 5, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wątroba, M.; Szukiewicz, D. The role of sirtuins in aging and age-related diseases. Adv. Med. Sci. 2016, 61, 52–62. [Google Scholar] [CrossRef]
- Bielak-Zmijewska, A.; Wnuk, M.; Przybylska, D.; Grabowska, W.; Lewinska, A.; Alster, O.; Korwek, Z.; Cmoch, A.; Myszka, A.; Pikula, S.; et al. A comparison of replicative senescence and doxorubicin-induced premature senescence of vascular smooth muscle cells isolated from human aorta. Biogerontology 2014, 15, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mohan, N.; Upadhyay, A.D.; Singh, A.P.; Sahu, V.; Dwivedi, S.; Dey, A.B.; Dey, S. Identification of serum sirtuins as novel noninvasive protein markers for frailty. Aging Cell 2014, 13, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z.X. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.; Witkin, K.L.; Cohen-Fix, O. Sizing up the nucleus: Nuclear shape, size and nuclear-envelope assembly. J. Cell Sci. 2009, 122, 1477–1486. [Google Scholar] [CrossRef]
- Wilson, K.L.; Dawson, S.C. Functional evolution of nuclear structure. J. Cell Biol. 2011, 195, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Mckay, J.A.; Mathers, J.C. Diet induced epigenetic changes and their implications for health. Acta Physiol. 2011, 202, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Csoka, A.B.; Szyf, M. Epigenetic side-effects of common pharmaceuticals: A potential new field in medicine and pharmacology. Med. Hypotheses 2009, 73, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, E.; Dimauro, I.; Mercatelli, N.; Wang, G.; Pitsiladis, Y.; Di Luigi, L. Physical activity in the prevention of human diseases: Role of epigenetic modifications. BMC Genom. 2017, 18, 802. [Google Scholar] [CrossRef] [PubMed]
- Szarc vel Szic, K.; Ndlovu, M.N.; Haegeman, G.; Vanden Berghe, W. Nature or nurture: Let food be your epigenetic medicine in chronic inflammatory disorders. Biochem. Pharm. 2010, 15, 1816–1832. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wade, P.A. Crosstalk between the microbiome and epigenome: Messages from bugs. J. Biochem. 2018, 163, 105–112. [Google Scholar] [CrossRef]
- Kim, M.; Costello, J. DNA methylation: An epigenetic mark of cellular memory. Exp. Mol. Med. 2017, 49, e322. [Google Scholar] [CrossRef]
- Bird, A. The essentials of DNA methylation. Cell 1992, 70, 5–8. [Google Scholar] [CrossRef]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Paabo, S.; Rebhan, M.; Schubeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Issa, J.P.J.; Ottaviano, Y.L.; Celano, P.; Hamilton, S.R.; Davidson, N.E.; Baylin, S.B. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat. Genet. 1994, 7, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Tazi, M.; Caution, K.; Ahmed, A.; Kanneganti, A.; Assani, K.; Kopp, B.; Marsh, C.; Dakhlallah, D.; Amer, A.O. Aging is assoiated with hypermethylation of autophagy genes in macrophages. Epigenetics 2016, 11, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Wilson, V.L.; Jones, P.A. DNA methylation decreases in aging but not in immortal cells. Science 1983, 220, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Li, N.; Ferreira, H.J.; Moran, S.; Pisano, D.G.; Gomez, A.; Diez, J.; Sanchez-Mut, J.V.; Setien, F.; Carmona, F.J.; et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. USA 2012, 109, 10522–10527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, B.; Lyko, F. DNA methyltransferase inhibitors: Old and new drugs for an epigenetic cancer therapy. Trends Pharm. Sci. 2004, 25, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Jurkowska, R.Z. New concepts in DNA methylation. Trends Biochem. Sci. 2014, 39, 310–318. [Google Scholar] [CrossRef]
- Ooi, S.K.T.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Hermann, A.; Schmitt, S.; Jeltsch, A. The human Dnmt2 has residual DNA-(Cytosine-C5) methyltransferase activity. J. Biol. Chem. 2003, 278, 31717–31721. [Google Scholar] [CrossRef]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2016, 14, 1108–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewinska, A.; Adamczyk-Grochala, J.; Kwasniewicz, E.; Deregowska, A.; Semik, E.; Zabek, T.; Wnuk, M. Reduced levels of methyltransferase DNMT2 sensitize human fibroblasts to oxidative stress and DNA damage that is accompanied by changes in proliferation-related miRNA expression. Redox Biol. 2018, 14, 20–34. [Google Scholar] [CrossRef]
- Lewinska, A.; Adamczyk-Grochala, J.; Kwasniewicz, E.; Wnuk, M. Downregulation of methyltransferase Dnmt2 results in condition-dependent telomere shortening and senescence or apoptosis in mouse fibroblasts. J. Cell Physiol. 2017, 232, 3714–3726. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Globisch, D.; Münzel, M.; Müller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Brückl, T.; Biel, M.; Carell, T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 2010, 5, e15367. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Jin, S.G.; Wu, X.; Li, A.X.; Pfeifer, G.P. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011, 39, 5015–5024. [Google Scholar] [CrossRef]
- Chen, H.; Dzitoyeva, S.; Manev, H. Effect of aging on 5-hydroxymethylcytosine in the mouse hippocampus. Restor. Neurol. Neurosci. 2012, 30, 237–245. [Google Scholar] [Green Version]
- Fasolino, M.; Liu, S.; Wang, Y.; Zhou, Z. Distinct cellular and molecular environments support aging-related DNA methylation changes in the substantia nigra. Epigenomics 2017, 9, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.H.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.; JenuweiN, T.; Peters, A. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003, 15, 1192–1200. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Smith, M.M. Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol. 2015, 7, a019364. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Garcia, B.A. Comprehensive catalog of currently documented histone modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef] [PubMed]
- Sterner, D.E.; Berger, S.L. Acetylation of Histones and Transcription-Related Factors. Microbiol. Mol. Biol. Rev. 2003, 64, 435–459. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raurell-Vila, H.; Bosch-Presegue, L.; Gonzalez, J.; Kane-Goldsmith, N.; Casal, C.; Brown, J.; Marazuela-Duque, A.; Singh, P.B.; Serrano, L.; Vaquero, A. An HP1 isoform-specific feedback mechanism regulates Suv39h1 activity under stress conditions. Epigenetics 2017, 12, 166–175. [Google Scholar] [CrossRef]
