The Autophagy Signaling Pathway: A Potential Multifunctional Therapeutic Target of Curcumin in Neurological and Neuromuscular Diseases

 , and
, and 
Abstract
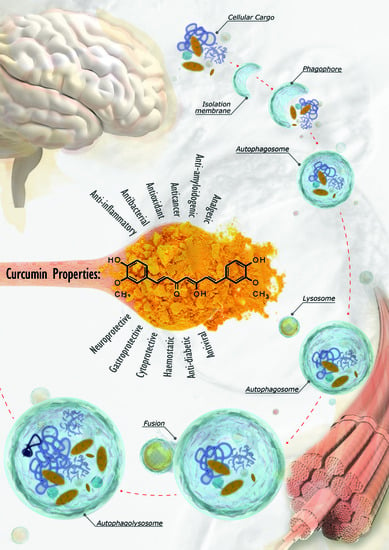
:
{kind=link}
{kind=link}
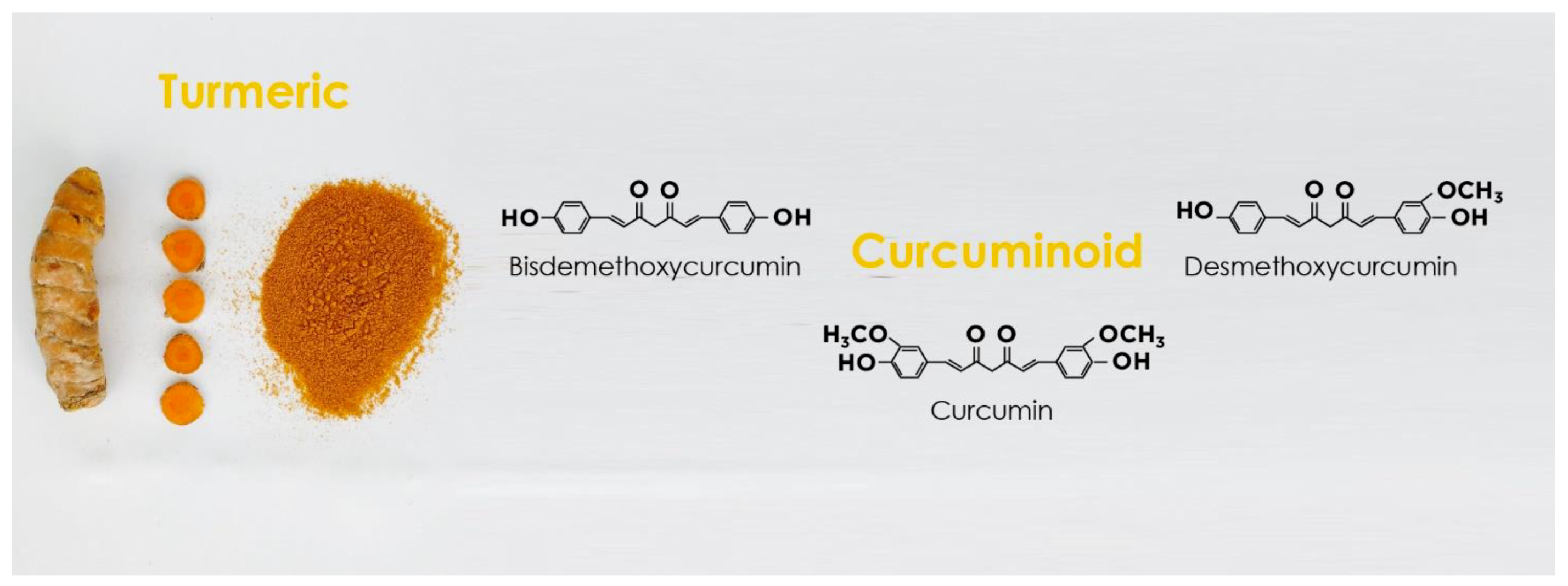{kind=link}
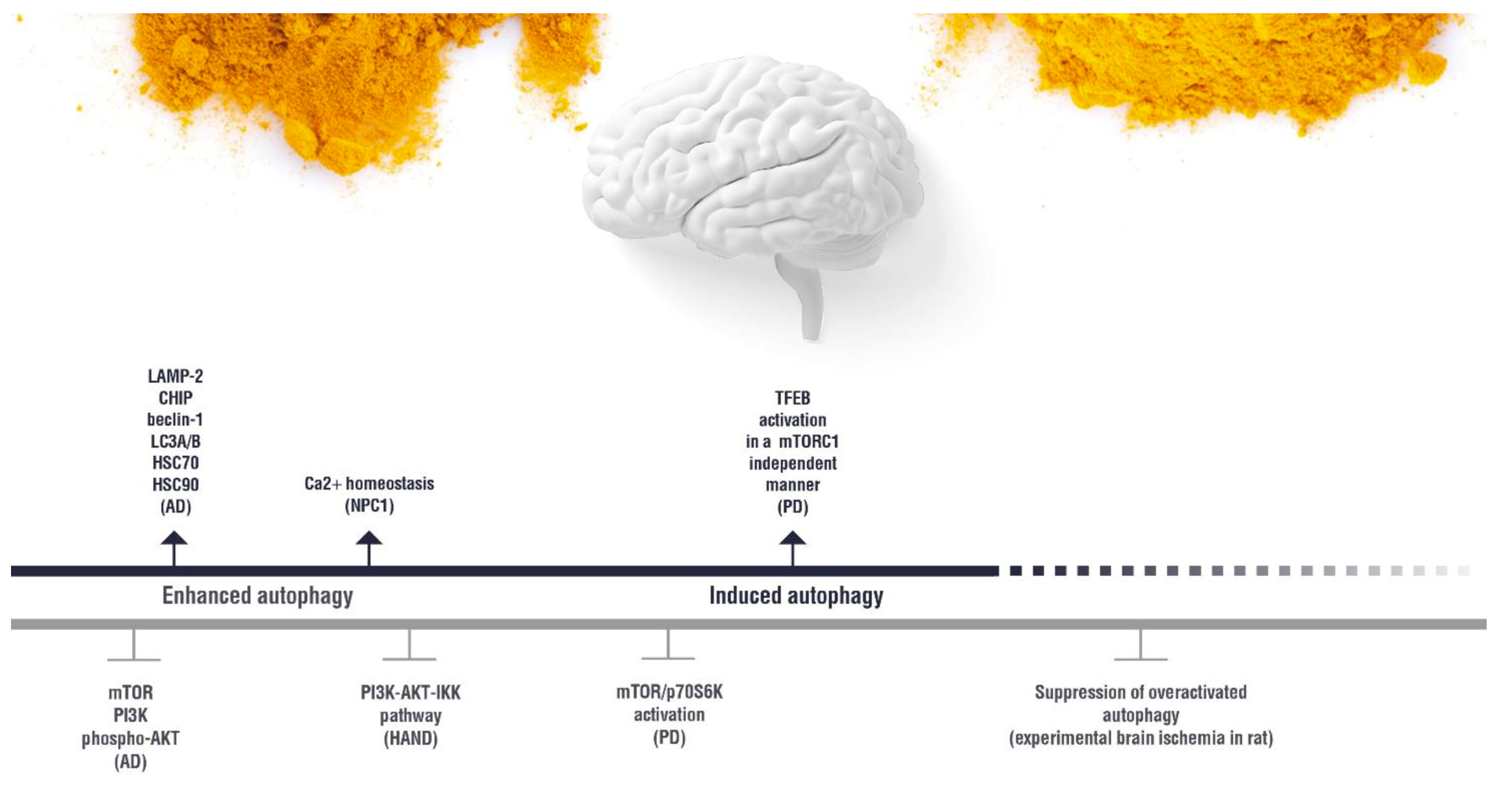{kind=link}
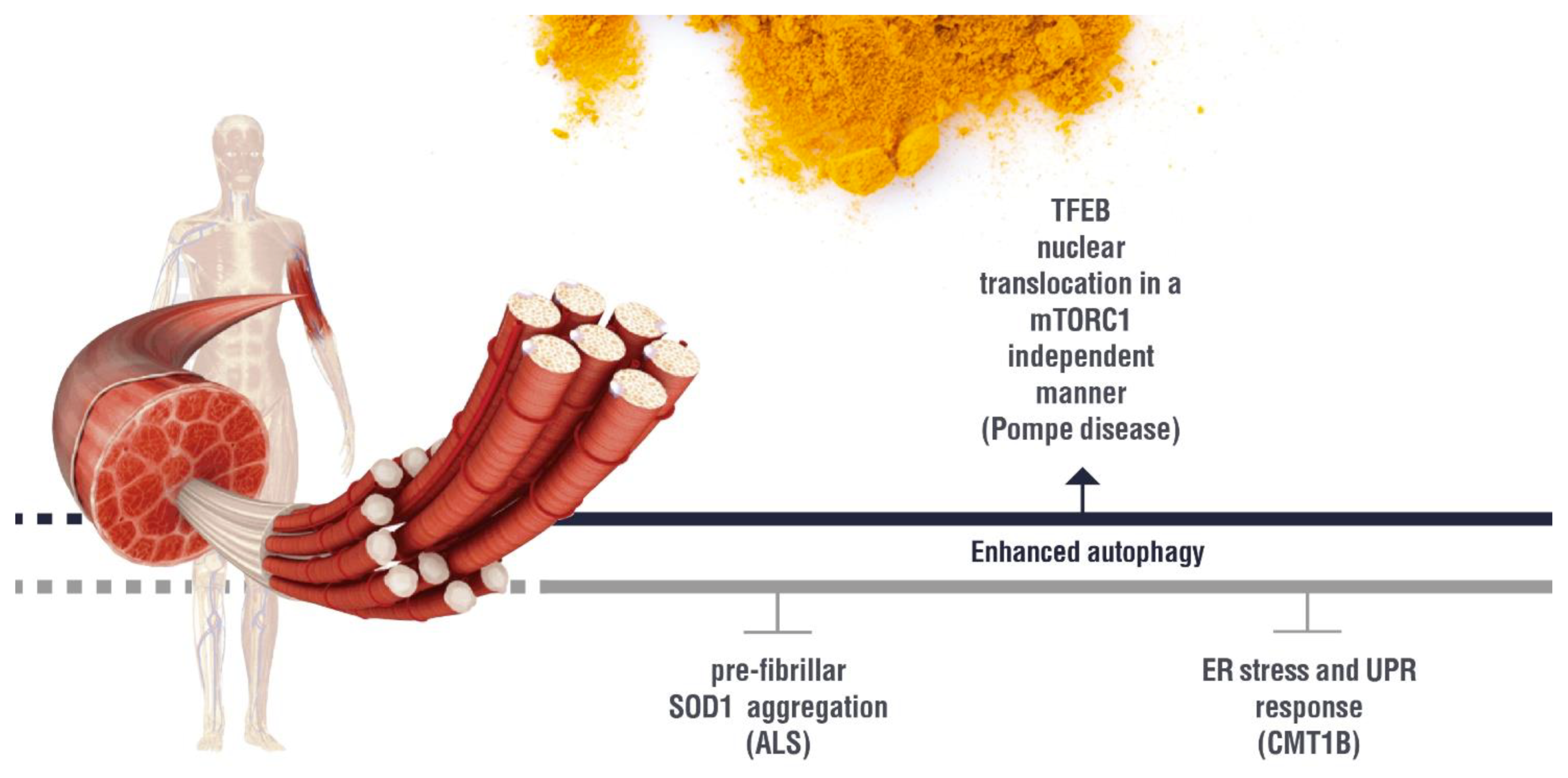{kind=link}
1. Introduction
2. Autophagy: Mechanisms and Regulation
3. Curcumin Structure and Activity
4. Autophagy Modulation and the Interplay between Autophagy and Curcumin as a Therapeutic Approach for Neurological Disorders
4.1. Neurodegenerative Disorders
4.2. Other Neurological Conditions
5. Effect of Curcumin on the Autophagy Pathways in Neuromuscular Diseases
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as “Curecumin”: From kitchen to clinic. Biochem. Pharmacol. 2008, 75, 787–809. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, A.; Hosseinzadeh, H. Antidotal or protective effects of Curcuma longa (turmeric) and its active ingredient, curcumin, against natural and chemical toxicities: A review. Biomed. Pharmacother. 2018, 99, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Frautschy, S.A.; Cole, G.M.; Masterman, D.L.; Cummings, J.L. A potential role of the curry spice curcumin in Alzheimer’s disease. Curr. Alzheimer Res. 2005, 2, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, A.; Cicero, A.; Panahi, Y.; Sahebkar, A. Curcumin: A naturally occurring autophagy modulator. J. Cell Physiol. 2019, 234, 5643–5654. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, V.; Sahebkar, A.; Hosseinzadeh, H. Turmeric (Curcuma longa) and its major constituent (curcumin) as nontoxic and safe substances: Review. Phytother.Res. 2018, 32, 985–995. [Google Scholar] [CrossRef]
- Aggarwal, B.; Sung, B. Pharmacological basis for the role of curcumin in chronic diseases: An age-old spice with modern targets. Trends Pharmacol. Sci. 2009, 30, 85–94. [Google Scholar] [CrossRef]
- Vidoni, C.; Castiglioni, A.; Seca, C.; Secomandi, E.; Melone, M.; Isidoro, C. Dopamine exacerbates mutant Huntingtin toxicity via oxidative-mediated inhibition of autophagy in SH-SY5Y neuroblastoma cells: Beneficial effects of anti-oxidant therapeutics. Neurochem. Int. 2016, 101, 132–143. [Google Scholar] [CrossRef]
- Vidoni, C.; Secomandi, E.; Castiglioni, A.; Melone, M.; Isidoro, C. Resveratrol protects neuronal-like cells expressing mutant Huntingtin from dopamine toxicity by rescuing ATG4-mediated autophagosome formation. Neurochem. Int. 2018, 117, 174–187. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.; Ballabio, A.; Boya, P.; Bravo, S.P.J.; Cecconi, F.; Choi, A.; Chu, C.; Codogno, P.; Colombo, M. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Guo, S.; Long, M.; Li, X.; Zhu, S.; Zhang, M.; Yang, Z. Curcumin activates autophagy and attenuates oxidative damage in EA.hy926 cells via the Akt/mTOR pathway. Mol.Med.Rep. 2016, 13, 2187–2193. [Google Scholar] [CrossRef]
- Salehi, B.; Stojanović, R.Z.; Matejić, J.; Sharifi, R.M.; Anil, K.N.; Martins, N.; Sharifi, R.J. The therapeutic potential of curcumin: A review of clinical trials. Eur.J.Med. Chem. 2019, 163, 527–545. [Google Scholar] [CrossRef]
- Sridhar, S.; Botbol, Y.; Macian, F.; Cuervo, A. Autophagy and disease: Always two sides to a problem. J. Pathol. 2012, 226, 255–273. [Google Scholar] [CrossRef]
