The Role of Glycosaminoglycans in Protection from Neonatal Necrotizing Enterocolitis: A Narrative Review


Abstract
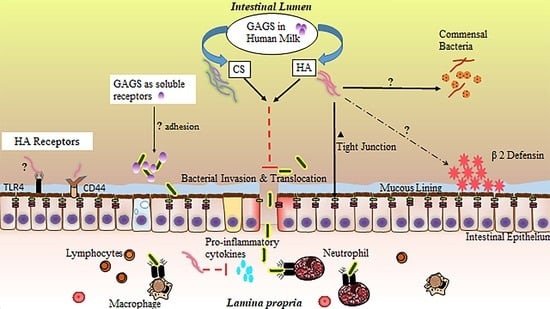
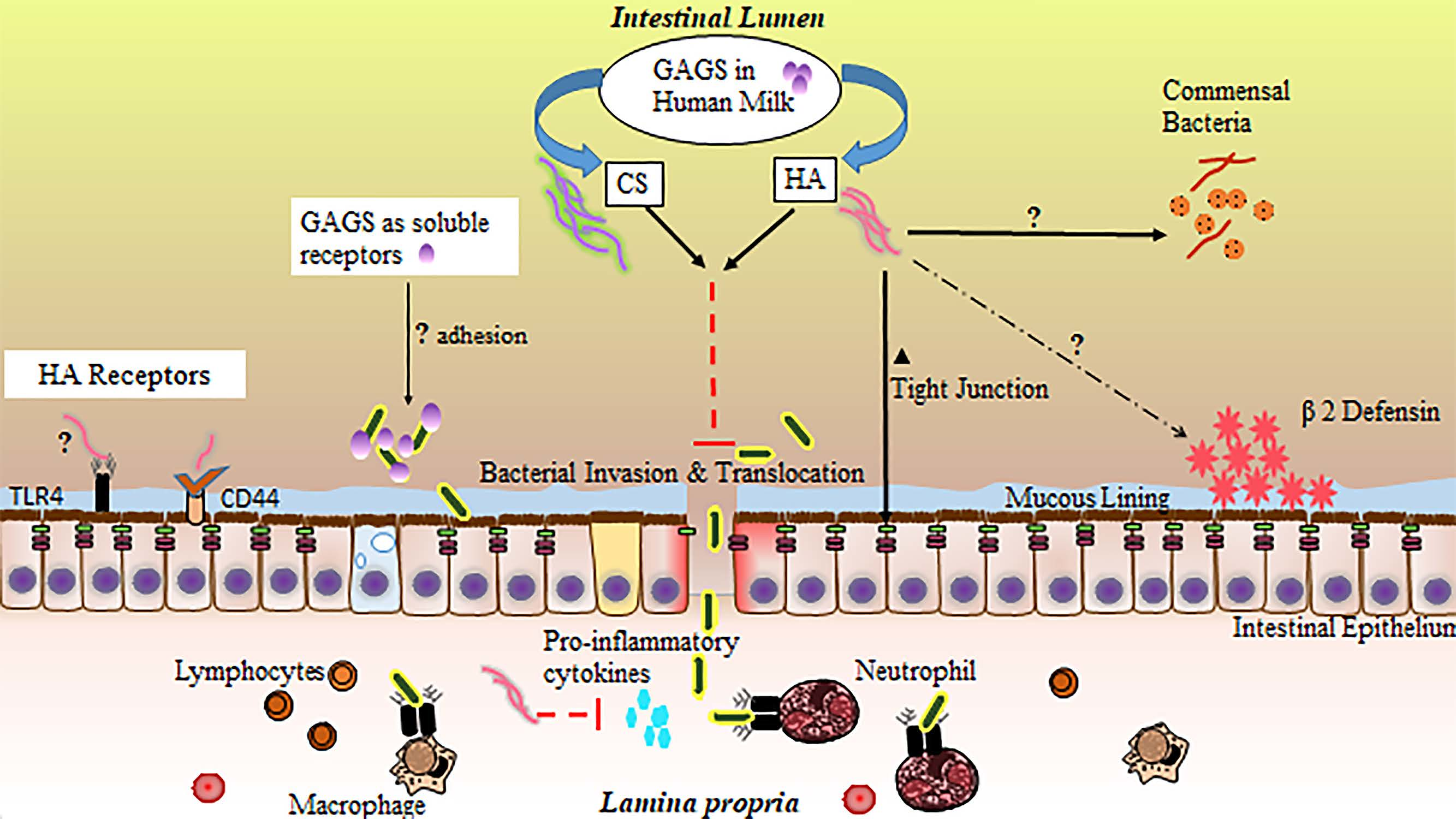
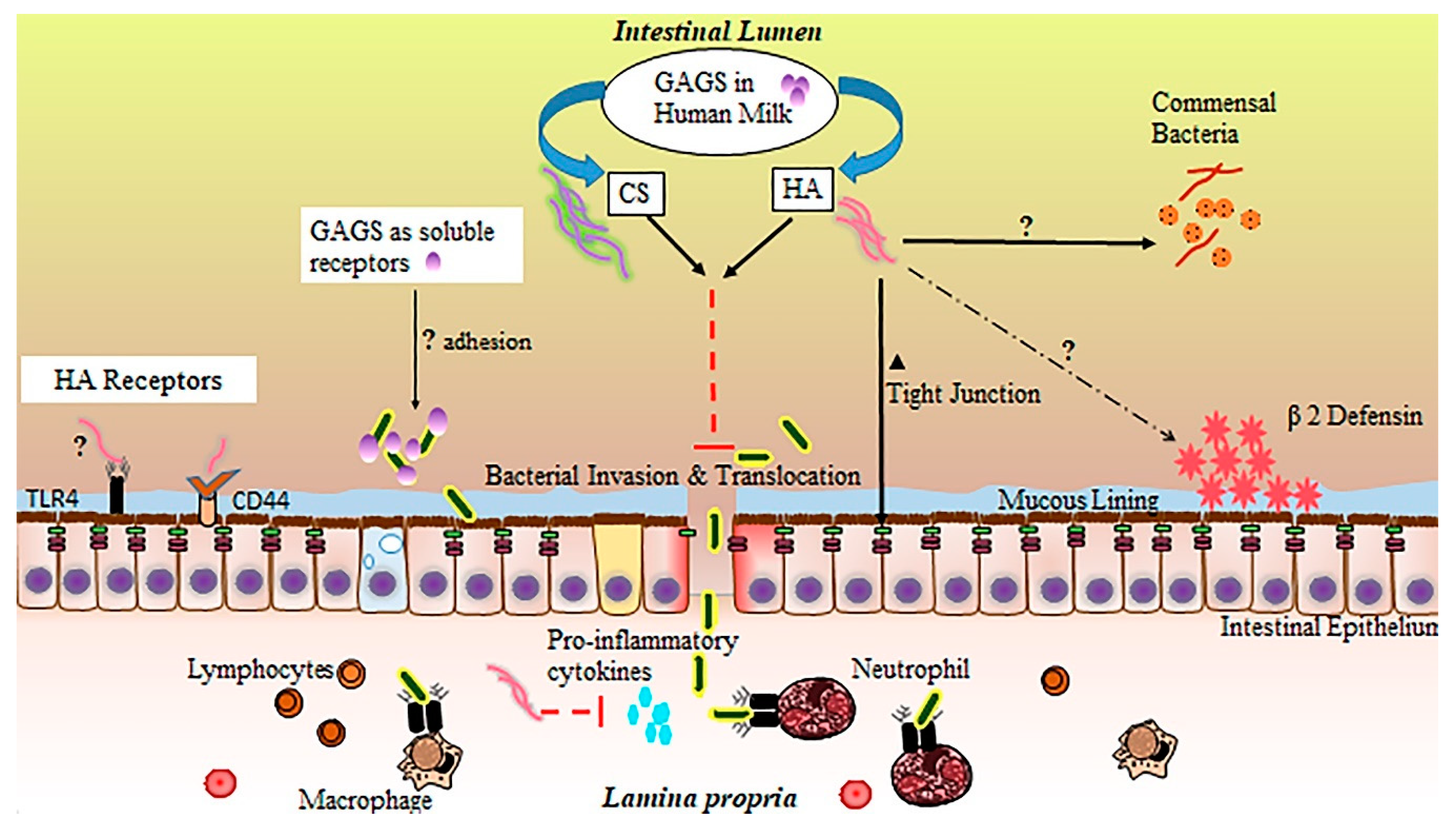
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Pathogenesis of Necrotizing Enterocolitis
2.1. Developmental Immaturity
2.1.1. Innate Immune System
2.1.2. Adaptive Immune System
2.2. Dysbiosis
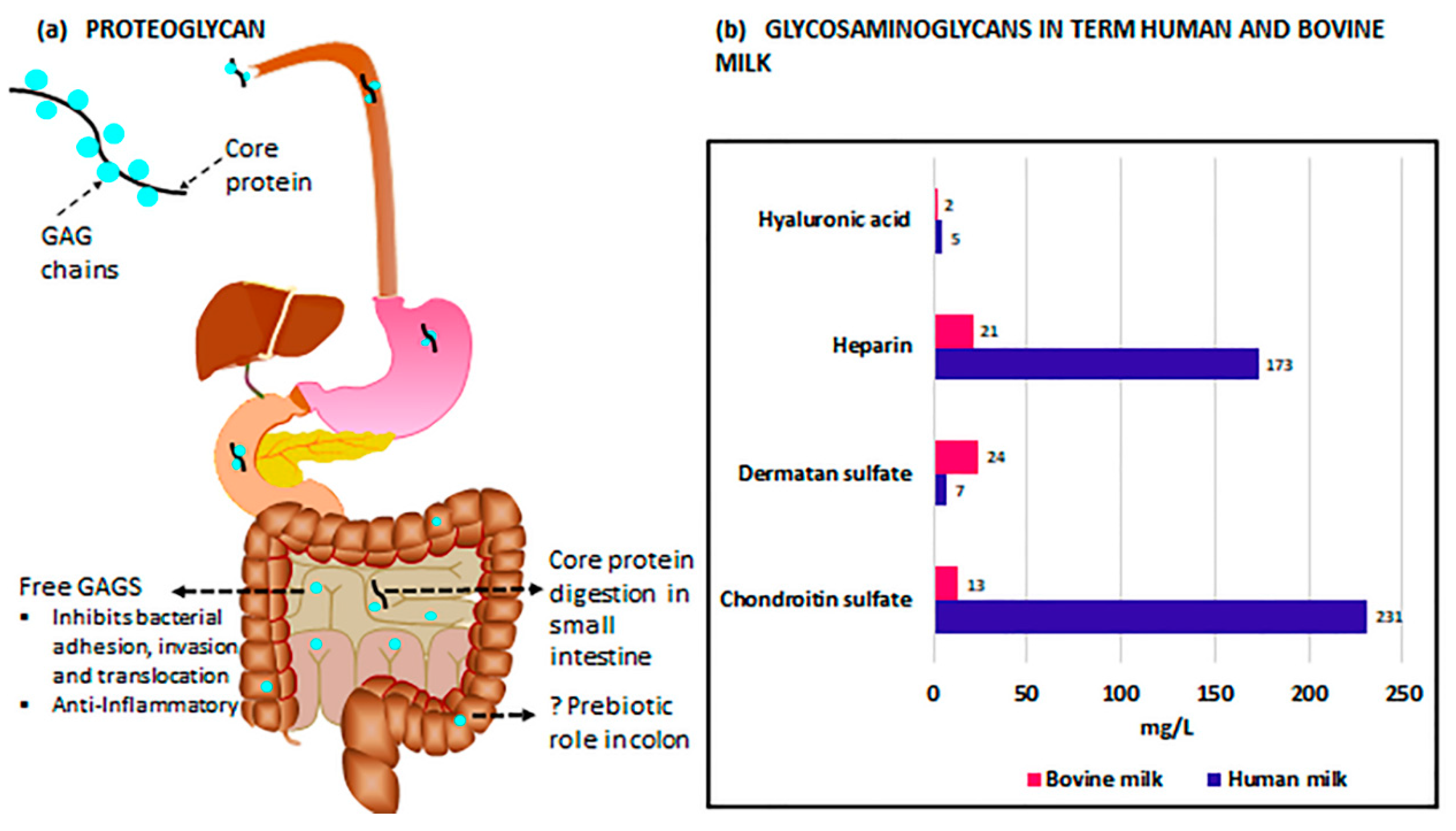
3. Glycosaminoglycans in Milk
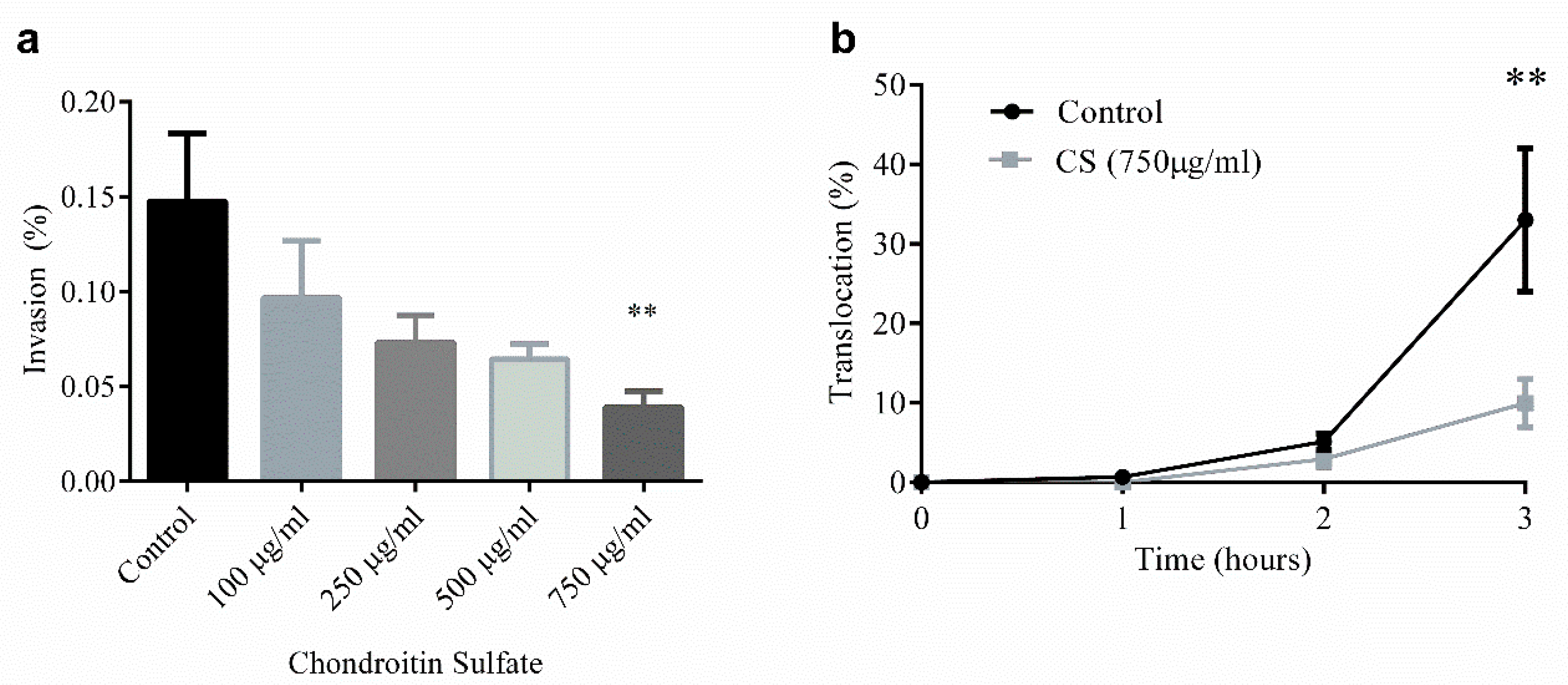
4. Glycosaminoglycan Protection against NEC
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Neu, J.; Walker, W.A. Necrotizing Enterocolitis. N. Engl. J. Med. 2011, 364, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, S.M.; Berryhill, T.F.; Ellenburg, J.L.; Jilling, T.; Cleveland, D.S.; Lorenz, R.G.; Martin, C.A. Pathogenesis of necrotizing enterocolitis: Modeling the innate immune response. Am. J. Pathol. 2015, 185, 4–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victora, J.D.; Silveira, M.F.; Tonial, C.T.; Victora, C.G.; Barros, F.C.; Horta, B.L.; Santos, I.S.D.; Bassani, D.G.; Garcia, P.C.R.; Scheeren, M.; et al. Prevalence, mortality and risk factors associated with very low birth weight preterm infants: An analysis of 33 years. J. Pediatr. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rich, B.S.; Dolgin, S.E. Necrotizing Enterocolitis. Pediatr. Rev. 2017, 38, 552–559. [Google Scholar] [CrossRef]
- Bering, S.B. Human Milk Oligosaccharides to Prevent Gut Dysfunction and Necrotizing Enterocolitis in Preterm Neonates. Nutrients 2018, 10, 1461. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, J. Human milk and the premature infant. Ann. Nutr. Metab. 2013, 62 (Suppl. 3), 8–14. [Google Scholar] [CrossRef]
- Neu, J.; Pammi, M. Pathogenesis of NEC: Impact of an altered intestinal microbiome. Semin. Perinatol. 2017, 41, 29–35. [Google Scholar] [CrossRef]
- Samuels, N.; van de Graaf, R.A.; de Jonge, R.C.J.; Reiss, I.K.M.; Vermeulen, M.J. Risk factors for necrotizing enterocolitis in neonates: A systematic review of prognostic studies. BMC Pediatr. 2017, 17, 105. [Google Scholar] [CrossRef]
- Cacho, N.T.; Parker, L.A.; Neu, J. Necrotizing Enterocolitis and Human Milk Feeding: A Systematic Review. Clin. Perinatol. 2017, 44, 49–67. [Google Scholar] [CrossRef]
- Miller, J.; Tonkin, E.; Damarell, R.A.; McPhee, A.J.; Suganuma, M.; Suganuma, H.; Middleton, P.F.; Makrides, M.; Collins, C.T. A Systematic Review and Meta-Analysis of Human Milk Feeding and Morbidity in Very Low Birth Weight Infants. Nutrients 2018, 10, 707. [Google Scholar] [CrossRef] [Green Version]
- Andreas, N.J.; Kampmann, B.; Mehring Le-Doare, K. Human breast milk: A review on its composition and bioactivity. Early Hum. Dev. 2015, 91, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Grulee, C.G.; Sanford, H.N.; Herron, P.H. Breast and artificial feeding: Influence on morbidity and mortality of twenty thousand infants. JAMA J. Am. Med. Assoc. 1934, 103, 735–738. [Google Scholar] [CrossRef]
- Howie, P.W.; Forsyth, J.S.; Ogston, S.A.; Clark, A.; Florey, C.D. Protective effect of breast feeding against infection. BMJ 1990, 300, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Abrams, S.A.; Schanler, R.J.; Lee, M.L.; Rechtman, D.J. Greater mortality and morbidity in extremely preterm infants fed a diet containing cow milk protein products. Breastfeed. Med. 2014, 9, 281–285. [Google Scholar] [CrossRef]
- Sisk, P.M.; Lovelady, C.A.; Dillard, R.G.; Gruber, K.J.; O’Shea, T.M. Early human milk feeding is associated with a lower risk of necrotizing enterocolitis in very low birth weight infants. J. Perinatol. 2007, 27, 428–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, A.; Cole, T.J. Breast milk and neonatal necrotising enterocolitis. Lancet 1990, 336, 1519–1523. [Google Scholar] [CrossRef]
- Meinzen-Derr, J.; Poindexter, B.; Wrage, L.; Morrow, A.L.; Stoll, B.; Donovan, E.F. Role of human milk in extremely low birth weight infants’ risk of necrotizing enterocolitis or death. J. Perinatol. 2009, 29, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corpeleijn, W.E.; Kouwenhoven, S.M.; Paap, M.C.; van Vliet, I.; Scheerder, I.; Muizer, Y.; Helder, O.K.; van Goudoever, J.B.; Vermeulen, M.J. Intake of own mother’s milk during the first days of life is associated with decreased morbidity and mortality in very low birth weight infants during the first 60 days of life. Neonatology 2012, 102, 276–281. [Google Scholar] [CrossRef]
- Arslanoglu, S.; Corpeleijn, W.; Moro, G.; Braegger, C.; Campoy, C.; Colomb, V.; Decsi, T.; Domellof, M.; Fewtrell, M.; Hojsak, I.; et al. Donor human milk for preterm infants: Current evidence and research directions. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 535–542. [Google Scholar] [CrossRef]
- Li, Y.; Nguyen, D.N.; de Waard, M.; Christensen, L.; Zhou, P.; Jiang, P.; Sun, J.; Bojesen, A.M.; Lauridsen, C.; Lykkesfeldt, J.; et al. Pasteurization Procedures for Donor Human Milk Affect Body Growth, Intestinal Structure, and Resistance against Bacterial Infections in Preterm Pigs. J. Nutr. 2017, 147, 1121–1130. [Google Scholar] [CrossRef] [Green Version]
