Effect of Konjac Mannan Oligosaccharides on Glucose Homeostasis via the Improvement of Insulin and Leptin Resistance In Vitro and In Vivo


Abstract
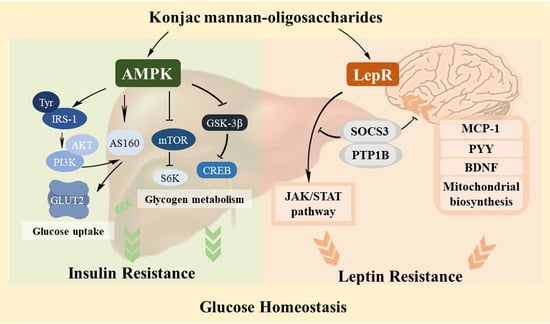
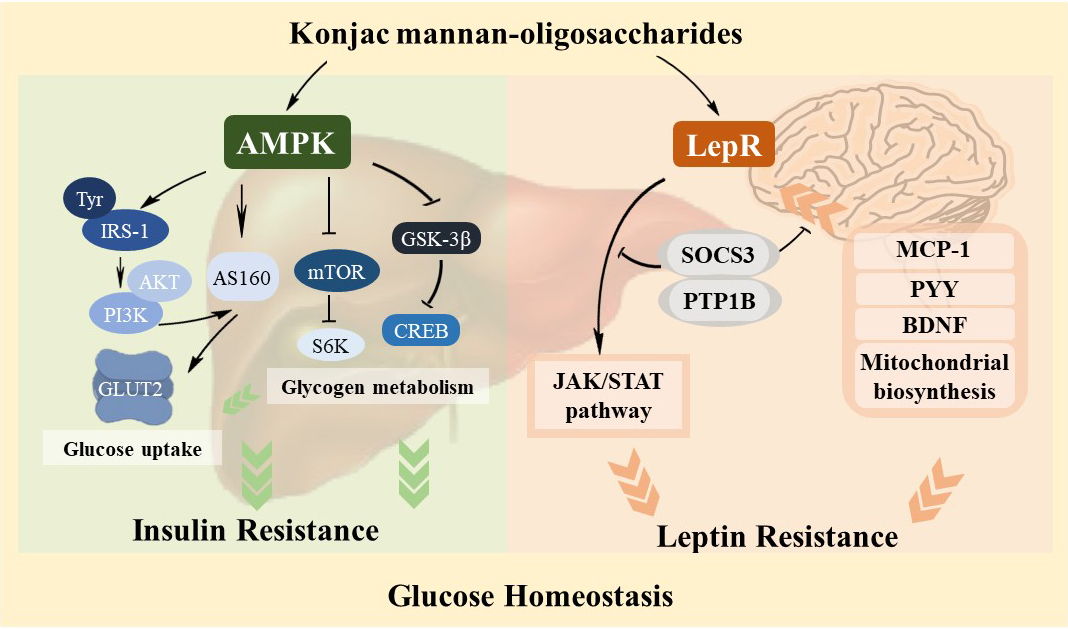
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Animal Experimental Design and Sample Collection of Serum and Tissues
2.3. Insulin Tolerance Test (ITT) and Oral Glucose Tolerance Test (OGTT)
2.4. Fasting Blood Glucose and Index of Insulin Resistance
2.5. Serum Biochemical Analysis
2.6. Biochemical Analysis of Liver and Brain Tissues
2.7. Histopathology
2.8. Cell Culture and Glucose Uptake
2.9. Protein Extraction and Western Blotting
2.10. mRNA Isolation and Gene Expression Quantification
2.11. Statistics
3. Results
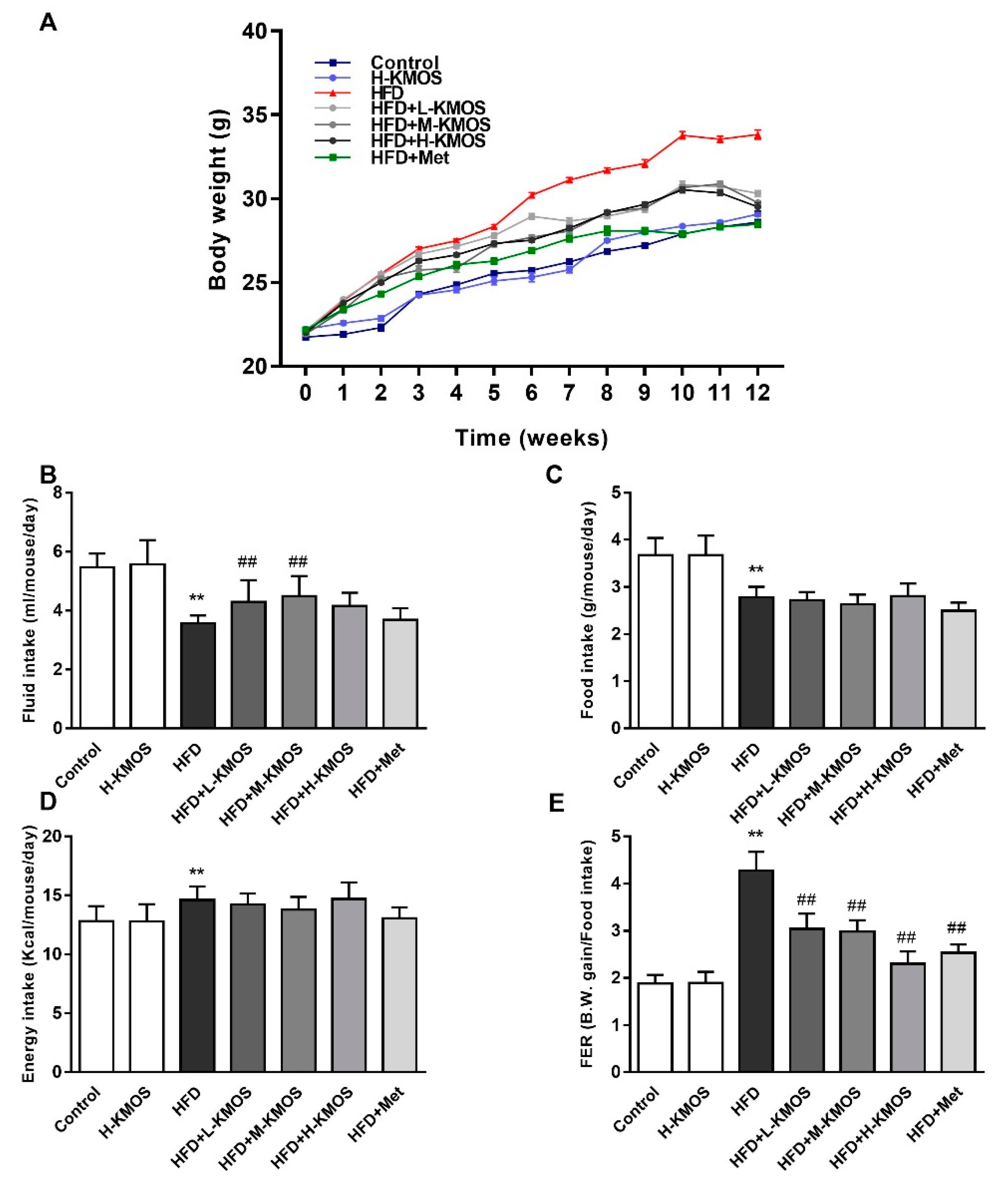
3.1. The Attenuation Effect of KMOS on Body Weight Gain in HFD-Fed C57BL/6J Mice
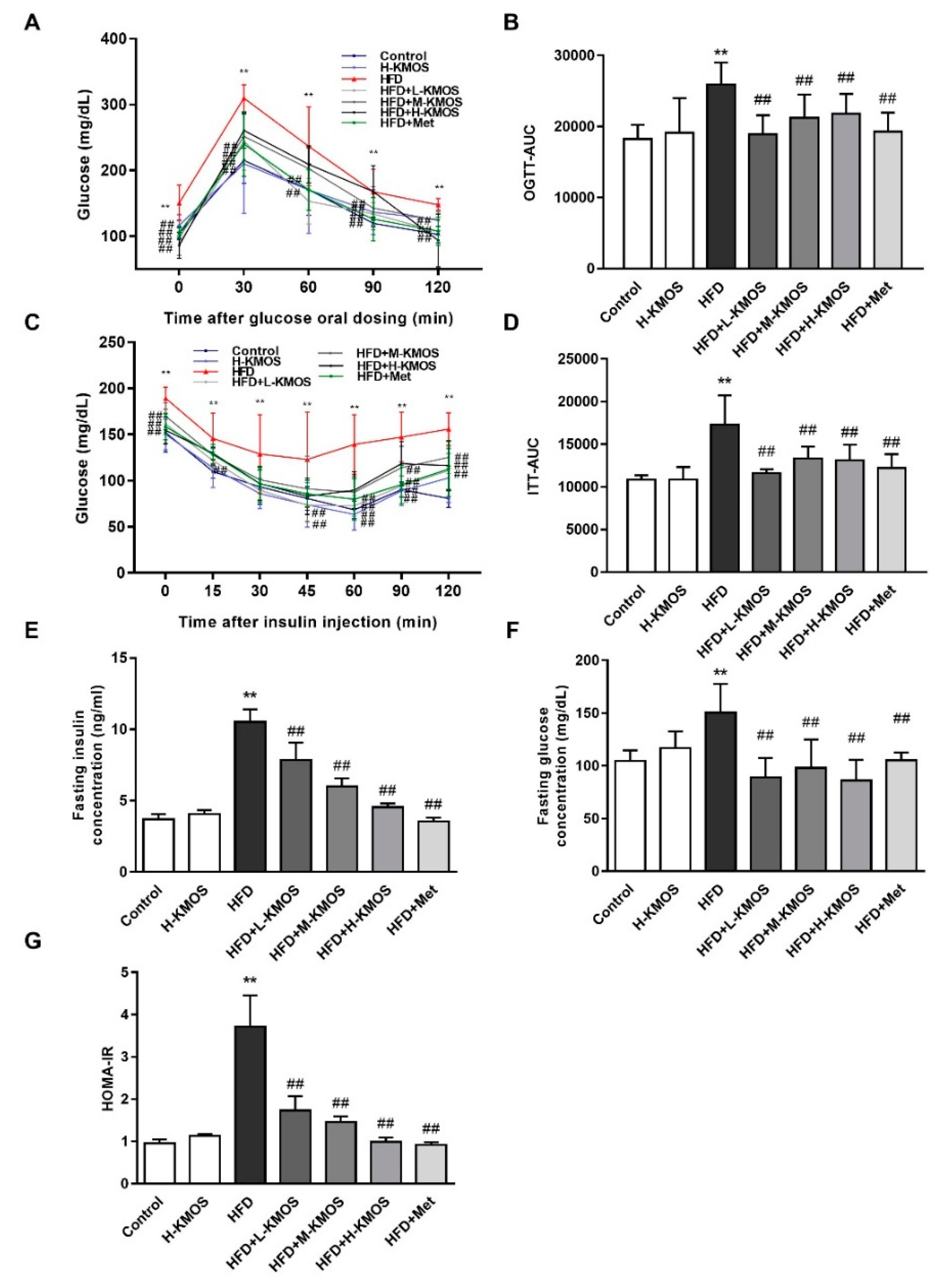
3.2. Improvement of Glucose Sensitivity and Insulin Sensitivity in HFD-Fed Mice Due to KMOS
3.3. Alleviation of Liver Damage and Hepatic Glucose Metabolism Disorder by KMOS
3.4. Regulation Effect of KMOS on Glucose Metabolism via the AMPK Pathway and Insulin Signaling Pathway
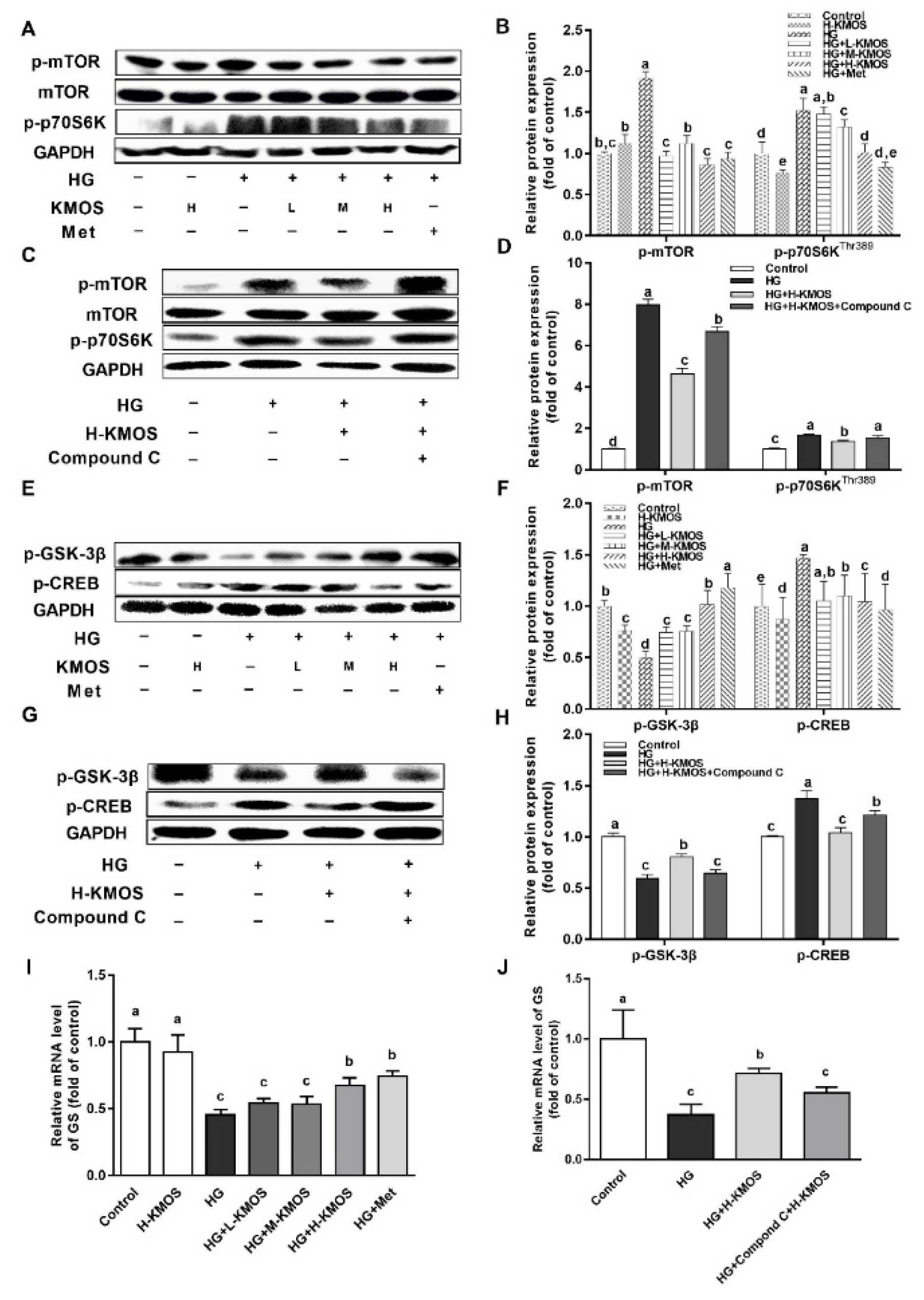
3.5. Management of KMOS with Respect to mTOR and the Glycogen Metabolic Pathway via the AMPK Pathway
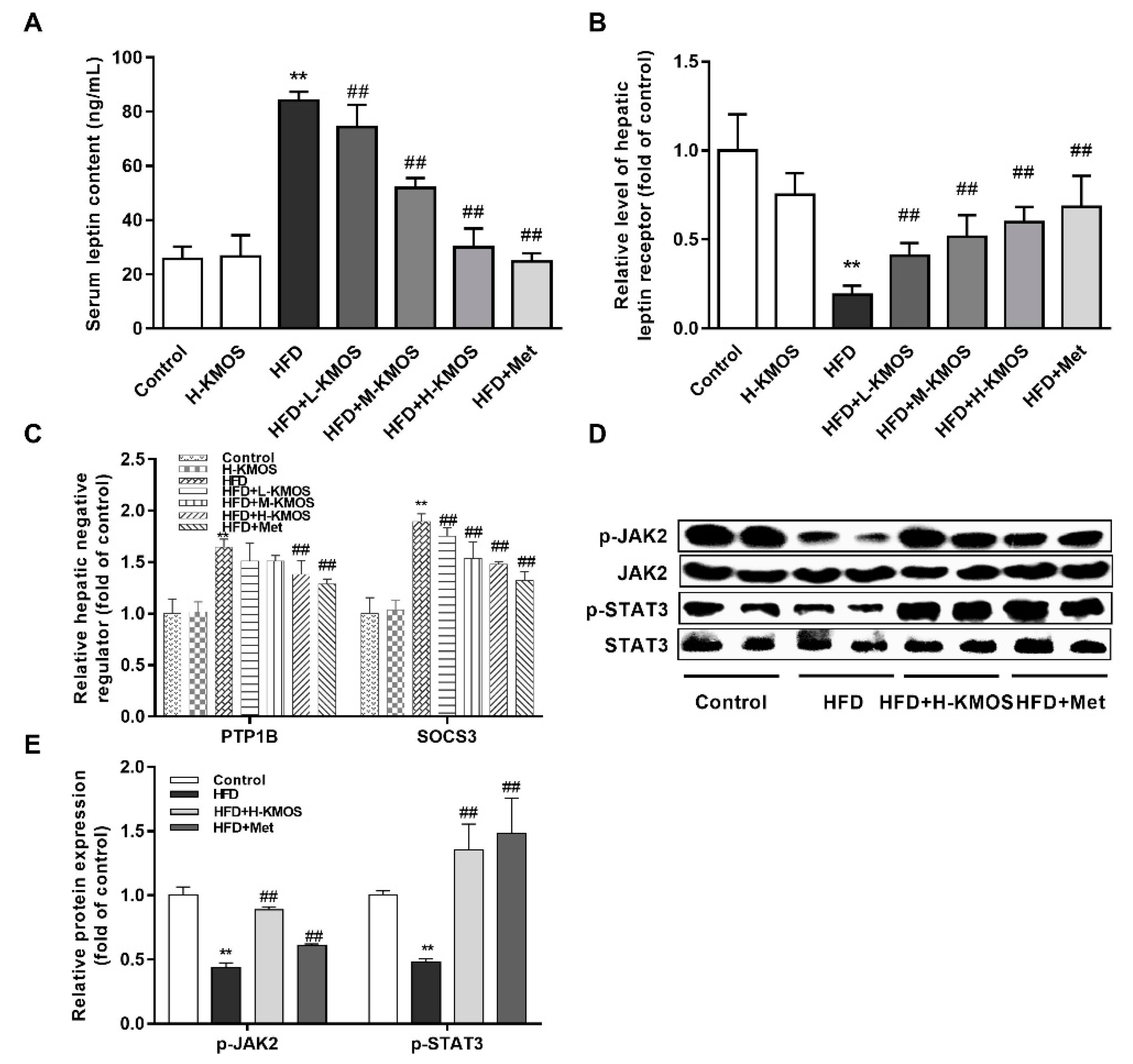
3.6. Alleviation of Hepatic Leptin Resistance by KMOS
3.7. Protective Effect of KMOS against Leptin Resistance and Abnormal Metabolism in the Brain
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredient | D12450B (Control) | D12492 (HFD) | ||
|---|---|---|---|---|
| g | kcal | g | kcal | |
| Casein, 80 Mesh | 200 | 800 | 200 | 800 |
| L-Cystine | 3 | 12 | 3 | 12 |
| Corn Starch | 315 | 1260 | 0 | 0 |
| Maltodextrin 10 | 35 | 1260 | 125 | 500 |
| Sucrose | 350 | 1400 | 68.8 | 275.2 |
| Cellulose, BW200 | 50 | 0 | 50 | 0 |
| Soybean Oil | 25 | 225 | 25 | 225 |
| Lard | 20 | 180 | 245 | 2205 |
| Mineral Mix, S10026 | 10 | 0 | 10 | 0 |
| Dicalcium Phosphate | 13 | 0 | 13 | 0 |
| Calcium Carbonate | 5.5 | 0 | 5.5 | 0 |