- Lin, S.; Tsai, M.; Cheng, H.; Weng, C. Natural Compounds from Herbs that can Potentially Execute as Autophagy Inducers for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1412. [Google Scholar] [CrossRef]
- Bielak-Zmijewska, A.; Grabowska, W.; Ciolko, A.; Bojko, A.; Mosieniak, G.; Bijoch, Ł.; Sikora, E. The Role of Curcumin in the Modulation of Ageing. Int. J. Mol. Sci. 2019, 20, 1239. [Google Scholar] [CrossRef]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: Pathogenesis and therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat.Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Baehrecke, E.H. Autophagy: Dual roles in life and death? Nat.Rev.Mol. Cell Biol. 2005, 6, 505–510. [Google Scholar] [CrossRef]
- Edinger, A.; Thompson, C. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. [Google Scholar] [CrossRef]
- Puri, C.; Renna, M.; Bento, C.; Moreau, K.; Rubinsztein, D. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 2013, 154, 1285–1299. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Ohsumi, Y.; Mizushima, N. Two ubiquitin-like conjugation systems essential for autophagy. Semin. Cell Dev. Biol. 2004, 15, 231–236. [Google Scholar] [CrossRef]
- Jung, C.; Jun, C.; Ro, S.; Kim, Y.; Otto, N.; Cao, J.; Kundu, M.; Kim, D. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell. 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Fan, W.; Nassiri, A.; Zhong, Q. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L). Proc. Natl. Acad. Sci. USA 2011, 108, 7769–7774. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef]
- Shao, Y.; Gao, Z.; Feldman, T.; Jiang, X. Stimulation of ATG12-ATG5 conjugation by ribonucleic acid. Autophagy 2007, 3, 10–16. [Google Scholar] [CrossRef]
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell. 2008, 19, 4651–4659. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Ravikumar, B.; Acevedo-Arozena, A.; Imarisio, S.; Berger, Z.; Vacher, C.; O’Kane, C.; Brown, S.; Rubinsztein, D. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat.Genet. 2005, 37, 771–776. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy: In sickness and in health. Trends Cell Biol. 2004, 14, 70–77. [Google Scholar] [CrossRef]
- Meléndez, A.; Tallóczy, Z.; Seaman, M.; Eskelinen, E.; Hall, D.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef]
- Hong, C.J.; Park, H.; Yu, S.W. Autophagy for the quality control of adult hippocampal neural stem cells. Brain Res. 2016, 1649, 166–172. [Google Scholar] [CrossRef]
- Scherz, S.R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12, 1509–1518. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.; Valenza, M.; Gennarino, V.; di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Perrone, L.; Devi, T.S.; Hosoya, K.; Terasaki, T.; Singh, L.P. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J. Cell Physiol. 2009, 221, 262–272. [Google Scholar] [CrossRef]
- Perrone, L.; Devi, T.S.; Hosoya, K.I.; Terasaki, T.; Singh, L.P. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis. 2010, 1, e65. [Google Scholar] [CrossRef]
- Perrone, L.; Sbai, O.; Nawroth, P.P.; Bierhaus, A. The Complexity of Sporadic Alzheimer’s Disease Pathogenesis: The Role of RAGE as Therapeutic Target to Promote Neuroprotection by Inhibiting Neurovascular Dysfunction. Int.J. Alzheimers Dis. 2012, 2012, 734956. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, Y.; Kelly, D.J.; Tan, C.Y.; Gill, A.; Cheng, D.; Braet, F.; Park, J.S.; Sue, C.; Pollock, C.A.; et al. Thioredoxin interacting protein (TXNIP) regulates tubular autophagy and mitophagy in diabetic nephropathy through the mTOR signaling pathway. Sci. Rep. 2016, 6, 29196. [Google Scholar] [CrossRef]