- Aksu, T.; Atalay, Y.; Turkyilmaz, C.; Gulbahar, O.; Hirfanoglu, I.M.; Demirel, N.; Onal, E.; Ergenekon, E.; Koc, E. The effects of breast milk storage and freezing procedure on interleukine-10 levels and total antioxidant activity. J. Matern. Fetal Neonatal Med. 2015, 28, 1799–1802. [Google Scholar] [CrossRef] [PubMed]
- Corpeleijn, W.E.; de Waard, M.; Christmann, V.; van Goudoever, J.B.; Jansen-van der Weide, M.C.; Kooi, E.M.; Koper, J.F.; Kouwenhoven, S.M.; Lafeber, H.N.; Mank, E.; et al. Effect of Donor Milk on Severe Infections and Mortality in Very Low-Birth-Weight Infants: The Early Nutrition Study Randomized Clinical Trial. JAMA Pediatr. 2016, 170, 654–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canizo Vazquez, D.; Salas Garcia, S.; Izquierdo Renau, M.; Iglesias-Platas, I. Availability of Donor Milk for Very Preterm Infants Decreased the Risk of Necrotizing Enterocolitis without Adversely Impacting Growth or Rates of Breastfeeding. Nutrients 2019, 11, 1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, S.L.; Lohmann, P.; Preidis, G.A.; Gordon, P.S.; O’Donnell, A.; Hagan, J.; Venkatachalam, A.; Balderas, M.; Luna, R.A.; Hair, A.B. Improved feeding tolerance and growth are linked to increased gut microbial community diversity in very-low-birth-weight infants fed mother’s own milk compared with donor breast milk. Am. J. Clin. Nutr. 2019, 109, 1088–1097. [Google Scholar] [CrossRef]
- Baethge, C.; Goldbeck-Wood, S.; Mertens, S. SANRA-a scale for the quality assessment of narrative review articles. Res. Integr. Peer Rev. 2019, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, R. Writing narrative style literature reviews. Med Writ. 2015, 24, 230–235. [Google Scholar] [CrossRef]
- Battersby, A.J.; Gibbons, D.L. The gut mucosal immune system in the neonatal period. Pediatr. Allergy Immunol. 2013, 24, 414–421. [Google Scholar] [CrossRef]
- Pammi, M.; Cope, J.; Tarr, P.I.; Warner, B.B.; Morrow, A.L.; Mai, V.; Gregory, K.E.; Kroll, J.S.; McMurtry, V.; Ferris, M.J.; et al. Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis: A systematic review and meta-analysis. Microbiome 2017, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Denning, N.L.; Prince, J.M. Neonatal intestinal dysbiosis in necrotizing enterocolitis. Mol. Med. 2018, 24, 4. [Google Scholar] [CrossRef]
- Moore, S.A.; Nighot, P.; Reyes, C.; Rawat, M.; McKee, J.; Lemon, D.; Hanson, J.; Ma, T.Y. Intestinal barrier dysfunction in human necrotizing enterocolitis. J. Pediatr. Surg. 2016, 51, 1907–1913. [Google Scholar] [CrossRef] [Green Version]
- Krappmann, D.; Wegener, E.; Sunami, Y.; Esen, M.; Thiel, A.; Mordmuller, B.; Scheidereit, C. The IkappaB kinase complex and NF-kappaB act as master regulators of lipopolysaccharide-induced gene expression and control subordinate activation of AP-1. Mol. Cell. Biol. 2004, 24, 6488–6500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodhi, C.P.; Neal, M.D.; Siggers, R.; Sho, S.; Ma, C.; Branca, M.F.; Prindle, T., Jr.; Russo, A.M.; Afrazi, A.; Good, M.; et al. Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology 2012, 143, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leaphart, C.L.; Cavallo, J.; Gribar, S.C.; Cetin, S.; Li, J.; Branca, M.F.; Dubowski, T.D.; Sodhi, C.P.; Hackam, D.J. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J. Immunol. 2007, 179, 4808–4820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, C.E.; Sodhi, C.P.; Good, M.; Lin, J.; Jia, H.; Yamaguchi, Y.; Lu, P.; Ma, C.; Branca, M.F.; Weyandt, S.; et al. Toll-like receptor 4-mediated lymphocyte influx induces neonatal necrotizing enterocolitis. J. Clin. Investig. 2016, 126, 495–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, M.; Siggers, R.H.; Sodhi, C.P.; Afrazi, A.; Alkhudari, F.; Egan, C.E.; Neal, M.D.; Yazji, I.; Jia, H.; Lin, J.; et al. Amniotic fluid inhibits Toll-like receptor 4 signaling in the fetal and neonatal intestinal epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, 11330–11335. [Google Scholar] [CrossRef] [Green Version]
- Good, M.; Sodhi, C.P.; Egan, C.E.; Afrazi, A.; Jia, H.; Yamaguchi, Y.; Lu, P.; Branca, M.F.; Ma, C.; Prindle, T., Jr.; et al. Breast milk protects against the development of necrotizing enterocolitis through inhibition of Toll-like receptor 4 in the intestinal epithelium via activation of the epidermal growth factor receptor. Mucosal. Immunol. 2015, 8, 1166–1179. [Google Scholar] [CrossRef]
- Neal, M.D.; Jia, H.; Eyer, B.; Good, M.; Guerriero, C.J.; Sodhi, C.P.; Afrazi, A.; Prindle, T., Jr.; Ma, C.; Branca, M.; et al. Discovery and validation of a new class of small molecule Toll-like receptor 4 (TLR4) inhibitors. PLoS ONE 2013, 8, e65779. [Google Scholar] [CrossRef] [Green Version]
- Good, M.; Sodhi, C.P.; Yamaguchi, Y.; Jia, H.; Lu, P.; Fulton, W.B.; Martin, L.Y.; Prindle, T.; Nino, D.F.; Zhou, Q.; et al. The human milk oligosaccharide 2’-fucosyllactose attenuates the severity of experimental necrotising enterocolitis by enhancing mesenteric perfusion in the neonatal intestine. Br. J. Nutr. 2016, 116, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Sodhi, C.P.; Hackam, D.J. Toll-like receptor regulation of intestinal development and inflammation in the pathogenesis of necrotizing enterocolitis. Pathophysiol. Off. J. Int. Soc. Pathophysiol. 2014, 21, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Burge, K.; Gunasekaran, A.; Eckert, J.; Chaaban, H. Curcumin and Intestinal Inflammatory Diseases: Molecular Mechanisms of Protection. Int. J. Mol. Sci. 2019, 20, 1912. [Google Scholar] [CrossRef] [Green Version]