| Potassium Citrate, 1 H2O | 16.5 | 0 | 16.5 | 0 |
| Vitamin Mix, V10001 | 10 | 40 | 10 | 40 |
| Choline Bitartrate | 2 | 0 | 2 | 0 |
| FD&C Blue Dye #1 | 0.05 | 0 | 0.05 | 0 |
| Total (g) and (kcal) | 1055.05 | 4057 | 773.85 | 4057 |
| Energy | % | % | % | % |
| Protein | 19.2 | 20 | 26.2 | 20 |
| Carbohydrate | 67.3 | 70 | 26.3 | 20 |
| Fat | 4.3 | 10 | 34.9 | 60 |
| Energy, kcal/g | 3.85 | 5.24 | ||
References
- Evert, A.B.; Boucher, J.L.; Cypress, M.; Dunbar, S.A.; Franz, M.J.; Mayer-Davis, E.J.; Neumiller, J.J.; Nwankwo, R.; Verdi, C.L.; Urbanski, P.; et al. Nutrition therapy recommendations for the management of adults with diabetes. Diabetes Care 2014, 37, S120–S143. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.A.; Keogh, J.B.; Clifton, P.M. Probiotics, prebiotics, synbiotics and insulin sensitivity. Nutr. Res. Rev. 2018, 31, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kubota, N.; Kadowaki, T. Imbalanced Insulin Actions in Obesity and Type 2 Diabetes: Key Mouse Models of Insulin Signaling Pathway. Cell Metab. 2017, 25, 797–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, A.; Handelsman, Y.; Heile, M.; Shannon, M. Burden of Illness in Type 2 Diabetes Mellitus. J. Manag. Care Spéc. Pharm. 2018, 24, S5–S13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y.; Dodd, G.T.; Tiganis, T. Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signaling. Trends Pharmacol. Sci. 2015, 36, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.S.; Kang, L.; Wasserman, D.H. The Extracellular Matrix and Insulin Resistance. Trends Endocrinol. Metab. 2015, 26, 357–366. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Abella, V.; Scotece, M.; Conde, J.; Pino, J.; Gonzalez-Gay, M.A.; Gómez-Reino, J.J.; Mera, A.; Lago, F.; Gómez, R.; Gualillo, O. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol. 2017, 13, 100–109. [Google Scholar] [CrossRef]
- Ye, Z.; Liu, G.; Guo, J.; Su, Z. Hypothalamic endoplasmic reticulum stress as a key mediator of obesity-induced leptin resistance. Obes. Rev. 2018, 19, 770–785. [Google Scholar] [CrossRef]