- Gillette, T.G.; Hill, J.A. Readers, writers, and erasers: Chromatin as the whiteboard of heart disease. Circ. Res. 2015, 116, 1245–1253. [Google Scholar] [CrossRef]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Zane, L.; Sharma, V.; Misteli, T. Common features of chromatin in aging and cancer: Cause or coincidence? Trends Cell Biol. 2014, 24, 686–694. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Sengupta, K.; Li, C.; Kim, H.S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; et al. Impaired DNA Damage Response, Genome Instability, and Tumorigenesis in SIRT1 Mutant Mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Poulose, N.; Raju, R. Sirtuin regulation in aging and injury. Biochim. Biophys. Acta 2015, 1852, 2442–2455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaquero, A.; Scher, M.; Erdjument-Bromage, H.; Tempst, P.; Serrano, L.; Reinberg, D. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature 2007, 450, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Presegué, L.; Vaquero, A. The dual role of sirtuins in cancer. Genes Cancer 2011, 2, 648–662. [Google Scholar] [CrossRef] [PubMed]
- Bouras, T.; Fu, M.; Sauve, A.A.; Wang, F.; Quong, A.A.; Perkins, N.D.; Hay, R.T.; Gu, W.; Pestell, R.G. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem. 2005, 280, 10264–10276. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef]
- Cardus, A.; Uryga, A.K.; Walters, G.; Erusalimsky, J.D. SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence. Cardiovasc. Res. 2013, 97, 571–579. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.; Slack, F. A developmental timing microRNA and its target regulate life span in C. elegans. Science 2005, 310, 1954–1957. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Drinkwater, R.D.; Blake, T.J.; Morley, A.A.; Turner, D.R. Human lymphocytes aged in vivo have reduced levels of methylation in transcriptionally active and inactive DNA. Mutat. Res. 1989, 219, 29–37. [Google Scholar] [CrossRef]
- Singhal, R.P.; Mays-Hoopes, L.L.; Eichhorn, G.L. DNA methylation in aging of mice. Mech. Ageing Dev. 1987, 41, 199–210. [Google Scholar] [CrossRef]
- Bollati, V.; Schwartz, J.; Wright, R.; Litonjua, A.; Tarantini, L.; Suh, H.; Sparrow, D.; Vokonas, P.; Baccarelli, A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech. Ageing Dev. 2009, 130, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Casillas, M.A.; Lopatina, N.; Andrews, L.G.; Tollefsbol, T.O. Transcriptional control of the DNA methyltransferases is altered in aging and neoplastically-transformed human fibroblasts. Mol. Cell Biochem. 2003, 252, 33–43. [Google Scholar] [CrossRef]
- Ahuja, N.; Issa, J.P.J. Aging, methylation and cancer. Histol. Histopathol. 2000, 15, 835–842. [Google Scholar]
- Hu, Z.; Chen, K.; Xia, Z.; Chavez, M.; Pal, S.; Seol, J.H.; . Chen, C.C.; Li, W.; Tyler, J.K. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014, 28, 396–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, R.J.; Karlseder, J. The great unravelling: Chromatin as a modulator of the aging process. Trends Biochem. Sci. 2012, 37, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; . Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Sekeri-Pataryas, K.E. Histone variants of H2A and H3 families are regulated during in vitro aging in the same manner as during differentiation. Exp. Gerontol. 1999, 34, 741–754. [Google Scholar] [CrossRef]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.Y.; Ma, Z.; et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 2017, 8, 4995. [Google Scholar] [CrossRef]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. γH2AX as a molecular marker of aging and disease. Epigenetics 2010, 5, 129–136. [Google Scholar] [CrossRef]
- Takahashi, A.; Imai, Y.; Yamakoshi, K.; Kuninaka, S.; Ohtani, N.; Yoshimoto, S.; Hori, S.; Tachibana, M.; Anderton, E.; Takeuchi, T.; et al. DNA damage signaling triggers degradation of histone methyltransferases through APC/C Cdh1 in senescent cells. Mol. Cell 2012, 45, 123–131. [Google Scholar] [CrossRef]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef]
- Kida, Y.; Goligorsky, M.S. Sirtuins, Cell Senescence, and Vascular Aging. Can. J. Cardiol. 2016, 32, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.K.; Berger, S.L. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009, 459, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.O.; Murphy, K.J.; Hayes, J.J.; Thiriet, C. Replication-independent nucleosome exchange is enhanced by local and specific acetylation of histone H4. Nucleic Acids Res. 2013, 41, 2228–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michishita, E.; McCord, R.A.; Berber, E.; Kioi, M.; Padilla-Nash, H.; Damian, M.; Cheung, P.; Kusumoto, R.; Kawahara, T.L.A.; Barrett, J.C.; et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 2008, 452, 492–496. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Vanhoutte, P.M.; Wang, Y. Loss-of-SIRT1 function during vascular ageing: Hyperphosphorylation mediated by cyclin-dependent kinase 5. Trends Cardiovasc. Med. 2014, 24, 81–84. [Google Scholar] [CrossRef]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [Green Version]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Lenain, C.; De Graaf, C.A.; Pagie, L.; Visser, N.L.; De Haas, M.; De Vries, S.S.; Peric-Hupkes, D.; van Steensel, B.; Peeper, D.S. Massive reshaping of genome-nuclear lamina interactions during oncogene-induced senescence. Genome Res. 2017, 27, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.L.; et al. Formation of macroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Kirschner, K.; Thuret, J.Y.; Pope, B.D.; Ryba, T.; Newman, S.; Ahmed, K.; Samarajiwa, S.A.; Salama, R.; Carroll, T.; et al. Independence of Repressive Histone Marks and Chromatin Compaction during Senescent Heterochromatic Layer Formation. Mol. Cell 2012, 47, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, T.; Kirschner, K. Chromosome organisation during ageing and senescence. Curr. Opin. Cell Biol. 2016, 40, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boumendil, C.; Hari, P.; Olsen, K.C.F.; Acosta, J.C.; Bickmore, W.A. Nuclear pore density controls heterochromatin reorganization during senescence. Genes Dev. 2019, 33, 144–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.L.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef]
- Burgess, R.C.; Burman, B.; Kruhlak, M.J.; Misteli, T. Activation of DNA Damage Response Signaling by Condensed Chromatin. Cell Rep. 2014, 9, 1703–1717. [Google Scholar] [CrossRef] [Green Version]
- Prieur, A.; Besnard, E.; Babled, A.; Lemaitre, J.M. P53 and p16INK4A independent induction of senescence by chromatin-dependent alteration of S-phase progression. Nat. Commun. 2011, 2, 473. [Google Scholar] [CrossRef]
- Olivieri, F.; Capri, M.; Bonafè, M.; Morsiani, C.; Jung, H.J.; Spazzafumo, L.; Viña, J.; Suh, Y. Circulating miRNAs and miRNA shuttles as biomarkers: Perspective trajectories of healthy and unhealthy aging. Mech. Ageing Dev. 2017, 165, 162–170. [Google Scholar] [CrossRef]
- Huang, X.; Luo, Y.; Mao, Y.S.; Ji, J.L. The link between long noncoding RNAs and depression. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2017, 73, 73–78. [Google Scholar] [CrossRef]
- Szafranski, K.; Abraham, K.J.; Mekhail, K. Non-coding RNA in neural function, disease, and aging. Front. Genet. 2015, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Karnati, H.K.; Panigrahi, M.K.; Gutti, R.K.; Greig, N.H.; Tamargo, I.A. MiRNAs: Key Players in Neurodegenerative Disorders and Epilepsy. J. Alzheimer’s Dis. 2015, 48, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Salta, E.; De Strooper, B. Non-coding RNAs with essential roles in neurodegenerative disorders. Lancet Neurol. 2012, 11, 189–200. [Google Scholar] [CrossRef]