- Mara, M.A.; Good, M.; Weitkamp, J.H. Innate and adaptive immunity in necrotizing enterocolitis. Semin. Fetal Neonatal Med. 2018, 23, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Udall, J.N.; Pang, K.; Fritze, L.; Kleinman, R.; Walker, W.A. Development of gastrointestinal mucosal barrier. I. The effect of age on intestinal permeability to macromolecules. Pediatr. Res. 1981, 15, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Managlia, E.; Liu, S.X.L.; Yan, X.; Tan, X.D.; Chou, P.M.; Barrett, T.A.; De Plaen, I.G. Blocking NF-kappaB Activation in Ly6c(+) Monocytes Attenuates Necrotizing Enterocolitis. Am. J. Pathol. 2019, 189, 604–618. [Google Scholar] [CrossRef] [Green Version]
- De Plaen, I.G.; Liu, S.X.; Tian, R.; Neequaye, I.; May, M.J.; Han, X.B.; Hsueh, W.; Jilling, T.; Lu, J.; Caplan, M.S. Inhibition of nuclear factor-kappaB ameliorates bowel injury and prolongs survival in a neonatal rat model of necrotizing enterocolitis. Pediatr. Res. 2007, 61, 716–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markel, T.A.; Crisostomo, P.R.; Wairiuko, G.M.; Pitcher, J.; Tsai, B.M.; Meldrum, D.R. Cytokines in necrotizing enterocolitis. Shock 2006, 25, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Yazji, I.; Sodhi, C.P.; Lee, E.K.; Good, M.; Egan, C.E.; Afrazi, A.; Neal, M.D.; Jia, H.; Lin, J.; Ma, C.; et al. Endothelial TLR4 activation impairs intestinal microcirculatory perfusion in necrotizing enterocolitis via eNOS-NO-nitrite signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 9451–9456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.J.; Besner, G.E. The role of the intestinal microcirculation in necrotizing enterocolitis. Semin. Pediatr. Surg. 2013, 22, 83–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoma, C. Preventing brain damage in necrotizing enterocolitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 75. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, B.O. Fight them or feed them: How the intestinal mucus layer manages the gut microbiota. Gastroenterol. Rep. 2019, 7, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ghosh, S.S.; Ghosh, S. Curcumin improves intestinal barrier function: Modulation of intracellular signaling, and organization of tight junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, C438–C445. [Google Scholar] [CrossRef]
- Martin, N.A.; Mount Patrick, S.K.; Estrada, T.E.; Frisk, H.A.; Rogan, D.T.; Dvorak, B.; Halpern, M.D. Active transport of bile acids decreases mucin 2 in neonatal ileum: Implications for development of necrotizing enterocolitis. PLoS ONE 2011, 6, e27191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.A.; Doelle, S.M.; Halpern, M.D.; Saunders, T.A.; Holubec, H.; Dvorak, K.; Boitano, S.A.; Dvorak, B. Intestinal barrier failure during experimental necrotizing enterocolitis: Protective effect of EGF treatment. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G938–G949. [Google Scholar] [CrossRef]
- McElroy, S.J.; Prince, L.S.; Weitkamp, J.H.; Reese, J.; Slaughter, J.C.; Polk, D.B. Tumor necrosis factor receptor 1-dependent depletion of mucus in immature small intestine: A potential role in neonatal necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G656–G666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Sherman, M.P.; Prince, L.S.; Bader, D.; Weitkamp, J.H.; Slaughter, J.C.; McElroy, S.J. Paneth cell ablation in the presence of Klebsiella pneumoniae induces necrotizing enterocolitis (NEC)-like injury in the small intestine of immature mice. Dis. Models Mech. 2012, 5, 522–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markasz, L.; Wanders, A.; Szekely, L.; Lilja, H.E. Diminished DEFA6 Expression in Paneth Cells Is Associated with Necrotizing Enterocolitis. Gastroenterol Res Pract. 2018, 2018, 7345426. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, H.B.; da Mota, H.C.; Coutinho, V.B.; Robalinho, T.I.; Furtado, A.F.; Walker, E.; King, G.; Mahida, Y.R.; Sewell, H.F.; Wakelin, D. Absence of lysozyme (muramidase) in the intestinal Paneth cells of newborn infants with necrotising enterocolitis. J. Clin. Pathol. 1998, 51, 512–514. [Google Scholar] [CrossRef] [Green Version]
- Salzman, N.H.; Bevins, C.L. Dysbiosis--a consequence of Paneth cell dysfunction. Semin. Immunol. 2013, 25, 334–341. [Google Scholar] [CrossRef]
- Strunk, T.; Temming, P.; Gembruch, U.; Reiss, I.; Bucsky, P.; Schultz, C. Differential maturation of the innate immune response in human fetuses. Pediatr. Res. 2004, 56, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Maheshwari, A.; Kelly, D.R.; Nicola, T.; Ambalavanan, N.; Jain, S.K.; Murphy-Ullrich, J.; Athar, M.; Shimamura, M.; Bhandari, V.; Aprahamian, C.; et al. TGF-beta2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology 2011, 140, 242–253. [Google Scholar] [CrossRef] [Green Version]