- Aittomaki, S.; Pesu, M. Therapeutic targeting of the Jak/STAT pathway. Basic Clin. Pharmacol. Toxicol. 2014, 114, 18–23. [Google Scholar] [CrossRef]
- Dodington, D.W.; Desai, H.R.; Woo, M. JAK/STAT—Emerging Players in Metabolism. Trends Endocrinol. Metab. 2018, 29, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Allison, M.B.; Myers, M.G., Jr. 20 years of leptin: Connecting leptin signaling to biological function. J. Endocrinol. 2014, 223, T25–T35. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. The Ascending GLP-1 Road from Clinical Safety to Reduction of Cardiovascular Complications. Diabetes 2018, 67, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Lipska, K.J.; Mayo, H.; Bailey, C.J.; McGuire, D.K. Metformin in patients with type 2 diabetes and kidney disease: A systematic review. JAMA 2014, 312, 2668–2675. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Canfora, E.E.; Blaak, E.E. Gastrointestinal Transit Time, Glucose Homeostasis and Metabolic Health: Modulation by Dietary Fibers. Nutrients 2018, 10, 275. [Google Scholar] [CrossRef]
- Adefegha, S.A. Functional foods and nutraceuticals as dietary intervention in chronic diseases; novel perspectives for health promotion and disease prevention. J. Diet. Suppl. 2018, 15, 977–1009. [Google Scholar] [CrossRef]
- Swennen, K.; Courtin, C.M.; Delcour, J.A. Non-digestible Oligosaccharides with Prebiotic Properties. Crit. Rev. Food Sci. Nutr. 2006, 46, 459–471. [Google Scholar] [CrossRef]
- Ibrahim, O.O. Functional Oligosaccharide: Chemicals Structure, Manufacturing, Health Benefits, Applications and Regulations. J. Food Chem. Nanotechnol. 2018, 4, 65–76. [Google Scholar] [CrossRef]
- Hong, S.J.; Lee, J.-H.; Kim, E.J.; Yang, H.J.; Park, J.-S.; Hong, S.-K. Anti-Obesity and Anti-Diabetic Effect of Neoagarooligosaccharides on High-Fat Diet-Induced Obesity in Mice. Mar. Drugs 2017, 15, 90. [Google Scholar] [CrossRef]
- Wu, X.; Wang, J.; Shi, Y.; Chen, S.; Yan, Q.; Jiang, Z.; Jing, H. N-Acetyl-chitobiose ameliorates metabolism dysfunction through Erk/p38 MAPK and histone H3 phosphorylation in type 2 diabetes mice. J. Funct. Foods 2017, 28, 96–105. [Google Scholar] [CrossRef]