- Caravia, X.M.; López-Otín, C. Regulatory roles of miRNAs in aging. Adv. Expmed. Biol. 2015, 887, 213–230. [Google Scholar]
- Kato, M.; Slack, F.J. Ageing and the small, non-coding RNA world. Ageing Res. Rev. 2013, 12, 429–435. [Google Scholar] [CrossRef] [PubMed]
- De Lencastre, A.; Pincus, Z.; Zhou, K.; Kato, M.; Lee, S.S.; Slack, F.J. MicroRNAs both promote and antagonize longevity in C. elegans. Curr. Biol. 2010, 20, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, J.; Xie, F.; Gao, X.; Ye, J.H.; Sun, L.Y.; Wei, R.; Ai, J. miR-124/ATF-6, a novel lifespan extension pathway of Astragalus polysaccharide in Caenorhabditis elegans. J. Cell Biochem. 2015, 116, 242–251. [Google Scholar] [CrossRef]
- Du, W.W.; Yang, W.; Fang, L.; Xuan, J.; Li, H.; Khorshidi, A.; Gupta, S.; Li, X.; Yang, B.B. MiR-17 extends mouse lifespan by inhibiting senescence signaling mediated by MKP7. Cell Death Dis. 2014, 5, e1355. [Google Scholar] [CrossRef]
- Boehm, M.; Slack, F.J. MicroRNA control of lifespan and metabolism. Cell Cycle 2006, 5, 837–840. [Google Scholar] [CrossRef]
- Poy, M.N.; Eliasson, L.; Krutzfeldt, J.; Kuwajima, S.; Ma, X.; Macdonald, P.E.; Pfeffer, S.; Tuschl, T.; Rajewsky, N.; Rorsman, P.; et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 2004, 432, 226–230. [Google Scholar] [CrossRef]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Cobb, A.M.; Larrieu, D.; Warren, D.T.; Liu, Y.; Srivastava, S.; Smith, A.J.O.; Bowater, R.P.; Jackson, S.P.; Shanahan, C.M. Prelamin A impairs 53BP1 nuclear entry by mislocalizing NUP153 and disrupting the Ran gradient. Aging Cell 2016, 15, 1039–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaina, S.; Heyn, H.; Carmona, F.J.; Varol, N.; Sayols, S.; Condom, E.; Ramirez-Ruz, J.; Gomez, A.; Goncalves, I.; Moran, S.; et al. DNA methylation map of human atherosclerosis. Circ. Cardiovasc. Genet. 2014, 7, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.G.; Teschendorff, A.E.; Rakyan, V.K.; Maxwell, A.P.; Beck, S.; Savage, D.A. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med. Genom. 2010, 3, 33. [Google Scholar] [CrossRef]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011, 43, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Long, T.I.; Arakawa, K.; Wang, R.; Yu, M.C.; Laird, P.W. DNA methylation as a biomarker for cardiovascular disease risk. PLoS ONE 2010, 5, e9692. [Google Scholar] [CrossRef]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of epigenetic drugs in disease: An overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Sheppard, A.; Gluckman, P.D.; Lillycrop, K.A.; Burdge, G.C.; McLean, C.; Rodford, J.; Slater-Jefferies, J.L.; Garratt, E.; Crozier, S.R.; et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes 2011, 60, 1528–1534. [Google Scholar] [CrossRef]
- Clarke-Harris, R.; Wilkin, T.J.; Hosking, J.; Pinkney, J.; Jeffery, A.N.; Metcalf, B.S.; Godfrey, K.M.; Voss, L.D.; Lillycrop, K.A.; Burdge, G.C. PGC1α promoter methylation in blood at 5-7 years predicts adiposity from 9 to 14 years (EarlyBird 50). Diabetes 2014, 63, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- Shiels, P.G.; McGuinness, D.; Eriksson, M.; Kooman, J.P.; Stenvinkel, P. The role of epigenetics in renal ageing. Nat. Rev. Nephrol 2017, 13, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasiulionis, M.G. Abnormal epigenetic regulation of immune system during aging. Front. Immunol. 2018, 9, 197. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Natoli, G. Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev. 2002, 16, 2219–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidler, C.; Wóycicki, R.; Ilnytskyy, Y.; Metz, G.; Kovalchuk, I.; Kovalchuk, O. Immunosenescence is associated with altered gene expression and epigenetic regulation in primary and secondary immune organs. Front. Genet. 2013, 4, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, K.; Ryan, D.P.; Pearson, B.L.; Henzel, K.S.; Neff, F.; Vidal, R.O.; Hennion, M.; Lehmann, I.; Schleif, M.; Schröder, S.; et al. Epigenetic alterations in longevity regulators, reduced life span, and exacerbated aging-related pathology in old father offspring mice. Proc. Natl. Acad. Sci. USA 2018, 115, E2348–E2357. [Google Scholar] [CrossRef] [Green Version]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [Green Version]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef] [PubMed]
- Steegers-Theunissen, R.P.; Obermann-Borst, S.A.; Kremer, D.; Lindemans, J.; Siebel, C.; Steegers, E.A.; Slagboom, P.E.; Heijmans, B.T. Periconceptional maternal folic acid use of 400 μg per day is related to increased methylation of the IGF2 gene in the very young child. PLoS ONE 2009, 4, e7845. [Google Scholar] [CrossRef] [PubMed]
- Hahn, O.; Grönke, S.; Stubbs, T.M.; Ficz, G.; Hendrich, O.; Krueger, F.; Andrews, S.; Zhang, Q.; Wakelam, J.M.; Beyer, A.; et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 2017, 18, 56. [Google Scholar] [CrossRef]
- Hass, B.S.; Hart, R.W.; Lu, M.H.; Lyn-Cook, B.D. Effects of caloric restriction in animals on cellular function, oncogene expression, and DNA methylation in vitro. Mutat. Res. 1993, 295, 281–289. [Google Scholar] [CrossRef]
- Chouliaras, L.; van den Hove, D.L.A.; Kenis, G.; Dela Cruz, J.; Lemmens, M.A.; van Os, J.; Steinbusch, H.W.; Schmitz, C.; Rutten, B.P. Caloric restriction attenuates age-related changes of DNA methyltransferase 3a in mouse hippocampus. Brain Behav. Immun. 2011, 25, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Tollefsbol, T.O. Glucose restriction can extend normal cell lifespan and impair precancerous cell growth through epigenetic control of hTERT and p16 expression. FASEB J. 2010, 24, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C. Epigenetics of aging and Alzheimer’s disease: Implications for pharmacogenomics and drug response. Int. J. Mol. Sci. 2015, 16, 30483–30543. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, Y.; Tawa, R.; Koizumi, A.; Uehara, Y.; Kurishita, A.; Sakurai, H.; Kamiyama, S.; Ono, T. Effects of energy restriction on age-associated changes of DNA methylation in mouse liver. Mutat. Res. 1993, 295, 63–69. [Google Scholar] [CrossRef]
- Van Straten, E.M.; Bloks, V.W.; Huijkman, N.C.; Baller, J.F.; Meer, H.; Lutjohann, D.; Kuipers, F.; Plosch, T. The liver X-receptor gene promoter is hypermethylated in a mouse model of prenatal protein restriction. Am. J. Physiol. Integr. Comp. Physiol. 2010, 298, R275–R282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.D.; Uthus, E.O. Dietary Folate and Selenium Affect Dimethylhydrazine-Induced Aberrant Crypt Formation, Global DNA Methylation and One-Carbon Metabolism in Rats. J. Nutr. 2003, 133, 2907–2914. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.D.; Uthus, E.O.; Finley, J.W. Dietary Selenium and Arsenic Affect DNA Methylation In Vitro in Caco-2 Cells and In Vivo in Rat Liver and Colon. J. Nutr. 2000, 130, 2903–2909. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (−)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef]