- Qiao, S.; Dennis, M.; Song, X.; Vadysirisack, D.D.; Salunke, D.; Nash, Z.; Yang, Z.; Liesa, M.; Yoshioka, J.; Matsuzawa, S.; et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat. Commun. 2015, 6, 7014. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri, M.M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Cakouros, D. Transcriptional control of the core cell-death machinery. Trends Biochem. Sci. 2004, 29, 193–199. [Google Scholar] [CrossRef]
- Polager, S.; Ofir, M.; Ginsberg, D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008, 27, 4860–4864. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Scherz, S.R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Yan, Y.; Finkel, T. Autophagy as a regulator of cardiovascular redox homeostasis. Free Radic. Biol. Med. 2012, 109, 108–113. [Google Scholar] [CrossRef]
- Massey, A.C.; Zhang, C.; Cuervo, A.M. Chaperone-mediated autophagy in aging and disease. Curr. Top. Dev. Biol. 2006, 73, 205–235. [Google Scholar]
- Pellacani, C.; Costa, L.G. Role of autophagy in environmental neurotoxicity. Environ. Pollut. 2018, 235, 791–805. [Google Scholar] [CrossRef]
- Vidoni, C.; Follo, C.; Savino, M.; Melone, M.A.; Isidoro, C. The Role of Cathepsin D in the Pathogenesis of Human Neurodegenerative Disorders. Med.Res.Rev. 2016, 36, 845–870. [Google Scholar] [CrossRef]
- Priyadarsini, K.I. The chemistry of curcumin: From extraction to therapeutic agent. Molecules 2014, 19, 20091–20112. [Google Scholar] [CrossRef]
- Sharma, O.P. Antioxidant activity of curcumin and related compounds. Biochem. Pharmacol. 1976, 25, 1811–1812. [Google Scholar] [CrossRef]
- Hewlings, S.J.; Kalman, D.S. Curcumin: A Review of Its’ Effects on Human Health. Foods 2017, 6, 92. [Google Scholar] [CrossRef]
- Manson, M.M. Inhibition of survival signalling by dietary polyphenols and indole-3-carbinol. Eur.J. Cancer 2005, 41, 1842–1853. [Google Scholar] [CrossRef]
- Esposito, T.; Schettino, C.; Polverino, P.; Allocca, S.; Adelfi, L.; D’Amico, A.; Capaldo, G.; Varriale, B.; Di Salle, A.; Peluso, G.; et al. Synergistic Interplay between Curcumin and Polyphenol-Rich Foods in the Mediterranean Diet: Therapeutic Prospects for Neurofibromatosis 1 Patients. Nutrients 2017, 9, 783. [Google Scholar] [CrossRef]
- Squillaro, T.; Schettino, C.; Sampaolo, S.; Galderis, I.U.; Di Iorio, G.; Giordano, A.; Melone, M.A.B. Adult-onset brain tumors and neurodegeneration: Are polyphenols protective? J. Cell Physiol. 2017, 233, 3955–3967. [Google Scholar] [CrossRef]
- Wojcik, M.; Krawczyk, M.; Wojcik, P.; Cypryk, K.; Wozniak, L.A. Molecular Mechanisms Underlying Curcumin-Mediated Therapeutic Effects in Type 2 Diabetes and Cancer. Oxid.Med. Cell Longev. 2018, 2018, 9698258. [Google Scholar] [CrossRef]
- Sharma, R.A.; McLelland, H.R.; Hill, K.A.; Ireson, C.R.; Euden, S.A.; Manson, M.M.; Pirmohamed, M.; Marnett, L.J.; Gescher, A.J.; Steward, W.P. Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin. Cancer Res. 2001, 7, 1894–1900. [Google Scholar]
- Garcia, A.M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacska, I.B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef]