- Behera, S.S.; Ray, R.C. Konjac glucomannan, a promising polysaccharide of Amorphophallus konjac K. Koch in health care. Int. J. Biol. Macromol. 2016, 92, 942–956. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Watanabe, T.; Matsuda, K. Studies on the Chemical Structure of Konjac Mannan. Agric. Biol. Chem. 1970, 34, 532–539. [Google Scholar]
- Wang, H.; Zhang, X.; Wang, S.; Li, H.; Lu, Z.; Shi, J.; Xu, Z. Mannan-oligosaccharide modulates the obesity and gut microbiota in high-fat diet-fed mice. Food Funct. 2018, 9, 3916–3929. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, Y.; Zhang, B. The effects of konjac oligosaccharide on TNBS-induced colitis in rats. Int. Immunopharmacol. 2016, 40, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Kiyohara, H.; Matsuzaki, T.; Yamada, H. Intestinal Peyer’s patch-immunomodulating glucomannans from rhizomes of Anemarrhena asphodeloides Bunge. Phytochemistry 2013, 96, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, S.; Yan, Q.; Li, Y.; Jiang, Z. Effect of Konjac mannan oligosaccharides on diphenoxylate-induced constipation in mice. J. Funct. Foods 2019, 57, 399–407. [Google Scholar] [CrossRef]
- Lu, X.-J.; Chen, X.-M.; Fu, D.-X.; Cong, W.; Ouyang, F. Effect of Amorphophallus Konjac oligosaccharides on STZ-induced diabetes model of isolated islets. Life Sci. 2002, 72, 711–719. [Google Scholar] [CrossRef]
- Zheng, J.; Li, H.; Zhang, X.; Jiang, M.; Luo, C.; Lü, Z.; Xu, Z.-H.; Shi, J.-S. Prebiotic Mannan-Oligosaccharides Augment the Hypoglycemic Effects of Metformin in Correlation with Modulating Gut Microbiota. J. Agric. Food Chem. 2018, 66, 5821–5831. [Google Scholar] [CrossRef]
- Liu, Z.; Patil, I.Y.; Jiang, T.; Sancheti, H.; Walsh, J.P.; Stiles, B.L.; Yin, F.; Cadenas, E. High-Fat Diet Induces Hepatic Insulin Resistance and Impairment of Synaptic Plasticity. PLoS ONE 2015, 10, e0128274. [Google Scholar] [CrossRef]