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in cellular functions: Role of polyphenols. Arch Biochem. Biophys. 2010, 501, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Xie, Z.; Jones, W.; Pavlovicz, R.E.; Liu, S.; Yu, J.; Li, P.K.; Lin, J.; Fuchs, J.R.; Marcucci, G.; et al. Curcumin is a potent DNA hypomethylation agent. Bioorganic Med. Chem. Lett. 2009, 19, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Ayissi, V.B.O.; Ebrahimi, A.; Schluesenner, H. Epigenetic effects of natural polyphenols: A focus on SIRT1-mediated mechanisms. Mol. Nutr. Food Res. 2014, 58, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea Polyphenol (−)-Epigallocatechin-3-Gallate Inhibits DNA Methyltransferase and Reactivates Methylation-Silenced Genes in Cancer Cell Lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar] [PubMed]
- Handel, M.L.; Watts, C.K.; DeFazio, A.; Day, R.O.; Sutherland, R.L. Inhibition of AP-1 binding and transcription by gold and selenium involving conserved cysteine residues in Jun and Fos. Proc. Natl. Acad. Sci. USA 1995, 92, 4497–4501. [Google Scholar] [CrossRef] [PubMed]
- Spyrou, G.; Björnstedt, M.; Kumar, S.; Holmgren, A. AP-1 DNA-binding activity is inhibited by selenite and selenodiglutathione. FEBS Lett. 1995, 368, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Xiang, N.; Zhao, R.; Song, G.; Zhong, W. Selenite reactivates silenced genes by modifying DNA methylation and histones in prostate cancer cells. Carcinogenesis 2008, 29, 2175–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.J. Mechanisms for the Inhibition of DNA Methyltransferases by Tea Catechins and Bioflavonoids. Mol. Pharm. 2005, 68, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Wang, C.; Lu, C.; Zhao, B.; Cui, Y.; Shi, X. and Ma, X. Quercetin is able to demethylate the p16INK4a gene promoter. Chemotherapy 2009, 55, 6–10. [Google Scholar] [CrossRef]
- Wolff, G.L.; Kodell, R.L.; Moore, S.R.; Cooney, C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998, 12, 949–957. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Weidman, J.R.; Waterland, R.A.; Jirtle, R.L. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ. Health Perspect. 2006, 114, 567–572. [Google Scholar] [CrossRef]
- Waterland, R.A.; Travisano, M.; Tahiliani, K.G.; Rached, M.T.; Mirza, S. Methyl donor supplementation prevents transgenerational amplification of obesity. Int. J. Obes. 2008, 32, 1373–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harkness, R.W.; Mittermaier, A.K. G-quadruplex dynamics. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- François, M.; Leifert, W.; Tellam, R.; Fenech, M. G-quadruplexes: A possible epigenetic target for nutrition. Mutat. Res. 2015, 764, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Zhu, W.; Shi, H.; Hewett, J.E.; Ruhlen, R.L.; MacDonald, R.S.; Rottinghaus, G.E.; Chen, Y.C.; Sauter, E.R. Soy isoflavones have an antiestrogenic effect and alter mammary promoter hypermethylation in healthy premenopausal women. Nutr. Cancer 2009, 61, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Majid, S.; Dar, A.A.; Shahryari, V.; Hirata, H.; Ahmad, A.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Dahiya, R. Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B-cell translocation gene 3 in prostate cancer. Cancer 2010, 116, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.K.; Ansari, M.Z.; Al Mutery, A.; Ashraf, M.; Nasab, R.; Rai, S.; Rais, N.; Hussain, A. Genistein induces alterations of epigenetic modulatory signatures in human cervical cancer cells. Anticancer Agents Med. Chem. 2018, 18, 412–421. [Google Scholar] [CrossRef]
- Priyadarsini, R.V.; Vinothini, G.; Murugan, R.S.; Manikandan, P.; Nagini, S. The flavonoid quercetin modulates the hallmark capabilities of hamster buccal pouch tumors. Nutr. Cancer 2011, 63, 218–226. [Google Scholar] [CrossRef]
- Hong, K.S.; Park, J.I.; Kim, M.J.; Kim, H.B.; Lee, J.W.; Dao, T.T.; Oh, W.K.; Kang, C.D.; Kim, S.H. Involvement of SIRT1 in hypoxic down-regulation of c-Myc and β-catenin and hypoxic preconditioning effect of polyphenols. Toxicol. Appl. Pharm. 2012, 259, 210–218. [Google Scholar] [CrossRef]
- Attoub, S.; Hassan, A.H.; Vanhoecke, B.; Iratni, R.; Takahashi, T.; Gaben, A.M.; Bracke, M.; Awad, S.; John, A.; Kamalboor, H.A.; et al. Inhibition of cell survival, invasion, tumor growth and histone deacetylase activity by the dietary flavonoid luteolin in human epithelioid cancer cells. Eur. J. Pharm. 2011, 651, 18–25. [Google Scholar] [CrossRef]
- Kang, S.K.; Cha, S.H.; Jeon, H.G. Curcumin-induced Histone Hypoacetylation Enhances Caspase-3-dependent Glioma Cell Death and Neurogenesis of Neural Progenitor Cells. Stem Cells Dev. 2006, 15, 165–174. [Google Scholar] [CrossRef]
- Teiten, M.H.; Dicato, M.; Diederich, M. Curcumin as a regulator of epigenetic events. Mol. Nutr. Food Res. 2013, 57, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, W.; Suszek, M.; Wnuk, M.; Lewinska, A.; Wasiak, E.; Sikora, E.; Bielak-Zmijewska, A. Curcumin elevates sirtuin level but does not postpone in vitro senescence of human cells building the vasculature. Oncotarget 2016, 7, 19201–19213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.C.; Myung, G.J.; Lee, Y.H.; Yoon, J.C.; Kwon, S.H.; Kang, H.B.; Kim, M.J.; Cha, J.H.; Kim, Y.J.; Jun, W.J.; et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009, 69, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Shukla, S.; Gupta, S. Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells. Int. J. Cancer 2010, 126, 2520–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Shi, D.; Liu, L.; Wang, J.; Xie, X.; Kang, T.; Deng, W. Quercetin suppresses cyclooxygenase-2 expression and angiogenesis through inactivation of P300 signaling. PLoS ONE 2011, 6, e22934. [Google Scholar] [CrossRef]
- Lee, W.J.; Chen, Y.R.; Tseng, T.H. Quercetin induces FasL-related apoptosis, in part, through promotion of histone H3 acetylation in human leukemia HL-60 cells. Oncol. Rep. 2011, 25, 583–591. [Google Scholar] [PubMed]
- Lin, C.; Kang, J.; Zheng, R. Oxidative stress is involved in inhibition of copper on histone acetylation in cells. Chem. Biol. Interact 2005, 151, 167–176. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Adhikary, G.; Eckert, R.L. The Bmi-1 polycomb protein antagonizes the (-)-epigallocatechin-3-gallate-dependent suppression of skin cancer cell survival. Carcinogenesis 2010, 31, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Tryndyak, V.P.; Muskhelishvili, L.; Rusyn, I.; Ross, S.A. Methyl deficiency, alterations in global histone modifications, and carcinogenesis. J. Nutr. 2007, 137, 216S–222S. [Google Scholar] [CrossRef]
- Giblin, W.; Skinner, M.E.; Lombard, D.B. Sirtuins: Guardians of mammalian healthspan. Trends Genet. 2014, 30, 271–286. [Google Scholar] [CrossRef]