- Maiti, P.; Hall, T.C.; Paladugu, L.; Kolli, N.; Learman, C.; Rossignol, J.; Dunbar, G.L. A comparative study of dietary curcumin, nanocurcumin, and other classical amyloid-binding dyes for labeling and imaging of amyloid plaques in brain tissue of 5×-familial Alzheimer’s disease mice. Histochem. Cell Biol. 2016, 146, 609–625. [Google Scholar] [CrossRef]
- Masuelli, L.; Di Stefano, E.; Fantini, M.; Mattera, R.; Benvenuto, M.; Marzocchella, L.; Sacchetti, P.; Focaccetti, C.; Bernardini, R.; Tresoldi, I.; et al. Resveratrol potentiates the in vitro and in vivo anti-tumoral effects of curcumin in head and neck carcinomas. Oncotarget 2014, 5, 10745–10762. [Google Scholar] [CrossRef]
- Mehanny, M.; Hathout, R.M.; Geneidi, A.S.; Mansour, S. Exploring the use of nanocarrier systems to deliver the magical molecule; Curcumin and its derivatives. J.Control. Release 2016, 225, 1–30. [Google Scholar] [CrossRef]
- Squillaro, T.; Cimini, A.; Peluso, G.; Giordano, A.; Melone, M.A.B. Nano-delivery systems for encapsulation of dietary polyphenols: An experimental approach for neurodegenerative diseases and brain tumors. Biochem. Pharmacol. 2018, 154, 303–317. [Google Scholar] [CrossRef]
- Rajitha, B.; Belalcazar, A.; Nagaraju, G.P.; Shaib, W.L.; Snyder, J.P.; Shoji, M.; Pattnaik, S.; Alam, A.; El-Rayes, B.F. Inhibition of NF-κB translocation by curcumin analogs induces G0/G1 arrest and downregulates thymidylate synthase in colorectal cancer. Cancer Lett. 2016, 373, 227–233. [Google Scholar] [CrossRef]
- Rajitha, B.; Nagaraju, G.P.; Shaib, W.L.; Alese, O.B.; Snyder, J.P.; Shoji, M.; Pattnaik, S.; Alam, A.; El-Rayes, B.F. Novel synthetic curcumin analogs as potent antiangiogenic agents in colorectal cancer. Mol. Carcinog. 2017, 56, 288–299. [Google Scholar] [CrossRef]
- Finicelli, M.; Squillaro, T.; Di Cristo, F.; Di Salle, A.; Melone, M.A.B.; Galderisi, U.; Peluso, G. Metabolic syndrome, Mediterranean diet, and polyphenols: Evidence and perspectives. J. Cell Physiol. 2019, 234, 5807–5826. [Google Scholar] [CrossRef]
- Ye, B.; Wang, Q.; Hu, H.; Shen, Y.; Fan, C.; Chen, P.; Ma, Y.; Wu, H.; Xiang, M. Restoring autophagic flux attenuates cochlear spiral ganglion neuron degeneration by promoting TFEB nuclear translocation via inhibiting MTOR. Autophagy 2019, 15, 998–1016. [Google Scholar] [CrossRef]
- Song, J.X.; Sun, Y.R.; Peluso, I.; Zeng, Y.; Yu, X.; Lu, J.H.; Xu, Z.; Wang, M.Z.; Liu, L.F.; Huang, Y.Y.; et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 2016, 12, 1372–1389. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Mazargui, H.; Lévêque, C.; Bartnik, D.; Fantini, J.; Gouget, T.; Melone, M.A.; Funke, S.A.; Willbold, D.; Perrone, L. A synthetic amino acid substitution of Tyr10 in Aβ peptide sequence yields a dominant negative variant in amyloidogenesis. Aging Cell 2012, 11, 530–541. [Google Scholar] [CrossRef]
- Perrone, L.; Mothes, E.; Vignes, M.; Mockel, A.; Figueroa, C.; Miquel, M.C.; Maddelei, N.M.L.; Falle, R.P. Copper transfer from Cu-Abeta to human serum albumin inhibits aggregation, radical production and reduces Abeta toxicity. Chembiochem 2010, 11, 110–118. [Google Scholar] [CrossRef]
- La Rosa, L.R.; Perrone, L.; Nielsen, M.S.; Calissano, P.; Andersen, O.M.; Matrone, C. Y682G Mutation of Amyloid Precursor Protein Promotes Endo-Lysosomal Dysfunction by Disrupting APP-SorLA Interaction. Front. Cell Neurosci. 2015, 9, 109. [Google Scholar] [CrossRef]