- Mokadem, M.; Zechner, J.F.; Margolskee, R.F.; Drucker, D.J.; Aguirre, V. Effects of Roux-en-Y gastric bypass on energy and glucose homeostasis are preserved in two mouse models of functional glucagon-like peptide-1 deficiency. Mol. Metab. 2013, 3, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Wang, Y.; Du, Q.; Liu, Z.; Liu, X. Cichoric Acid Reverses Insulin Resistance and Suppresses Inflammatory Responses in the Glucosamine-Induced HepG2 Cells. J. Agric. Food Chem. 2015, 63, 10903–10913. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Song, J.; Du, M.; Mao, X. Bovine α-Lactalbumin Hydrolysates (α-LAH) Ameliorate Adipose Insulin Resistance and Inflammation in High-Fat Diet-Fed C57BL/6J Mice. Nutrients 2018, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Tierney, A.C.; McMonagle, J.; Shaw, D.I.; Gulseth, H.L.; Helal, O.; Saris, W.H.; Paniagua, J.A.; Golabek-Leszczynska, I.; Defoort, C.; Williams, C.M.; et al. Effects of dietary fat modification on insulin sensitivity and on other risk factors of the metabolic syndrome—LIPGENE: A European randomized dietary intervention study. Int. J. Obes. 2011, 35, 800–809. [Google Scholar] [CrossRef]
- Kahn, S.E.; Prigeon, R.L.; Schwartz, R.S.; Fujimoto, W.Y.; Knopp, R.H.; Brunzell, J.D.; Porte, D. Obesity, body fat distribution, insulin sensitivity and Islet beta-cell function as explanations for metabolic diversity. J. Nutr. 2001, 131, 354S–360S. [Google Scholar] [CrossRef]
- Rosqvist, F.; Iggman, D.; Kullberg, J.; Cedernaes, J.; Johansson, H.-E.; Larsson, A.; Johansson, L.; Ahlström, H.; Arner, P.; Dahlman, I.; et al. Overfeeding Polyunsaturated and Saturated Fat Causes Distinct Effects on Liver and Visceral Fat Accumulation in Humans. Diabetes 2014, 63, 2356–2368. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes Association. 2. Classification and diagnosis of diabetes. Diabetes Care 2015, 38 (Suppl. 1), S8–S16. [Google Scholar] [CrossRef]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [Green Version]
- Asemi, Z.; Bahmani, S.; Shakeri, H.; Jamal, A.; Faraji, A.M. Effect of multispecies probiotic supplements on serum minerals, liver enzymes and blood pressure in patients with type 2 diabetes. Int. J. Diabetes Dev. C. 2015, 35, 90–95. [Google Scholar] [CrossRef]