- Jayasena, T.; Poljak, A.; Smythe, G.; Braidy, N.; Münch, G.; Sachdev, P. The role of polyphenols in the modulation of sirtuins and other pathways involved in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharm. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greathouse, K.L.; Samuels, M.; DiMarco, N.M.; Criswell, D.S. Effects of increased dietary fat and exercise on skeletal muscle lipid peroxidation and antioxidant capacity in male rats. Eur. J. Nutr. 2005, 44, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Chung, H.Y.; Koltai, E.; Taylor, A.W.; Goto, S. Exercise, oxidative stress and hormesis. Ageing Res. Rev. 2008, 44, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Chung, H.Y.; Goto, S. Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic Biol. Med. 2008, 44, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.M.; Lushchak, O.V.; Koliada, A.K. Anti-aging pharmacology: Promises and pitfalls. Ageing Res. Rev. 2016, 31, 9–35. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.L.; Benzer, S.; Min, K.T. Life extension in Drosophila by feeding a drug. Proc. Natl. Acad. Sci. USA 2002, 99, 838–843. [Google Scholar] [CrossRef]
- Tao, D.; Lu, J.; Sun, H.; Zhao, Y.M.; Yuan, Z.G.; Li, X.X.; Huang, B.Q. Trichostatin A Extends the Lifespan of Drosophila melanogaster by Elevating hsp22 Expression. Acta Biochim. Biophys. Sin. 2004, 36, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Sun, H.; Lu, J.; Li, X.; Chen, X.; Tao, D.; Huang, W.; Huang, B. Lifespan extension and elevated hsp gene expression in Drosophila caused by histone deacetylase inhibitors. J. Exp. Biol. 2005, 208, 697–705. [Google Scholar] [CrossRef]
- Christensen, D.P.; Dahllöf, M.; Lundh, M.; Rasmussen, D.N.; Nielsen, M.D.; Billestrup, N.; Grunnet, L.G.; Mandrup-Poulsen, T. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol. Med. Camb. Mass 2011, 17, 378–390. [Google Scholar] [CrossRef]
- Penney, J.; Tsai, L.H. Histone deacetylases in memory and cognition. Sci. Signal 2014, 7, re12. [Google Scholar] [CrossRef]
- Ganai, S.A.; Ramadoss, M.; Mahadevan, V. Histone Deacetylase (HDAC) Inhibitors—Emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr. Neuropharmacol. 2016, 14, 55–71. [Google Scholar] [CrossRef]
- Wang, L.; Yu, C.; Yu, G.; Lu, X.; Yu, D. Histone Acetylation Modifiers in the Pathogenesis of Alzheimer’s Disease. Front. Cell Neurosci. 2015, 9, 226. [Google Scholar]
- Sharma, S.; Taliyan, R. Targeting Histone Deacetylases: A Novel Approach in Parkinson’s Disease. Parkinsons Dis. 2015, 2015, 303294. [Google Scholar] [CrossRef]
- Chakrabarti, M.; Khandkar, M.; Banik, N.L.; Ray, S.K. Alterations in expression of specific microRNAs by combination of 4-HPR and EGCG inhibited growth of human malignant neuroblastoma cells. Brain Res. 2012, 1454, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Boesch-Saadatmandi, C.; Wagner, A.E.; Wolffram, S.; Rimbach, G. Effect of quercetin on inflammatory gene expression in mice liver in vivo—Role of redox factor 1, miRNA-122 and miRNA-125b. Pharm. Res. 2012, 65, 523–530. [Google Scholar] [CrossRef]
- Li, Y.; Vandenboom, T.G.; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009, 69, 6. [Google Scholar] [CrossRef]
- Sun, M.; Estrov, Z.; Ji, Y.; Coombes, K.R.; Harris, D.H.; Kurzrock, R. Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Mol. Cancer 2008, 7, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.Z.; Zang, J.N.; Deng, L.L.; Wang, Z.Y.; Yu, C.H. Pentamethylquercetin reduces fat deposition via Sirt1-mediated pathways in male obese mice induced by a high fat diet. Food Chem. Toxicol. 2013, 62, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Barrès, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Barrès, R.; Kirchner, H.; Rasmussen, M.; Yan, J.; Kantor, F.R.; Krook, A.; Näslund, E.; Zierath, J.R. Weight Loss after Gastric Bypass Surgery in Human Obesity Remodels Promoter Methylation. Cell Rep. 2013, 3, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, S.C.; Gillberg, L.; Bork-Jensen, J.; Ribel-Madsen, R.; Lara, E.; Calvanese, V.; Ling, C.; Fernandez, A.F.; Fraga, M.F.; Poulsen, P.; et al. Young men with low birthweight exhibit decreased plasticity of genome-wide muscle DNA methylation by high-fat overfeeding. Diabetologia 2014, 57, 1154–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rönn, T.; Volkov, P.; Davegárdh, C.; Dayeh, T.; Hall, E.; Olsson, A.H.; Nilsson, E.; Tornberg, A.; Dekker Nitert, M.; Eriksson, K.F.; et al. A Six Months Exercise Intervention Influences the Genome-wide DNA Methylation Pattern in Human Adipose Tissue. PLoS Genet. 2013, 9, e1003572. [Google Scholar] [CrossRef] [PubMed]
- Weems, J.C.; Griesel, B.A.; Olson, A.L. Class II histone deacetylases downregulate GLUT4 transcription in response to increased cAMP signaling in cultured adipocytes and fasting mice. Diabetes 2012, 61, 1404–1414. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.L.; Fairlie, E.; Garnham, A.P.; Hargreaves, M. Exercise-induced histone modifications in human skeletal muscle. J. Physiol. 2009, 587, 5951–5958. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Nuclear hormone receptor co-repressors: Structure and function. Mol. Cell Endocrinol. 2012, 348, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Franks, A.L.; Slansky, J.E. Multiple associations between a broad spectrum of autoimmune diseases, chronic inflammatory diseases and cancer. Anticancer Res. 2012, 32, 41119–41136. [Google Scholar]
- Flowers, E.; Won, G.Y.; Fukuoka, Y. MicroRNAs associated with exercise and diet: A systematic review. Physiol. Genom. 2014, 47, 1–11. [Google Scholar] [CrossRef]
- Cummins, J.M.; He, Y.; Leary, R.J.; Pagliarini, R.; Diaz, L.A.; Sjoblom, T.; Barad, O.; Bentwich, Z.; Szafranska, A.E.; Labourier, E.; et al. The colorectal microRNAome. Proc. Natl. Acad. Sci. USA 2006, 103, 3687–3692. [Google Scholar] [CrossRef] [Green Version]
- Rowlands, D.S.; Page, R.A.; Sukala, W.R.; Giri, M.; Ghimbovschi, S.D.; Hayat, I.; Cheema, B.S.; Lys, I.; Leikis, M.; Sheard, P.W.; et al. Multi-omic integrated networks connect DNA methylation and miRNA with skeletal muscle plasticity to chronic exercise in Type 2 diabetic obesity. Physiol. Genom. 2014, 46, 747–765. [Google Scholar] [CrossRef]
- Denham, J.; O’Brien, B.J.; Marques, F.Z.; Charchar, F.J. Changes in the leukocyte methylome and its effect on cardiovascular-related genes after exercise. J. Appl. Physiol. 2015, 118, 475–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laker, R.C.; Wlodek, M.E.; Connelly, J.J.; Yan, Z. Epigenetic origins of metabolic disease: The impact of the maternal condition to the offspring epigenome and later health consequences. Food Sci. Hum. Wellness 2013, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chen, N. Regulatory role of microRNAs in muscle atrophy during exercise intervention. Int. J. Mol. Sci. 2018, 19, 405. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.; Vitucci, D.; Labruna, G.; Imperlini, E.; Randers, M.B.; Schmidt, J.F.; Hagman, M.; Andersen, T.R.; Russo, R.; Orrù, S.; et al. Effect of lifelong football training on the expression of muscle molecular markers involved in healthy longevity. Eur. J. Appl. Physiol. 2017, 117, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Lew, J.K.S.; Pearson, J.T.; Schwenke, D.O.; Katare, R. Exercise mediated protection of diabetic heart through modulation of microRNA mediated molecular pathways. Cardiovasc. Diabetol. 2017, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; Kumsta, C.; Sandri, M.; Ballabio, A.; Hansen, M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 2015, 6, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. SIRT1: Regulation of longevity via autophagy. Cell Signal 2009, 21, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Artal-Martinez de Narvajas, A.; Gomez, T.S.; Zhang, J.J.S.; Mann, A.O.; Taoda, Y.; Gorman, J.A.; Herreros-Villanueva, M.; Gress, T.M.; Ellenrieder, V.; Bujanda, L.; et al. Epigenetic Regulation of Autophagy by the Methyltransferase G9a. Mol. Cell Biol. 2013, 33, 3983–3993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef]