- MohanKumar, K.; Namachivayam, K.; Chapalamadugu, K.C.; Garzon, S.A.; Premkumar, M.H.; Tipparaju, S.M.; Maheshwari, A. Smad7 interrupts TGF-beta signaling in intestinal macrophages and promotes inflammatory activation of these cells during necrotizing enterocolitis. Pediatr. Res. 2016, 79, 951–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namachivayam, K.; Blanco, C.L.; MohanKumar, K.; Jagadeeswaran, R.; Vasquez, M.; McGill-Vargas, L.; Garzon, S.A.; Jain, S.K.; Gill, R.K.; Freitag, N.E.; et al. Smad7 inhibits autocrine expression of TGF-beta2 in intestinal epithelial cells in baboon necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G167–G180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuller, S.S.; Sadeghi, K.; Wisgrill, L.; Dangl, A.; Diesner, S.C.; Prusa, A.R.; Klebermasz-Schrehof, K.; Greber-Platzer, S.; Neumuller, J.; Helmer, H.; et al. Preterm neonates display altered plasmacytoid dendritic cell function and morphology. J. Leukoc. Biol. 2013, 93, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Emami, C.N.; Mittal, R.; Wang, L.; Ford, H.R.; Prasadarao, N.V. Recruitment of dendritic cells is responsible for intestinal epithelial damage in the pathogenesis of necrotizing enterocolitis by Cronobacter sakazakii. J. Immunol. 2011, 186, 7067–7079. [Google Scholar] [CrossRef] [Green Version]
- Jilling, T.; Simon, D.; Lu, J.; Meng, F.J.; Li, D.; Schy, R.; Thomson, R.B.; Soliman, A.; Arditi, M.; Caplan, M.S. The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J. Immunol. 2006, 177, 3273–3282. [Google Scholar] [CrossRef] [Green Version]
- Le Mandat Schultz, A.; Bonnard, A.; Barreau, F.; Aigrain, Y.; Pierre-Louis, C.; Berrebi, D.; Peuchmaur, M. Expression of TLR-2, TLR-4, NOD2 and pNF-kappaB in a neonatal rat model of necrotizing enterocolitis. PLoS ONE 2007, 2, e1102. [Google Scholar] [CrossRef] [Green Version]
- Dimmitt, R.A.; Staley, E.M.; Chuang, G.; Tanner, S.M.; Soltau, T.D.; Lorenz, R.G. Role of postnatal acquisition of the intestinal microbiome in the early development of immune function. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 262–273. [Google Scholar] [CrossRef] [Green Version]
- Nanthakumar, N.; Meng, D.; Goldstein, A.M.; Zhu, W.; Lu, L.; Uauy, R.; Llanos, A.; Claud, E.C.; Walker, W.A. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: An immature innate immune response. PLoS ONE 2011, 6, e17776. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhu, L.; Fatheree, N.Y.; Liu, X.; Pacheco, S.E.; Tatevian, N.; Rhoads, J.M. Changes in intestinal Toll-like receptors and cytokines precede histological injury in a rat model of necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G442–G450. [Google Scholar] [CrossRef] [Green Version]
- Dowling, D.J.; Levy, O. Ontogeny of early life immunity. Trends Immunol. 2014, 35, 299–310. [Google Scholar] [CrossRef]
- Gutzeit, C.; Magri, G.; Cerutti, A. Intestinal IgA production and its role in host-microbe interaction. Immunol. Rev. 2014, 260, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Borges, M.C.; Sesso, M.L.; Roberti, L.R.; de Menezes Oliveira, M.A.; Nogueira, R.D.; Geraldo-Martins, V.R.; Ferriani, V.P. Salivary antibody response to streptococci in preterm and fullterm children: A prospective study. Arch. Oral Biol. 2015, 60, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, R.D.; Sesso, M.L.; Borges, M.C.; Mattos-Graner, R.O.; Smith, D.J.; Ferriani, V.P. Salivary IgA antibody responses to Streptococcus mitis and Streptococcus mutans in preterm and fullterm newborn children. Arch. Oral Biol. 2012, 57, 647–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, A.S.; Behrendt, C.L.; Hooper, L.V. Reciprocal interactions between commensal bacteria and gamma delta intraepithelial lymphocytes during mucosal injury. J. Immunol. 2009, 182, 3047–3054. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Haque, S.F.; Silberzahn, T.; Hamilton, K.; Langford, C.; Ellis, P.; Carr, R.; Hayday, A.C. Neonates harbour highly active gammadelta T cells with selective impairments in preterm infants. Eur. J. Immunol. 2009, 39, 1794–1806. [Google Scholar] [CrossRef] [PubMed]
- Weitkamp, J.H.; Rosen, M.J.; Zhao, Z.; Koyama, T.; Geem, D.; Denning, T.L.; Rock, M.T.; Moore, D.J.; Halpern, M.D.; Matta, P.; et al. Small intestinal intraepithelial TCRgammadelta+ T lymphocytes are present in the premature intestine but selectively reduced in surgical necrotizing enterocolitis. PLoS ONE 2014, 9, e99042. [Google Scholar] [CrossRef] [PubMed]
- Weitkamp, J.H.; Koyama, T.; Rock, M.T.; Correa, H.; Goettel, J.A.; Matta, P.; Oswald-Richter, K.; Rosen, M.J.; Engelhardt, B.G.; Moore, D.J.; et al. Necrotising enterocolitis is characterised by disrupted immune regulation and diminished mucosal regulatory (FOXP3)/effector (CD4, CD8) T cell ratios. Gut 2013, 62, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Elias, K.M.; Laurence, A.; Davidson, T.S.; Stephens, G.; Kanno, Y.; Shevach, E.M.; O’Shea, J.J. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 2008, 111, 1013–1020. [Google Scholar] [CrossRef] [Green Version]
- Nino, D.F.; Sodhi, C.P.; Egan, C.E.; Zhou, Q.; Lin, J.; Lu, P.; Yamaguchi, Y.; Jia, H.; Martin, L.Y.; Good, M.; et al. Retinoic Acid Improves Incidence and Severity of Necrotizing Enterocolitis by Lymphocyte Balance Restitution and Repopulation of LGR5+ Intestinal Stem Cells. Shock 2017, 47, 22–32. [Google Scholar] [CrossRef]
- Grizotte-Lake, M.; Zhong, G.; Duncan, K.; Kirkwood, J.; Iyer, N.; Smolenski, I.; Isoherranen, N.; Vaishnava, S. Commensals Suppress Intestinal Epithelial Cell Retinoic Acid Synthesis to Regulate Interleukin-22 Activity and Prevent Microbial Dysbiosis. Immunity 2018, 49, 1103–1115. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6, 237ra265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stinson, L.F.; Boyce, M.C.; Payne, M.S.; Keelan, J.A. The Not-so-Sterile Womb: Evidence That the Human Fetus Is Exposed to Bacteria Prior to Birth. Front. Microbiol. 2019, 10, 1124. [Google Scholar] [CrossRef] [PubMed]