- Lallukka, S.; Yki-Järvinen, H. Non-alcoholic fatty liver disease and risk of type 2 diabetes. Best Pr. Res. Clin. Endocrinol. Metab. 2016, 30, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Tsai, L.T.-Y.; Rosen, E.D. Nuclear Mechanisms of Insulin Resistance. Trends Cell Biol. 2016, 26, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Hu, J.; Zhao, W.; Gao, X.; Jiang, C.; Liu, K.; Liu, B.; Huang, F. Quercetin differently regulates insulin-mediated glucose transporter 4 translocation under basal and inflammatory conditions in adipocytes. Mol. Nutr. Food Res. 2014, 58, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Ducommun, S.; Quan, C.; Xie, B.; Li, M.; Wasserman, D.H.; Sakamoto, K.; Mackintosh, C.; Chen, S. AS160 deficiency causes whole-body insulin resistance via composite effects in multiple tissues. Biochem. J. 2013, 449, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Koay, A.; Rimmer, K.A.; Mertens, H.D.; Gooley, P.R.; Stapleton, D. Oligosaccharide recognition and binding to the carbohydrate binding module of AMP-activated protein kinase. FEBS Lett. 2007, 581, 5055–5059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mobbs, J.I.; Koay, A.; Di Paolo, A.; Bieri, M.; Petrie, E.J.; Gorman, M.A.; Doughty, L.; Parker, M.W.; Stapleton, D.I.; Griffin, M.D.; et al. Determinants of oligosaccharide specificity of the carbohydrate-binding modules of AMP-activated protein kinase. Biochem. J. 2015, 468, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Thong, F.S.; Bilan, P.J.; Klip, A. The Rab GTPase-Activating Protein AS160 Integrates Akt, Protein Kinase C, and AMP-Activated Protein Kinase Signals Regulating GLUT4 Traffic. Diabetes 2007, 56, 414–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.-P.; Le, L.; Zhai, W.; Wan, W.; Hu, K.; Yong, P.; He, C.; Xu, L.; Xiao, P. Protective effects of marein on high glucose-induced glucose metabolic disorder in HepG2 cells. Phytomedicine 2016, 23, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Kezic, A.; Popovic, L.; Lalic, K. mTOR inhibitor therapy and metabolic consequences: Where do we stand? Oxid. Med. Cell. Longev. 2018, 2018, 2640342. [Google Scholar] [CrossRef] [PubMed]