- Eisenberg, T.; Schroeder, S.; Andryushkova, A.; Pendl, T.; Küttner, V.; Bhukel, A.; Mariño, G.; Pietrocola, F.; Harger, A.; Zimmermann, S.; et al. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme A stimulates autophagy and prolongs lifespan. Cell Metab. 2014, 19, 431–444. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Muthusamy, S.; Liang, R.; Sarojini, H.; Wang, E. Gain of survival signaling by down-regulation of three key miRNAs in brain of calorie-restricted mice. Aging 2011, 3, 223–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Chen, D.; He, Y.; Melendez, A.; Feng, Z.; Hong, Q.; Bai, X.; Li, Q.; Cai, G.; Wang, J.; et al. MiR-34 modulates Caenorhabditis elegans lifespan via repressing the autophagy gene atg9. Age 2013, 35, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Muñoz, M.E.; Arrieta, M.C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6, 23129. [Google Scholar] [CrossRef]
- Walker, R.W.; Clemente, J.C.; Peter, I.; Loos, R.J.F. The prenatal gut microbiome: Are we colonized with bacteria in utero? Pediatr. Obes. 2017, 12, 3–17. [Google Scholar] [CrossRef]
- Lim, E.S.; Rodriguez, C.; Holtz, L.R. Amniotic fluid from healthy term pregnancies does not harbor a detectable microbial community. Microbiome 2018, 6, 87. [Google Scholar] [CrossRef]
- Rehbinder, E.M.; Lødrup Carlsen, K.C.; Staff, A.C.; Angell, I.L.; Landrø, L.; Hilde, K.; Gaustad, P.; Rudi, K. Is amniotic fluid of women with uncomplicated term pregnancies free of bacteria? Am. J. Obs. Gynecol. 2018, 219, e1–e289. [Google Scholar] [CrossRef]
- Mackie, R.I.; Sghir, A.; Gaskins, H.R. Developmental microbial ecology of the neonatal gastrointestinal tract. Am. J. Clin. Nutr. 1999, 69, 1035S–1045S. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Wu, L.; Luo, J.; Liang, X.; Xiao, B.; Zhu, Y. The impacts of delivery mode on infant’s oral microflora. Sci. Rep. 2018, 8, 11938. [Google Scholar] [CrossRef]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Heal Dis. 2015, 26, 26050. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Zeng, D.N.; Chi, L.; Tan, Y.; Galzote, C.; Cardona, C.; Lax, S.; Gilbert, J.; Quan, Z.X. The influence of age and gender on skin-associated microbial communities in urban and rural human populations. PLoS ONE 2015, 10, e0141842. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Codella, R.; Luzi, L.; Terruzzi, I. Exercise has the guts: How physical activity may positively modulate gut microbiota in chronic and immune-based diseases. Dig. Liver Dis. 2018, 50, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mach, N.; Fuster-Botella, D. Endurance exercise and gut microbiota: A review. J. Sport Heal Sci. 2017, 6, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Palleja, A.; Mikkelsen, K.H.; Forslund, S.K.; Kashani, A.; Allin, K.H.; Nielsen, T.; Hansen, T.H.; Liang, S.; Feng, Q.; Zhang, C.; et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 2018, 3, 1255–1265. [Google Scholar] [CrossRef]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Tasnim, N.; Abulizi, N.; Pither, J.; Hart, M.M.; Gibson, D.L. Linking the gut microbial ecosystem with the environment: Does gut health depend on where we live? Front. Microbiol. 2017, 8, 1935. [Google Scholar] [CrossRef] [PubMed]
- Salazar, N.; Arboleya, S.; Valdés, L.; Stanton, C.; Ross, P.; Ruiz, L.; Gueimonde, M.; de los Reyes-Gavilan, C.G. The human intestinal microbiome at extreme ages of life. Dietary intervention as a way to counteract alterations. Front. Genet. 2014, 5, 406. [Google Scholar] [CrossRef]
- Rolhion, N.; Chassaing, B. When pathogenic bacteria meet the intestinal microbiota. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascal, M.; Perez-Gordo, M.; Caballero, T.; Escribese, M.M.; Lopez Longo, M.N.; Luengo, O.; Manso, L.; Matheu, V.; Seoane, E.; Zamorano, M.; et al. Microbiome and allergic diseases. Front. Immunol. 2018, 9, 1584. [Google Scholar] [CrossRef] [PubMed]
- De la Cuesta-Zuluaga, J.; Corrales-Agudelo, V.; Velásquez-Mejía, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Gut microbiota is associated with obesity and cardiometabolic disease in a population in the midst of Westernization. Sci. Rep. 2018, 8, 11356. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ling, Z.; Zhang, Y.; Mao, H.; Ma, Z.; Yin, Y.; Wang, W.; Tang, W.; Tan, Z.; Shi, J.; et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav. Immun. 2015, 48, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna, R.A.; Foster, J.A. Gut brain axis: Diet microbiota interactions and implications for modulation of anxiety and depression. Curr. Opin. Biotechnol. 2015, 32, 35–41. [Google Scholar] [CrossRef]
- Kataoka, K. The intestinal microbiota and its role in human health and disease. J. Med. Investig. 2016, 63, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.; Mach, N. The crosstalk between the gut microbiota and mitochondria during exercise. Front. Physiol. 2017, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Gordon, J.I. The core gut microbiome, energy balance and obesity. J. Physiol. 2009, 587, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ticinesi, A.; Lauretani, F.; Milani, C.; Nouvenne, A.; Tana, C.; Del Rio, D.; Maggio, M.; Ventura, M.; Meschi, T.; Lauretani, F.; et al. Aging gut microbiota at the cross-road between nutrition, physical frailty, and sarcopenia: Is there a gut–muscle axis? Nutrients 2017, 9, 1303. [Google Scholar] [CrossRef]
- Tran, L.; Greenwood-Van Meerveld, B. Age-associated remodeling of the intestinal epithelial barrier. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Holt, P.R. Intestinal malabsorption in the elderly. Dig. Dis. 2007, 25, 144–150. [Google Scholar] [CrossRef]
- Bottazzi, B.; Riboli, E.; Mantovani, A. Aging, inflammation and cancer. Semin Immunol. 2018, 40, 74–82. [Google Scholar] [CrossRef]
- Zinger, A.; Cho, W.C.; Ben-Yehuda, A. Cancer and Aging—The Inflammatory Connection. Aging Dis. 2018, 8, 611–627. [Google Scholar] [CrossRef]