- Leiby, J.S.; McCormick, K.; Sherrill-Mix, S.; Clarke, E.L.; Kessler, L.R.; Taylor, L.J.; Hofstaedter, C.E.; Roche, A.M.; Mattei, L.M.; Bittinger, K.; et al. Lack of detection of a human placenta microbiome in samples from preterm and term deliveries. Microbiome 2018, 6, 196. [Google Scholar] [CrossRef] [PubMed]
- de Goffau, M.C.; Lager, S.; Sovio, U.; Gaccioli, F.; Cook, E.; Peacock, S.J.; Parkhill, J.; Charnock-Jones, D.S.; Smith, G.C.S. Human placenta has no microbiome but can contain potential pathogens. Nature 2019, 572, 329–334. [Google Scholar] [CrossRef]
- Patel, R.M.; Denning, P.W. Intestinal microbiota and its relationship with necrotizing enterocolitis. Pediatr. Res. 2015, 78, 232–238. [Google Scholar] [CrossRef] [Green Version]
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the human infant intestinal microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef] [Green Version]
- Jost, T.; Lacroix, C.; Braegger, C.P.; Chassard, C. New insights in gut microbiota establishment in healthy breast fed neonates. PLoS ONE 2012, 7, e44595. [Google Scholar] [CrossRef]
- Elgin, T.G.; Kern, S.L.; McElroy, S.J. Development of the Neonatal Intestinal Microbiome and Its Association with Necrotizing Enterocolitis. Clin. Ther. 2016, 38, 706–715. [Google Scholar] [CrossRef] [Green Version]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; Gonzalez, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- La Rosa, P.S.; Warner, B.B.; Zhou, Y.; Weinstock, G.M.; Sodergren, E.; Hall-Moore, C.M.; Stevens, H.J.; Bennett, W.E., Jr.; Shaikh, N.; Linneman, L.A.; et al. Patterned progression of bacterial populations in the premature infant gut. Proc. Natl. Acad. Sci. USA 2014, 111, 12522–12527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, M.K.; Wang, B.; Ahmadi, S.; Burnham, C.A.; Tarr, P.I.; Warner, B.B.; Dantas, G. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat. Microbiol. 2016, 1, 16024. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.V.; Scholz, M.; Zolfo, M.; Taft, D.H.; Schibler, K.R.; Tett, A.; Segata, N.; Morrow, A.L. Metagenomic Sequencing with Strain-Level Resolution Implicates Uropathogenic E. coli in Necrotizing Enterocolitis and Mortality in Preterm Infants. Cell Rep. 2016, 14, 2912–2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlisle, E.M.; Morowitz, M.J. The intestinal microbiome and necrotizing enterocolitis. Curr. Opin. Pediatr. 2013, 25, 382–387. [Google Scholar] [CrossRef]
- Wang, Y.; Hoenig, J.D.; Malin, K.J.; Qamar, S.; Petrof, E.O.; Sun, J.; Antonopoulos, D.A.; Chang, E.B.; Claud, E.C. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 2009, 3, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4, 14. [Google Scholar] [CrossRef]
- Afrazi, A.; Sodhi, C.P.; Richardson, W.; Neal, M.; Good, M.; Siggers, R.; Hackam, D.J. New insights into the pathogenesis and treatment of necrotizing enterocolitis: Toll-like receptors and beyond. Pediatr. Res. 2011, 69, 183–188. [Google Scholar] [CrossRef]
- Musemeche, C.A.; Kosloske, A.M.; Bartow, S.A.; Umland, E.T. Comparative effects of ischemia, bacteria, and substrate on the pathogenesis of intestinal necrosis. J. Pediatr. Surg. 1986, 21, 536–538. [Google Scholar] [CrossRef]
- Morowitz, M.J.; Poroyko, V.; Caplan, M.; Alverdy, J.; Liu, D.C. Redefining the role of intestinal microbes in the pathogenesis of necrotizing enterocolitis. Pediatrics 2010, 125, 777–785. [Google Scholar] [CrossRef]
- Mai, V.; Young, C.M.; Ukhanova, M.; Wang, X.; Sun, Y.; Casella, G.; Theriaque, D.; Li, N.; Sharma, R.; Hudak, M.; et al. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS ONE 2011, 6, e20647. [Google Scholar] [CrossRef] [PubMed]
- Torrazza, R.M.; Neu, J. The altered gut microbiome and necrotizing enterocolitis. Clin. Perinatol. 2013, 40, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Warner, B.B.; Deych, E.; Zhou, Y.; Hall-Moore, C.; Weinstock, G.M.; Sodergren, E.; Shaikh, N.; Hoffmann, J.A.; Linneman, L.A.; Hamvas, A.; et al. Gut bacteria dysbiosis and necrotising enterocolitis in very low birthweight infants: A prospective case-control study. Lancet 2016, 387, 1928–1936. [Google Scholar] [CrossRef] [Green Version]
- Neu, J.; Pammi, M. Necrotizing enterocolitis: The intestinal microbiome, metabolome and inflammatory mediators. Semin. Fetal Neonatal Med. 2018, 23, 400–405. [Google Scholar] [CrossRef]
- Torrazza, R.M.; Ukhanova, M.; Wang, X.; Sharma, R.; Hudak, M.L.; Neu, J.; Mai, V. Intestinal microbial ecology and environmental factors affecting necrotizing enterocolitis. PLoS ONE 2013, 8, e83304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbler, P.T.; Procianoy, R.S.; Mai, V.; Silveira, R.C.; Corso, A.L.; Rojas, B.S.; Roesch, L.F.W. Low Microbial Diversity and Abnormal Microbial Succession Is Associated with Necrotizing Enterocolitis in Preterm Infants. Front. Microbiol. 2017, 8, 2243. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishna, K.P.; Macadangdang, B.R.; Rogers, M.B.; Tometich, J.T.; Firek, B.A.; Baker, R.; Ji, J.; Burr, A.H.P.; Ma, C.; Good, M.; et al. Maternal IgA protects against the development of necrotizing enterocolitis in preterm infants. Nat. Med. 2019, 25, 1110–1115. [Google Scholar] [CrossRef]