- Melone, M.A.B.; Dato, C.; Paladino, S.; Coppola, C.; Trebini, C.; Giordana, M.T.; Perrone, L. Verapamil Inhibits Ser202/Thr205 Phosphorylation of Tau by Blocking TXNIP/ROS/p38 MAPK Pathway. Pharm.Res. 2018, 35, 44. [Google Scholar] [CrossRef]
- Perrone, L.; Grant, W.B. Observational and ecological studies of dietary advanced glycation end products in national diets and Alzheimer’s disease incidence and prevalence. J. Alzheimers Dis. 2015, 45, 965–979. [Google Scholar] [CrossRef]
- Takalo, M.; Salminen, A.; Soininen, H.; Hiltunen, M.; Haapasalo, A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am.J. Neurodegener. Dis. 2013, 2, 1–14. [Google Scholar]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Kim, D.S. Discovery of natural products from Curcuma longa that protect cells from beta- amyloid insult: A drug discovery effort against Alzheimer’s disease. J.Nat.Prod. 2002, 65, 1227–1231. [Google Scholar] [CrossRef]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef]
- Baum, L.; Ng, A. Curcumin interaction with copper and iron suggests one possible mechanism of action in Alzheimer’s disease animal models. J. Alzheimers Dis. 2004, 6, 367–377. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, X.; Teng, Z.; Zhang, T.; Li, Y. Downregulation of PI3K/Akt/mTOR signaling pathway in curcumin-induced autophagy in APP/PS1 double transgenic mice. Eur.J. Pharmacol. 2014, 740, 312–320. [Google Scholar] [CrossRef]
- Howell, J.C.; Chun, E.; Farrell, A.N.; Hur, E.Y.; Caroti, C.M.; Iuvone, P.M.; Haque, R. Global microRNA expression profiling: Curcumin (diferuloylmethane) alters oxidative stress-responsive microRNAs in human ARPE-19 cells. Mol. Vis. 2013, 19, 544–560. [Google Scholar]
- Tiribuzi, R.; Crispoltoni, L.; Porcellati, S.; Di Lullo, M.; Florenzano, F.; Pirro, M.; Orlacchio, A. MiR128 up-regulation correlates with impaired amyloid β(1–42) degradation in monocytes from patients with sporadic Alzheimer’s disease. Neurobiol. Aging 2014, 35, 345–356. [Google Scholar] [CrossRef]
- Maiti, P.; Rossignol, J.; Dunbar, G.L. Curcumin Modulates Molecular Chaperones and Autophagy-Lysosomal Pathways In Vitro after Exposure to Aβ42. J. Alzheimers Dis. Parkinsonism 2017, 7, 299. [Google Scholar] [CrossRef]
- Jaroonwitchawan, T.; Chaicharoenaudomrung, N.; Namkaew, J.; Noisa, P. Curcumin attenuates paraquat-induced cell death in human neuroblastoma cells through modulating oxidative stress and autophagy. Neurosci. Lett. 2017, 636, 40–47. [Google Scholar] [CrossRef]
- Ma, Q.L.; Zuo, X.; Yang, F.; Ubeda, O.J.; Gant, D.J.; Alaverdyan, M.; Teng, E.; Hu, S.; Chen, P.P.; Maiti, P.; et al. Curcumin suppresses soluble tau dimers and corrects molecular chaperone, synaptic, and behavioral deficits in aged human tau transgenic mice. J. Biol. Chem. 2013, 288, 4056–4065. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, Y.; Cheng, X.; Lian, Y.; Zeng, Z.; Wu, C.; Zhu, H.; Xu, H. The Potential Protective Effect of Curcumin on Amyloid-β-42 Induced Cytotoxicity in HT-22 Cells. Biomed.Res. Int 2018, 2018, 8134902. [Google Scholar] [CrossRef]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS ONE 2010, 5, e9313. [Google Scholar] [CrossRef]