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis in Health and Disease. Gastroenterol. Clin. North Am. 2017, 46, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Claesson, M.J.; Cusack, S.; O’Sullivan, O.; Greene-Diniz, R.; de Weerd, H.; Flannery, E.; Marchesi, J.R.; Falush, D.; Dinan, T.; Fitzgerald, G.; et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. USA 2011, 108, 4586–4591. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef]
- Jackson, M.; Jeffery, I.B.; Beaumont, M.; Bell, J.T.; Clark, A.G.; Ley, R.E.; O’Toole, P.W.; Spector, T.D.; Steves, C.J. Signatures of early frailty in the gut microbiota. Genome Med. 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Aidy, S.E.; van den Bogert, B.; Kleerebezem, M. The small intestine microbiota, nutritional modulation and relevance for health. Curr. Opin. Biotechnol. 2015, 32, 14–20. [Google Scholar] [CrossRef]
- Amer, S.; Manzar, H.S. Small intestinal bacterial overgrowth in older people. Rev. Clin. Gerontol. 2015, 25, 81–85. [Google Scholar] [CrossRef]
- Sicard, J.F.; Le Bihan, G.; Vogeleer, P.; Jacques, M.; Harel, J. Interactions of Intestinal Bacteria with Components of the Intestinal Mucus. Front. Cell Infect. Microbiol. 2017, 7, 387. [Google Scholar] [CrossRef]
- Quigley, E.M.M. Gut bacteria in health and disease. Gastroenterol. Hepatol. 2013, 9, 560–569. [Google Scholar]
- O’Toole, P.W.; Claesson, M.J. Gut microbiota: Changes throughout the lifespan from infancy to elderly. Int. Dairy J. 2010, 20, 281–291. [Google Scholar] [CrossRef]
- Hopkins, M.J.; Sharp, R.; Macfarlane, G.T. Variation in human intestinal microbiota with age. Dig. Liver Dis. 2002, 34, S12–S18. [Google Scholar] [CrossRef]
- König, J.; Wells, J.; Cani, P.D.; García-Ródenas, C.L.; MacDonald, T.; Mercenier, A.; Whyte, J.; Troost, F.; Brummer, R.J. Human intestinal barrier function in health and disease. Clin. Transl. Gastroenterol. 2016, 7, e196. [Google Scholar] [CrossRef] [PubMed]
- Herrou, J.; Choi, V.M.; Bubeck Wardenburg, J.; Crosson, S. Activation Mechanism of the Bacteroides fragilis Cysteine Peptidase, Fragipain. Biochemistry 2016, 55, 4077–4084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoni, L.; Nuding, S.; Wehkamp, J.; Stange, E.F. Intestinal barrier in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Mulak, A.; Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609–10620. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A. Brain-gut-microbiota axis in Alzheimer’s disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Burcharth, J.; Pommergaard, H.C. Linking gut microbiota to colorectal cancer. J. Cancer 2017, 8, 3378–3395. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability—A new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [PubMed]
- Mihajlovski, A.; Doré, J.; Levenez, F.; Alric, M.; Brugère, J.F. Molecular evaluation of the human gut methanogenic archaeal microbiota reveals an age-associated increase of the diversity. Environ. Microbiol. Rep. 2010, 2, 272–280. [Google Scholar] [CrossRef]
- Pimentel, M.; Lin, H.C.; Enayati, P.; van den Burg, B.; Lee, H.R.; Chen, J.H.; Park, S.; Kong, Y.; Conklin, J. Methane, a gas produced by enteric bacteria, slows intestinal transit and augments small intestinal contractile activity. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1089–G1095. [Google Scholar] [CrossRef] [Green Version]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef]
- Jugé, R.; Rouaud-Tinguely, P.; Breugnot, J.; Servaes, K.; Grimaldi, C.; Roth, M.P.; Coppin, H.; Closs, B. Shift in skin microbiota of Western European women across aging. J. Appl. Microbiol. 2018, 125, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Shibagaki, N.; Suda, W.; Clavaud, C.; Bastien, P.; Takayasu, L.; Iioka, E.; Kurokawa, R.; Yamashita, N.; Hattori, Y.; Shindo, C.; et al. Aging-related changes in the diversity of women’s skin microbiomes associated with oral bacteria. Sci. Rep. 2017, 7, 10567. [Google Scholar] [CrossRef] [PubMed]
- Leyden, J.J.; McGinley, K.J.; Mills, O.H.; Kligman, A.M. Age related changes in the resident bacterial flora of the human face. J. Investig. Derm. 1975, 65, 379–381. [Google Scholar] [CrossRef]
- Ravn, A.H.; Thyssen, J.P.; Egeberg, A. Skin disorders in Parkinson’s disease: Potential biomarkers and risk factors. Clin. Cosmet. Investig. Derm. 2017, 10, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, D.K.; Sinclair, E.; Xu, Y.; Sarkar, D.; Walton-Doyle, C.; Liscio, C.; Banks, P.; Milne, J.; Silverdale, M.; Kunath, T.; et al. Discovery of volatile biomarkers of Parkinson ’ s disease from sebum. ACS Central Sci. 2019, 5, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Besa, V.; Teschler, H.; Kurth, I.; Khan, A.M.; Zarogoulidis, P.; Baumbach, J.I.; Sommerwerck, U.; Freitag, L.; Darwiche, K. Exhaled volatile organic compounds discriminate patients with chronic obstructive pulmonary disease from healthy subjects. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 399–406. [Google Scholar] [Green Version]
- Bari, M.R.; Hiron, M.M.; Zaman, S.M.; Rahman, M.M.; Ganguly, K.C. Microbes responsible for acute exacerbation of COPD. Mymensingh Med. J. 2010, 19, 576–585. [Google Scholar]
- Shaw, J.G.; Vaughan, A.; Dent, A.G.; O’Hare, P.E.; Goh, F.; Bowman, R.V.; Fong, K.M.; Yang, I.A. Biomarkers of progression of chronic obstructive pulmonary disease (COPD). J. Thorac. Dis. 2014, 6, 1532–1547. [Google Scholar]
- Shukla, S.D.; Budden, K.F.; Neal, R.; Hansbro, P.M. Microbiome effects on immunity, health and disease in the lung. Clin. Transl. Immunol. 2017, 6, e133. [Google Scholar] [CrossRef]
- Coburn, B.; Wang, P.W.; Diaz Caballero, J.; Clark, S.T.; Brahma, V.; Donaldson, S.; Zhang, Y.; Surendra, A.; Gong, Y.; Elizabeth Tullis, D.; et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci. Rep. 2015, 5, 10241. [Google Scholar] [CrossRef]
- Preza, D.; Olsen, I.; Willumsen, T.; Grinde, B.; Paster, B.J. Diversity and site-specificity of the oral microflora in the elderly. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef]
- Feres, M.; Teles, F.; Teles, R.; Figueiredo, L.C.; Faveri, M. The subgingival periodontal microbiota of the aging mouth. Periodontol. 2000 2016, 72, 30–53. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Xia, M.; Yan, X.; Li, D.; Wang, L.; Xu, Y.; Jin, T.; Ling, W. Gut microbiota metabolism of anthocyanin promotes reverse cholesterol transport in mice via repressing miRNA-10b. Circ. Res. 2012, 111, 967–981. [Google Scholar] [CrossRef]
- Ohno, M.; Nishida, A.; Sugitani, Y.; Nishino, K.; Inatomi, O.; Sugimoto, M.; Kawahara, M.; Andoh, A. Nanoparticle curcumin ameliorates experimental colitis via modulation of gut microbiota and induction of regulatory T cells. PLoS ONE 2017, 12, e0185999. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Y.; Xiang, L.; Wang, Z.; Xiao, G.G.; Hu, J. Effect of curcumin on the diversity of gut microbiota in ovariectomized rats. Nutrients 2017, 9, E1146. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liu, L.; Ji, H.F. Regulative effects of curcumin spice administration on gut microbiota and its pharmacological implications. Food Nutr. Res. 2017, 61, 1361780. [Google Scholar] [CrossRef] [PubMed]