- Morrow, A.L.; Lagomarcino, A.J.; Schibler, K.R.; Taft, D.H.; Yu, Z.; Wang, B.; Altaye, M.; Wagner, M.; Gevers, D.; Ward, D.V.; et al. Early microbial and metabolomic signatures predict later onset of necrotizing enterocolitis in preterm infants. Microbiome 2013, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Bizzarro, M.J. Avoiding Unnecessary Antibiotic Exposure in Premature Infants: Understanding When (Not) to Start and When to Stop. JAMA Netw. Open 2018, 1, e180165. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, R.G.; Chowdhury, D.; Hansen, N.I.; Smith, P.B.; Stoll, B.J.; Sanchez, P.J.; Das, A.; Puopolo, K.M.; Mukhopadhyay, S.; Higgins, R.D.; et al. Prolonged duration of early antibiotic therapy in extremely premature infants. Pediatr. Res. 2019, 85, 994–1000. [Google Scholar] [CrossRef]
- Cotten, C.M. Adverse consequences of neonatal antibiotic exposure. Curr. Opin. Pediatr. 2016, 28, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, V.N.; Northrup, V.; Bizzarro, M.J. Antibiotic exposure in the newborn intensive care unit and the risk of necrotizing enterocolitis. J. Pediatr. 2011, 159, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouhy, F.; Guinane, C.M.; Hussey, S.; Wall, R.; Ryan, C.A.; Dempsey, E.M.; Murphy, B.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C.; et al. High-throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicillin and gentamicin. Antimicrob. Agents Chemother. 2012, 56, 5811–5820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Kobayashi, T.; Songjinda, P.; Tateyama, A.; Tsubouchi, M.; Kiyohara, C.; Shirakawa, T.; Sonomoto, K.; Nakayama, J. Influence of antibiotic exposure in the early postnatal period on the development of intestinal microbiota. FEMS Immunol. Med. Microbiol. 2009, 56, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, C.; Morrow, A.L.; Lagomarcino, A.J.; Altaye, M.; Taft, D.H.; Yu, Z.; Newburg, D.S.; Ward, D.V.; Schibler, K.R. Early empiric antibiotic use in preterm infants is associated with lower bacterial diversity and higher relative abundance of Enterobacter. J. Pediatr. 2014, 165, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [Green Version]
- Biasucci, G.; Rubini, M.; Riboni, S.; Morelli, L.; Bessi, E.; Retetangos, C. Mode of delivery affects the bacterial community in the newborn gut. Early Hum. Dev. 2010, 86 (Suppl. 1), 13–15. [Google Scholar] [CrossRef]
- Singh, N.; Dhayade, A.; Mohamed, A.L.; Chaudhari, T.V. Morbidity and Mortality in Preterm Infants following Antacid Use: A Retrospective Audit. Int. J. Pediatr. 2016, 2016, 9649162. [Google Scholar] [CrossRef] [Green Version]
- Terrin, G.; Passariello, A.; De Curtis, M.; Manguso, F.; Salvia, G.; Lega, L.; Messina, F.; Paludetto, R.; Canani, R.B. Ranitidine is associated with infections, necrotizing enterocolitis, and fatal outcome in newborns. Pediatrics 2012, 129, e40–e45. [Google Scholar] [CrossRef] [Green Version]
- Bilali, A.; Galanis, P.; Bartsocas, C.; Sparos, L.; Velonakis, E. H2-blocker therapy and incidence of necrotizing enterocolitis in preterm infants: A case-control study. Pediatr. Neonatol. 2013, 54, 141–142. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.W.; Tran, L.; Norori, J.; Ferris, M.J.; Eren, A.M.; Taylor, C.M.; Dowd, S.E.; Penn, D. Histamine-2 receptor blockers alter the fecal microbiota in premature infants. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Lueschow, S.R.; Stumphy, J.; Gong, H.; Kern, S.L.; Elgin, T.G.; Underwood, M.A.; Kalanetra, K.M.; Mills, D.A.; Wong, M.H.; Meyerholz, D.K.; et al. Loss of murine Paneth cell function alters the immature intestinal microbiome and mimics changes seen in neonatal necrotizing enterocolitis. PLoS ONE 2018, 13, e0204967. [Google Scholar] [CrossRef] [PubMed]
- Pannaraj, P.S.; Li, F.; Cerini, C.; Bender, J.M.; Yang, S.; Rollie, A.; Adisetiyo, H.; Zabih, S.; Lincez, P.J.; Bittinger, K.; et al. Association Between Breast Milk Bacterial Communities and Establishment and Development of the Infant Gut Microbiome. JAMA Pediatr. 2017, 171, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.M.; Foster, J.A.; Forney, L.J.; Schutte, U.M.; Beck, D.L.; Abdo, Z.; Fox, L.K.; Williams, J.E.; McGuire, M.K.; McGuire, M.A. Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS ONE 2011, 6, e21313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, Z.T.; Totten, S.M.; Smilowitz, J.T.; Popovic, M.; Parker, E.; Lemay, D.G.; Van Tassell, M.L.; Miller, M.J.; Jin, Y.S.; German, J.B.; et al. Maternal fucosyltransferase 2 status affects the gut bifidobacterial communities of breastfed infants. Microbiome 2015, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Putignani, L.; Del Chierico, F.; Petrucca, A.; Vernocchi, P.; Dallapiccola, B. The human gut microbiota: A dynamic interplay with the host from birth to senescence settled during childhood. Pediatr. Res. 2014, 76, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Oozeer, R.; van Limpt, K.; Ludwig, T.; Ben Amor, K.; Martin, R.; Wind, R.D.; Boehm, G.; Knol, J. Intestinal microbiology in early life: Specific prebiotics can have similar functionalities as human-milk oligosaccharides. Am. J. Clin. Nutr. 2013, 98, 561s–571s. [Google Scholar] [CrossRef]