- Bautista, S.J.; Boras, I.; Vissa, A.; Mecica, N.; Yip, C.M.; Kim, P.K.; Antonescu, C.N. mTOR complex 1 controls the nuclear localization and function of glycogen synthase kinase 3β. J. Biol. Chem. 2018, 293, 14723–14739. [Google Scholar] [CrossRef]
- Yuan, H.-D.; Piao, G.-C. An Active Part of Artemisia sacrorum Ledeb. Suppresses Gluconeogenesis through AMPK Mediated GSK3β and CREB Phosphorylation in Human HepG2 Cells. Biosci. Biotechnol. Biochem. 2011, 75, 1079–1084. [Google Scholar] [CrossRef]
- Coppari, R.; Ichinose, M.; Lee, C.E.; Pullen, A.E.; Kenny, C.D.; McGovern, R.A.; Tang, V.; Liu, S.M.; Ludwig, T.; Chua, S.C.; et al. The hypothalamic arcuate nucleus: A key site for mediating leptin’s effects on glucose homeostasis and locomotor activity. Cell Metab. 2005, 1, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Wang, M.Y.; Kakuma, T.; Wang, Z.W.; Babcock, E.; McCorkle, K.; Higa, M.; Zhou, Y.T.; Unger, R.H. Liporegulation in diet-induced obesity. The antisteatotic role of hyperleptinemia. J. Biol. Chem. 2001, 276, 5629–5635. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Fu, C.; Huang, L.; Jiang, Y.; Deng, X.; Guo, J.; Su, Z. Anti-Obesity Effect of Chitosan Oligosaccharide Capsules (COSCs) in Obese Rats by Ameliorating Leptin Resistance and Adipogenesis. Mar. Drugs 2018, 16, 198. [Google Scholar] [CrossRef] [PubMed]
- Aberdein, N.; Dambrino, R.J.; do Carmo, J.M.; Wang, Z.; Mitchell, L.E.; Drummond, H.A.; Hall, J.E. Role of PTP1B in POMC neurons during chronic high-fat diet: Sex differences in regulation of liver lipids and glucose tolerance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R478–R488. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Wang, Z.; Wan, W. Suppressor of Cytokine Signaling 3: Emerging Role Linking Central Insulin Resistance and Alzheimer’s Disease. Front. Mol. Neurosci. 2018, 12, 417. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.; Herzog, H.; Shi, Y.-C. Regulation of energy homeostasis by the NPY system. Trends Endocrinol. Metab. 2015, 26, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Norman, E.D.; Lee, K.; Cutler, R.G.; Telljohann, R.S.; Egan, J.M.; Mattson, M.P. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 2008, 18, 1085–1088. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Sun, Y.; Qiao, Q.; Zhao, T.; Zhang, W.; Ren, B.; Liu, Q.; Liu, X. Sesamol ameliorates high-fat and high-fructose induced cognitive defects via improving insulin signaling disruption in the central nervous system. Food Funct. 2017, 8, 710–719. [Google Scholar] [CrossRef]







| Forward Primer | Reverse Primer | |
|---|---|---|
| Mouse-Sirt1 | TCTGTCTCCTGTGGGATTCC | GATGCTGTTGCAAAGGAACC |
| Mouse-Tfam | GCTTCCAGGGGGCTAAGGAT | CCCAATCCCAATGACAACTC |
| Mouse-Pgc1-α | GAAAGGGCCAAACAGAGAGA | GTAAATCACACGGCGCTCTT |
| Mouse-Gs | CCATGAACAGCAAGGGTTGTAA | TGGAAGTGGGCAACCACATA |
| Mouse-Glut2 | TCAGAAGACAAGATCACCGGA | GCTGGTGTGACTGTAAGTGGG |
| Mouse-Gapdh | TGGAGAAACCTGCCAAGTATGA | TGGAGAAACCTGCCAAGTATGA |
| Human-Gs | GCCTTTCCAGAGCACTTCAC | CTCCTCGTCCTCATCGTAGC |
| Human-β-actin | TGGATCAGCAAGCAGGAGTA | TCGGCCACATTGTGAACTTT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, D.; Yan, Q.; Li, Y.; Liu, J.; Liu, H.; Jiang, Z. Effect of Konjac Mannan Oligosaccharides on Glucose Homeostasis via the Improvement of Insulin and Leptin Resistance In Vitro and In Vivo. Nutrients 2019, 11, 1705. https://doi.org/10.3390/nu11081705
Zhu D, Yan Q, Li Y, Liu J, Liu H, Jiang Z. Effect of Konjac Mannan Oligosaccharides on Glucose Homeostasis via the Improvement of Insulin and Leptin Resistance In Vitro and In Vivo. Nutrients. 2019; 11(8):1705. https://doi.org/10.3390/nu11081705
Chicago/Turabian StyleZhu, Di, Qiaojuan Yan, Yanxiao Li, Jun Liu, Haijie Liu, and Zhengqiang Jiang. 2019. "Effect of Konjac Mannan Oligosaccharides on Glucose Homeostasis via the Improvement of Insulin and Leptin Resistance In Vitro and In Vivo" Nutrients 11, no. 8: 1705. https://doi.org/10.3390/nu11081705
APA StyleZhu, D., Yan, Q., Li, Y., Liu, J., Liu, H., & Jiang, Z. (2019). Effect of Konjac Mannan Oligosaccharides on Glucose Homeostasis via the Improvement of Insulin and Leptin Resistance In Vitro and In Vivo. Nutrients, 11(8), 1705. https://doi.org/10.3390/nu11081705