- Mayta-Apaza, A.C.; Pottgen, E.; De Bodt, J.; Papp, N.; Marasini, D.; Howard, L.; Abranko, L.; Van de Wiele, T.; Lee, S.O.; Carbonero, F. Impact of tart cherries polyphenols on the human gut microbiota and phenolic metabolites in vitro and in vivo. J. Nutr. Biochem. 2018, 59, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C. Interactions of gut microbiota with dietary polyphenols and consequences to human health. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Dueñas, M.; Muñoz-González, I.; Cueva, C.; Jiménez-Girón, A.; Sánchez-Patán, F.; Santos-Buelga, C.; Moreno-Arribas, M.V.; Bartolomé, B. A survey of modulation of gut microbiota by dietary polyphenols. BioMed Res. Int. 2015, 2015, 850902. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.T.; Vaughn, A.R.; Sharma, V.; Chopra, D.; Mills, P.J.; Peterson, S.N.; Sivamani, R.K. Effects of Turmeric and Curcumin Dietary Supplementation on Human Gut Microbiota: A Double-Blind, Randomized, Placebo-Controlled Pilot Study. J. Evid. Based Integr. Med. 2018, 23, 399–406. [Google Scholar] [CrossRef]
- Zam, W. Gut Microbiota as a Prospective Therapeutic Target for Curcumin: A Review of Mutual Influence. J. Nutr. Metab. 2018, 2018, 1367984. [Google Scholar] [CrossRef]
- Perez, A.; Gonzalez-Manzano, S.; Jimene, z.R.; Perez-Abud, R.; Haro, J.M.; Osuna, A.; Santos-Buelga, C.; Duarte, J.; Perez-Vizcaino, F. The flavonoid quercetin induces acute vasodilator effects in healthy volunteers: Correlation with beta-glucuronidase activity. Pharm. Res. 2014, 89, 11–18. [Google Scholar] [CrossRef]
- Menendez, C.; Dueñas, M.; Galindo, P.; González-Manzano, S.; Jimenez, R.; Moreno, L.; Zarzuelo, M.J.; Rodríguez-Gómez, I.; Duarte, J.; Santos-Buelga, C.; et al. Vascular deconjugation of quercetin glucuronide: The flavonoid paradox revealed? Mol. Nutr. Food Res. 2011, 55, 1780–1790. [Google Scholar] [CrossRef]
- Sasaki, H.; Sunagawa, Y.; Takahashi, K.; Imaizumi, A.; Fukuda, H.; Hashimoto, T.; Wada, H.; Katanasaka, Y.; Kakeya, H.; Fujita, M.; et al. Innovative preparation of curcumin for improved oral bioavailability. Biol. Pharm. Bull. 2011, 34, 660–665. [Google Scholar] [CrossRef]
- Mukkavilli, R.; Yang, C.; Tanwar, R.S.; Saxena, R.; Gundala, S.R.; Zhang, Y.; Ghareeb, A.; Floyd, S.D.; Vangala, S.; Kuo, W.W.; et al. Pharmacokinetic-pharmacodynamic correlations in the development of ginger extract as an anticancer agent. Sci. Rep. 2018, 8, 3056. [Google Scholar] [CrossRef]
- Kaneko, A.; Matsumoto, T.; Matsubara, Y.; Sekiguchi, K.; Koseki, J.; Yakabe, R.; Aoki, K.; Aiba, S.; Yamasaki, K. Glucuronides of phytoestrogen flavonoid enhance macrophage function via conversion to aglycones by β-glucuronidase in macrophages. Immun. Inflamm. Dis. 2017, 5, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Szymusiak, M.; Hu, X.; Leon Plata, P.A.; Ciupinski, P.; Wang, Z.J.; Liu, Y. Bioavailability of curcumin and curcumin glucuronide in the central nervous system of mice after oral delivery of nano-curcumin. Int. J. Pharm. 2016, 511, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosh, F.M.; Maison, N.; Holtrop, G.; Young, P.; Stevens, V.J.; Ince, J.; Johnstone, A.M.; Lobley, G.E.; Flint, H.J.; Louis, P. Phylogenetic distribution of genes encoding β-glucuronidase activity in human colonic bacteria and the impact of diet on faecal glycosidase activities. Environ. Microbiol. 2012, 14, 1876–1887. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Uechi, S.; Takara, K.; Asikin, Y.; Wada, K. Evaluation of an oral carrier system in rats: Bioavailability and antioxidant properties of liposome-encapsulated curcumin. J. Agric Food Chem. 2009, 57, 9141–9146. [Google Scholar] [CrossRef] [PubMed]
- Wellman, A.S.; Metukuri, M.R.; Kazgan, N.; Xu, X.; Xu, Q.; Ren, N.S.X.; Czopik, A.; Shanahan, M.T.; Kang, A.; Chen, W.; et al. Intestinal Epithelial Sirtuin 1 Regulates Intestinal Inflammation During Aging in Mice by Altering the Intestinal Microbiota. Gastroenterology 2017, 153, 772–786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.L.; Zhou, M.; Kang, C.; Lang, H.D.; Chen, M.T.; Hui, S.C.; Wang, B.; Mi, M.T. Crosstalk between gut microbiota and Sirtuin-3 in colonic inflammation and tumorigenesis. Exp. Mol. Med. 2018, 50, 21. [Google Scholar] [CrossRef]
- Vikram, A.; Kim, Y.R.; Kumar, S.; Li, Q.; Kassan, M.; Jacobs, J.S.; Irani, K. Vascular microRNA-204 is remotely governed by the microbiome and impairs endothelium-dependent vasorelaxation by downregulating Sirtuin1. Nat. Commun. 2016, 7, 12565. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Kirchgessner, A. Gut inflammation in chronic fatigue syndrome. Nutr. Metab. 2010, 7, 79. [Google Scholar] [CrossRef]
- Den Besten, G.; Gerding, A.; Van Dijk, T.H.; Ciapaite, J.; Bleeker, A.; van Eunen, K.; Havinga, R.; Groen, A.K.; Reijngoud, D.J.; Bakker, B.M. Protection against the metabolic syndrome by guar gum-derived short-chain fatty acids depends on peroxisome proliferator-activated receptorγ And Glucagon-like peptide-1. PLoS ONE 2015, 10, e0136364. [Google Scholar] [CrossRef]
- Walsh, M.E.; Bhattacharya, A.; Sataranatarajan, K.; Qaisar, R.; Sloane, L.; Rahman, M.M.; Kinter, M.; Van Remmen, H. The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging. Aging Cell 2015, 14, 957–970. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef]
- Dukes, A.; Davis, C.; El Refaey, M.; Upadhyay, S.; Mork, S.; Arounleut, P.; Johnson, M.H.; Hill, W.D.; Isales, C.M.; Hamrick, M.W. The aromatic amino acid tryptophan stimulates skeletal muscle IGF1/p70s6k/mTor signaling in vivo and the expression of myogenic genes in vitro. Nutrition 2015, 31, 1018–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, J.; Xu, H.; Zhang, Y.; Chu, L.; Liu, Q.; Song, N.; Yang, C. Novel tumor-targeting, self-assembling peptide nanofiber as a carrier for effective curcumin delivery. Int. J. Nanomed. 2013, 9, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Salazar, N.; López, P.; Valdés, L.; Margolles, A.; Suárez, A.; Patterson, A.M.; Cuervo, A.; de los Reyes-Gavilán, C.G.; Ruas-Madiedo, P.; Gonzalez, S.; et al. Microbial Targets for the Development of Functional Foods Accordingly with Nutritional and Immune Parameters Altered in the Elderly. J. Am. Coll. Nutr. 2013, 32, 399–406. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gadecka, A.; Bielak-Zmijewska, A. Slowing Down Ageing: The Role of Nutrients and Microbiota in Modulation of the Epigenome. Nutrients 2019, 11, 1251. https://doi.org/10.3390/nu11061251
Gadecka A, Bielak-Zmijewska A. Slowing Down Ageing: The Role of Nutrients and Microbiota in Modulation of the Epigenome. Nutrients. 2019; 11(6):1251. https://doi.org/10.3390/nu11061251
Chicago/Turabian StyleGadecka, Agnieszka, and Anna Bielak-Zmijewska. 2019. "Slowing Down Ageing: The Role of Nutrients and Microbiota in Modulation of the Epigenome" Nutrients 11, no. 6: 1251. https://doi.org/10.3390/nu11061251
APA StyleGadecka, A., & Bielak-Zmijewska, A. (2019). Slowing Down Ageing: The Role of Nutrients and Microbiota in Modulation of the Epigenome. Nutrients, 11(6), 1251. https://doi.org/10.3390/nu11061251