- Lu, J.H.; Tan, J.Q.; Durairajan, S.S.; Liu, L.F.; Zhang, Z.H.; Ma, L.; Shen, H.M.; Chan, H.Y.; Li, M. Isorhynchophylline, a natural alkaloid, promotes the degradation of alpha-synuclein in neuronal cells via inducing autophagy. Autophagy 2012, 8, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Yu, Y.; Li, X.; Ross, C.A.; Smith, W.W. Curcumin protects against A53T alpha-synuclein-induced toxicity in a PC12 induc- ible cell model for Parkinsonism. Pharmacol.Res. 2011, 63, 439–444. [Google Scholar] [CrossRef]
- Jiang, T.F.; Zhang, Y.; Zhou, H.Y.; Wang, H.M.; Tian, L.P.; Liu, J.; Ding, J.Q.; Chen, S.D. Curcumin ameliorates the neurodegenerative pathology in A53T α-synuclein cell model of Parkinson’s disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J. Neuroimmune Pharmacol. 2013, 8, 356–469. [Google Scholar] [CrossRef]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef]
- Ordoñez, M.P.; Steele, J.W. Modeling Niemann Pick type C1 using human embryonic and induced pluripotent stem cells. Brain Res. 2017, 1656, 63–67. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat.Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Ordonez, M.P. Defective mitophagy in human Niemann Pick type C1 neurons is due to abnormal autophagy activation. Autophagy 2012, 8, 1157–1158. [Google Scholar] [CrossRef]
- Lim, J.A.; Kakhlon, O.; Li, L.; Myerowitz, R.; Raben, N. Pompe disease: Shared and unshared features of lysosomal storage disorders. Rare Dis. 2015, 3, e1068978. [Google Scholar] [CrossRef]
- Williams, I.M.; Wallom, K.L.; Smith, D.A.; Al Eisa, N.; Smith, C.; Platt, F.M. Improved neuroprotection using miglustat, curcumin and ibuprofen as a triple combination therapy in Niemann-Pick disease type C1 mice. Neurobiol. Dis. 2014, 67, 9–17. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek, K.M.; Czuczwar, S.J. Neuroprotective and Neurological/Cognitive Enhancement Effects of Curcumin after Brain Ischemia Injury with Alzheimer’s Disease Phenotype. Int. J. Mol. Sci. 2018, 19, 4002. [Google Scholar] [CrossRef]
- Zhang, Y.; Fang, M.; Sun, Y.; Zhang, T.; Shi, N.; Li, J.; Jin, L.; Liu, K.; Fu, J. Curcumin attenuates cerebral ischemia injury in Sprague-Dawley rats and PC12 cells by suppressing overactivated autophagy. J. Photochem. Photobiol. B 2018, 184, 1–6. [Google Scholar] [CrossRef]
- Chen, G.; Liu, S.; Pan, R.; Li, G.; Tang, H.; Jiang, M.; Xing, Y.; Jin, F.; Lin, L.; Dong, J. Curcumin Attenuates gp120-Induced Microglial Inflammation by Inhibiting Autophagy via the PI3K Pathway. Cell Mol. Neurobiol. 2018, 38, 1465–1477. [Google Scholar] [CrossRef]
- Brown, A. Understanding the MIND phenotype: Macrophage microglia inflammation in neurocognitive disorders related to human immunodeficiency virus infection. Clin. Transl.Med. 2015, 4, 7. [Google Scholar] [CrossRef]
- Gonzalez, S.F.; Martin, G.J. The neuropathogenesis of AIDS. Nat.Rev. Immunol. 2005, 5, 69–81. [Google Scholar] [CrossRef]
- Yang, Z.; Zhong, L.; Zhong, S.; Xian, R.; Yuan, B. Hypoxia induces microglia autophagy and neural inflammation injury in focal cer-ebral ischemia model. Exp.Mol. Pathol. 2015, 98, 219–224. [Google Scholar] [CrossRef]
- Bucchia, M.; Ramirez, A.; Parente, V.; Simone, C.; Nizzardo, M.; Magri, F.; Dametti, S.; Corti, S. Therapeutic development in amyotrophic lateral sclerosis. Clin. Ther. 2015, 37, 668–680. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Ezzi, S.A.; Urushitani, M.; Julien, J.P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007, 102, 170–178. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D.; Ohama, E.; Reaume, A.G.; Scott, R.W.; Cleveland, D.W. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef]