- Gomez-Llorente, C.; Plaza-Diaz, J.; Aguilera, M.; Munoz-Quezada, S.; Bermudez-Brito, M.; Peso-Echarri, P.; Martinez-Silla, R.; Vasallo-Morillas, M.I.; Campana-Martin, L.; Vives-Pinera, I.; et al. Three main factors define changes in fecal microbiota associated with feeding modality in infants. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 461–466. [Google Scholar] [CrossRef]
- Penders, J.; Thijs, C.; Vink, C.; Stelma, F.F.; Snijders, B.; Kummeling, I.; van den Brandt, P.A.; Stobberingh, E.E. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 2006, 118, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Penders, J.; Vink, C.; Driessen, C.; London, N.; Thijs, C.; Stobberingh, E.E. Quantification of Bifidobacterium spp.; Escherichia coli and Clostridium difficile in faecal samples of breast-fed and formula-fed infants by real-time PCR. FEMS Microbiol. Lett. 2005, 243, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, S.; Aydin, S.; Ozkan, Y.; Kumru, S. Ghrelin is present in human colostrum, transitional and mature milk. Peptides 2006, 27, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Michalski, M.C.; Briard, V.; Michel, F.; Tasson, F.; Poulain, P. Size distribution of fat globules in human colostrum, breast milk, and infant formula. J. Dairy Sci. 2005, 88, 1927–1940. [Google Scholar] [CrossRef] [Green Version]
- Ballard, O.; Morrow, A.L. Human milk composition: Nutrients and bioactive factors. Pediatr. Clin. N. Am. 2013, 60, 49–74. [Google Scholar] [CrossRef] [Green Version]
- Coppa, G.V.; Gabrielli, O.; Zampini, L.; Galeazzi, T.; Ficcadenti, A.; Padella, L.; Santoro, L.; Soldi, S.; Carlucci, A.; Bertino, E.; et al. Oligosaccharides in 4 different milk groups, Bifidobacteria, and Ruminococcus obeum. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 80–87. [Google Scholar] [CrossRef]
- Morrow, A.L.; Ruiz-Palacios, G.M.; Altaye, M.; Jiang, X.; Guerrero, M.L.; Meinzen-Derr, J.K.; Farkas, T.; Chaturvedi, P.; Pickering, L.K.; Newburg, D.S. Human milk oligosaccharides are associated with protection against diarrhea in breast-fed infants. J. Pediatr. 2004, 145, 297–303. [Google Scholar] [CrossRef]
- Goldman, A.S. Modulation of the gastrointestinal tract of infants by human milk. Interfaces and interactions. An evolutionary perspective. J. Nutr. 2000, 130, 426s–431s. [Google Scholar] [CrossRef]
- Santaolalla, R.; Fukata, M.; Abreu, M.T. Innate immunity in the small intestine. Curr. Opin. Gastroenterol. 2011, 27, 125–131. [Google Scholar] [CrossRef]
- Ricard-Blum, S. Glycosaminoglycans: Major biological players. Glycoconj. J. 2017, 34, 275–276. [Google Scholar] [CrossRef] [Green Version]
- Coppa, G.V.; Facinelli, B.; Magi, G.; Marini, E.; Zampini, L.; Mantovani, V.; Galeazzi, T.; Padella, L.; Marchesiello, R.L.; Santoro, L.; et al. Human milk glycosaminoglycans inhibit in vitro the adhesion of Escherichia coli and Salmonella fyris to human intestinal cells. Pediatr. Res. 2016, 79, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Stern, R.; Asari, A.A.; Sugahara, K.N. Hyaluronan fragments: An information-rich system. Eur. J. Cell Biol. 2006, 85, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Barthe, L.; Woodley, J.; Lavit, M.; Przybylski, C.; Philibert, C.; Houin, G. In vitro intestinal degradation and absorption of chondroitin sulfate, a glycosaminoglycan drug. Arzneimittelforschung 2004, 54, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Maccari, F.; Mantovani, V.; Gabrielli, O.; Carlucci, A.; Zampini, L.; Galeazzi, T.; Galeotti, F.; Coppa, G.V.; Volpi, N. Metabolic fate of milk glycosaminoglycans in breastfed and formula fed newborns. Glycoconj. J. 2016, 33, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Fallacara, A.; Baldini, E.; Manfredini, S.; Vertuani, S. Hyaluronic Acid in the Third Millennium. Polymers 2018, 10, 701. [Google Scholar] [CrossRef] [Green Version]
- Lee-Sayer, S.S.; Dong, Y.; Arif, A.A.; Olsson, M.; Brown, K.L.; Johnson, P. The where, when, how, and why of hyaluronan binding by immune cells. Front. Immunol. 2015, 6, 150. [Google Scholar] [CrossRef] [Green Version]
- Coppa, G.V.; Gabrielli, O.; Buzzega, D.; Zampini, L.; Galeazzi, T.; Maccari, F.; Bertino, E.; Volpi, N. Composition and structure elucidation of human milk glycosaminoglycans. Glycobiology 2011, 21, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Volpi, N.; Gabrielli, O.; Carlucci, A.; Zampini, L.; Santoro, L.; Padella, L.; Marchesello, R.L.; Maccari, F.; Coppa, G.V. Human milk glycosaminoglycans in feces of breastfed newborns: Preliminary structural elucidation and possible biological role. Breastfeed. Med. Off. J. Acad. Breastfeed. Med. 2014, 9, 105–106. [Google Scholar] [CrossRef]
- Coppa, G.V.; Gabrielli, O.; Zampini, L.; Galeazzi, T.; Maccari, F.; Buzzega, D.; Galeotti, F.; Bertino, E.; Volpi, N. Glycosaminoglycan content in term and preterm milk during the first month of lactation. Neonatology 2012, 101, 74–76. [Google Scholar] [CrossRef]
- Wang, C.; Lang, Y.; Li, Q.; Jin, X.; Li, G.; Yu, G. Glycosaminoglycanomic profiling of human milk in different stages of lactation by liquid chromatography-tandem mass spectrometry. Food Chem. 2018, 258, 231–236. [Google Scholar] [CrossRef]