- Stepp, M.W.; Folz, R.J.; Yu, J.; Zelko, I.N. The c10orf10 gene product is a new link between oxidative stress and autophagy. Biochim. Biophys. Acta. 2014, 1843, 1076–1088. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [Green Version]
- Buratti, E.; Brindisi, A.; Giombi, M.; Tisminetzky, S.; Ayala, Y.M.; Baralle, F.E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005, 280, 37572–37584. [Google Scholar] [CrossRef]
- Kawahara, Y.; Mieda, S.A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352. [Google Scholar] [CrossRef] [Green Version]
- Kuo, P.H.; Doudeva, L.G.; Wang, Y.T.; Shen, C.K.; Yuan, H.S. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009, 37, 1799–1808. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Kabuta, C.; Kono, K.; Wada, K.; Kabuta, T. 4-Hydroxynonenal induces persistent insolubilization of TDP-43 and alters its intracellular localization. Biochem. Biophys.Res. Commun. 2015, 463, 82–87. [Google Scholar] [CrossRef]
- Oral, O.; Akkoc, Y.; Bayraktar, O.; Gozuacik, D. Physiological and pathological significance of the molecular cross-talk between autophagy and apoptosis. Histol. Histopathol. 2016, 31, 479–498. [Google Scholar]
- Cashman, J.R.; Gagliardi, S.; Lanier, M.; Ghirmai, S.; Abel, K.J.; Fiala, M. Curcumins promote monocytic gene expression related to β-amyloid and superoxide dismutase clearance. Neurodegener. Dis. 2012, 10, 274–276. [Google Scholar] [CrossRef]
- Bhatia, N.K.; Srivastava, A.; Katyal, N.; Jain, N.; Khan, M.A.; Kundu, B.; Deep, S. Curcumin binds to the pre-fibrillar aggregates of Cu/Zn superoxide dismutase (SOD1) and alters its amyloidogenic pathway resulting in reduced cytotoxicity. Biochim. Biophys. Acta 2015, 2015, 426–436. [Google Scholar] [CrossRef]
- Ahmadi, M.; Agah, E.; Nafissi, S.; Jaafari, M.R.; Harirchian, M.H.; Sarraf, P.; Faghihi, K.S.; Hosseini, S.J.; Ghoreishi, A.; Aghamollaii, V.; et al. Safety and Efficacy of Nanocurcumin as Add-On Therapy to Riluzole in Patients With Amyotrophic Lateral Sclerosis: A Pilot Randomized Clinical Trial. Neurotherapeutics 2018, 15, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.B.; Love, J.M.; O’Neill, A.; Lovering, R.M.; Bloch, R.J. Influences of desmin and keratin 19 on passive biomechanical properties of mouse skeletal muscle. J.Biomed. Biotechnol. 2012, 2012, 704061. [Google Scholar] [CrossRef]
- Bär, H.; Mücke, N.; Kostareva, A.; Sjoberg, G.; Aebi, U.; Herrmann, H. Severe muscle disease-causing desmin mutations interfere with in vitro filament assembly at distinct stages. Proc. Natl. Acad. Sci. USA 2005, 102, 15099–15104. [Google Scholar] [CrossRef]
- Capetanaki, Y. Desmin cytoskeleton: A potential regulator of muscle mitochondrial behavior and func- tion. Trends Cardiovasc.Med. 2002, 12, 339–348. [Google Scholar] [CrossRef]
- Cabet, E.; Batonnet, P.S.; Delort, F.; Gausserès, B.; Vicart, P.; Lilienbaum, A. Antioxidant Treatment and Induction of Autophagy Cooperate to Reduce Desmin Aggregation in a Cellular Model of Desminopathy. PLoS ONE 2015, 10, e0137009. [Google Scholar] [CrossRef]
- Patzkó, A.; Bai, Y.; Saporta, M.A.; Katona, I.; Wu, X.; Vizzuso, D.; Feltri, M.L.; Wang, S.; Dillon, L.M.; Kamholz, J.; et al. Curcumin derivatives promote Schwann cell differentiation and improve neuropathy in R98C CMT1B mice. Brain 2012, 135, 3551–3566. [Google Scholar] [CrossRef] [Green Version]
- Khajavi, M.; Inoue, K.; Wiszniewski, W.; Ohyama, T.; Snipes, G.J.; Lupski, J.R. Curcumin treatment abrogates endoplasmic reticulum retention and aggregation-induced apoptosis associated with neuropathy-causing myelin protein zero-truncating mutants. Am.J.Hum.Genet. 2005, 77, 841–850. [Google Scholar] [CrossRef]
- Khajavi, M.; Shiga, K.; Wiszniewski, W.; He, F.; Shaw, C.A.; Yan, J.; Wensel, T.G.; Snipes, G.J.; Lupski, J.R. Oral curcumin mitigates the clinical and neuropathologic phenotype of the Trembler-J mouse: A potential therapy for inherited neuropathy. Am.J.Hum.Genet. 2007, 81, 438–453. [Google Scholar] [CrossRef]
- Spampanato, C.; Feeney, E.; Li, L.; Cardone, M.; Lim, J.A.; Annunziata, F.; Zare, H.; Polishchuk, R.; Puertollano, R.; Parenti, G.; et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 2013, 5, 691–706. [Google Scholar] [CrossRef]


 (induction);
(induction);  (inhibition). AD: Alzheimer’s disease; HAND: HIV-induced neurocognitive disorder; PD: Parkinson’s disease; NPC1: Niemann Pick C1.
(induction); (inhibition). AD: Alzheimer’s disease; HAND: HIV-induced neurocognitive disorder; PD: Parkinson’s disease; NPC1: Niemann Pick C1.
(inhibition). AD: Alzheimer’s disease; HAND: HIV-induced neurocognitive disorder; PD: Parkinson’s disease; NPC1: Niemann Pick C1.
(induction); (inhibition). AD: Alzheimer’s disease; HAND: HIV-induced neurocognitive disorder; PD: Parkinson’s disease; NPC1: Niemann Pick C1. (induction); (inhibition). ALS: Amyotrophic lateral sclerosis; ER:Endoplasmic Reticulum; UPR: Unfolded Protein Response; CMT1B: Charcot Marie Tooth 1B.
(induction); (inhibition). ALS: Amyotrophic lateral sclerosis; ER:Endoplasmic Reticulum; UPR: Unfolded Protein Response; CMT1B: Charcot Marie Tooth 1B.
(induction); (inhibition). ALS: Amyotrophic lateral sclerosis; ER:Endoplasmic Reticulum; UPR: Unfolded Protein Response; CMT1B: Charcot Marie Tooth 1B.
(induction); (inhibition). ALS: Amyotrophic lateral sclerosis; ER:Endoplasmic Reticulum; UPR: Unfolded Protein Response; CMT1B: Charcot Marie Tooth 1B.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrone, L.; Squillaro, T.; Napolitano, F.; Terracciano, C.; Sampaolo, S.; Melone, M.A.B. The Autophagy Signaling Pathway: A Potential Multifunctional Therapeutic Target of Curcumin in Neurological and Neuromuscular Diseases. Nutrients 2019, 11, 1881. https://doi.org/10.3390/nu11081881
Perrone L, Squillaro T, Napolitano F, Terracciano C, Sampaolo S, Melone MAB. The Autophagy Signaling Pathway: A Potential Multifunctional Therapeutic Target of Curcumin in Neurological and Neuromuscular Diseases. Nutrients. 2019; 11(8):1881. https://doi.org/10.3390/nu11081881
Chicago/Turabian StylePerrone, Lorena, Tiziana Squillaro, Filomena Napolitano, Chiara Terracciano, Simone Sampaolo, and Mariarosa Anna Beatrice Melone. 2019. "The Autophagy Signaling Pathway: A Potential Multifunctional Therapeutic Target of Curcumin in Neurological and Neuromuscular Diseases" Nutrients 11, no. 8: 1881. https://doi.org/10.3390/nu11081881
APA StylePerrone, L., Squillaro, T., Napolitano, F., Terracciano, C., Sampaolo, S., & Melone, M. A. B. (2019). The Autophagy Signaling Pathway: A Potential Multifunctional Therapeutic Target of Curcumin in Neurological and Neuromuscular Diseases. Nutrients, 11(8), 1881. https://doi.org/10.3390/nu11081881