- Soares da Costa, D.; Reis, R.L.; Pashkuleva, I. Sulfation of Glycosaminoglycans and Its Implications in Human Health and Disorders. Annu. Rev. Biomed. Eng. 2017, 19, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Volpi, N.; Maccari, F.; Galeotti, F.; Peila, C.; Coscia, A.; Zampini, L.; Monachesi, C.; Gabrielli, O.; Coppa, G. Human milk glycosaminoglycan composition from women of different countries: A pilot study. J. Matern. Fetal Neonatal Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mannello, F.; Maccari, F.; Santinelli, A.; Volpi, N. Chondroitin sulfate structure is modified in human milk produced by breast affected by invasive carcinoma. Breast 2011, 20, 586–587. [Google Scholar] [CrossRef] [PubMed]
- Cerdo, T.; Ruiz, A.; Jauregui, R.; Azaryah, H.; Torres-Espinola, F.J.; Garcia-Valdes, L.; Teresa Segura, M.; Suarez, A.; Campoy, C. Maternal obesity is associated with gut microbial metabolic potential in offspring during infancy. J. Physiol. Biochem. 2018, 74, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Wiederschain, G.Y.; Newburg, D.S. Glycoconjugate stability in human milk: Glycosidase activities and sugar release. J. Nutr. Biochem. 2001, 12, 559–564. [Google Scholar] [CrossRef]
- Coscia, A.; Peila, C.; Bertino, E.; Coppa, G.V.; Moro, G.E.; Gabrielli, O.; Zampini, L.; Galeazzi, T.; Maccari, F.; Volpi, N. Effect of holder pasteurisation on human milk glycosaminoglycans. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Cartmell, A.; Lowe, E.C.; Basle, A.; Firbank, S.J.; Ndeh, D.A.; Murray, H.; Terrapon, N.; Lombard, V.; Henrissat, B.; Turnbull, J.E.; et al. How members of the human gut microbiota overcome the sulfation problem posed by glycosaminoglycans. Proc. Natl. Acad. Sci. USA 2017, 114, 7037–7042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Liu, X.; Lyu, Z.; Gu, H.; Li, D.; Chen, H. Glycosaminoglycans (GAGs) and GAG mimetics regulate the behavior of stem cell differentiation. Colloids Surf. B Biointerfaces 2017, 150, 175–182. [Google Scholar] [CrossRef]
- Poterucha, T.J.; Libby, P.; Goldhaber, S.Z. More than an anticoagulant: Do heparins have direct anti-inflammatory effects? Thromb. Haemost. 2017, 117, 437–444. [Google Scholar] [CrossRef]
- Pomin, V.H. Sulfated glycans in inflammation. Eur. J. Med. Chem. 2015, 92, 353–369. [Google Scholar] [CrossRef]
- Bode, L.; Salvestrini, C.; Park, P.W.; Li, J.-P.; Esko, J.D.; Yamaguchi, Y.; Murch, S.; Freeze, H.H. Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J. Clin. Investig. 2008, 118, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Klein, N.J.; Shennan, G.I.; Heyderman, R.S.; Levin, M. Alteration in glycosaminoglycan metabolism and surface charge on human umbilical vein endothelial cells induced by cytokines, endotoxin and neutrophils. J. Cell Sci. 1992, 102, 821–832. [Google Scholar] [PubMed]
- Ade-Ajayi, N.; Spitz, L.; Kiely, E.; Drake, D.; Klein, N. Intestinal glycosaminoglycans in neonatal necrotizing enterocolitis. Br. J. Surg. 1996, 83, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Nakase, H.; Matsuura, M.; Honzawa, Y.; Matsumura, K.; Uza, N.; Yamaguchi, Y.; Mizoguchi, E.; Chiba, T. Heparan sulfate on intestinal epithelial cells plays a critical role in intestinal crypt homeostasis via Wnt/beta-catenin signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G241–G249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozin, A.P.; Goldstein, M.; Sprecher, H. Antibacterial activity of glucosamine sulfate and chondroitine sulfate? Clin. Exp. Rheumatol. 2008, 26, 509–510. [Google Scholar]
- Carlson, G.A.; Dragoo, J.L.; Samimi, B.; Bruckner, D.A.; Bernard, G.W.; Hedrick, M.; Benhaim, P. Bacteriostatic properties of biomatrices against common orthopaedic pathogens. Biochem. Biophys. Res. Commun. 2004, 321, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Pirnazar, P.; Wolinsky, L.; Nachnani, S.; Haake, S.; Pilloni, A.; Bernard, G.W. Bacteriostatic effects of hyaluronic acid. J. Periodontol. 1999, 70, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Kamochi, R.; Oiki, S.; Murata, K.; Hashimoto, W. Probiotics in human gut microbiota can degrade host glycosaminoglycans. Sci. Rep. 2018, 8, 10674. [Google Scholar] [CrossRef] [Green Version]
- Zuniga, M.; Monedero, V.; Yebra, M.J. Utilization of Host-Derived Glycans by Intestinal Lactobacillus and Bifidobacterium Species. Front. Microbiol. 2018, 9, 1917. [Google Scholar] [CrossRef] [Green Version]
- Sava, I.G.; Zhang, F.; Toma, I.; Theilacker, C.; Li, B.; Baumert, T.F.; Holst, O.; Linhardt, R.J.; Huebner, J. Novel interactions of glycosaminoglycans and bacterial glycolipids mediate binding of enterococci to human cells. J. Biol. Chem. 2009, 284, 18194–18201. [Google Scholar] [CrossRef] [Green Version]
- Hafez, M.M.; Aboulwafa, M.M.; Yassien, M.A.; Hassouna, N.A. Role of different classes of mammalian cell surface molecules in adherence of coagulase positive and coagulase negative staphylococci. J. Basic Microbiol. 2008, 48, 353–362. [Google Scholar] [CrossRef]
- Henry-Stanley, M.J.; Hess, D.J.; Erlandsen, S.L.; Wells, C.L. Ability of the heparan sulfate proteoglycan syndecan-1 to participate in bacterial translocation across the intestinal epithelial barrier. Shock 2005, 24, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Newburg, D.S.; Linhardt, R.J.; Ampofo, S.A.; Yolken, R.H. Human milk glycosaminoglycans inhibit HIV glycoprotein gp120 binding to its host cell CD4 receptor. J. Nutr. 1995, 125, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.R.; Rho, H.K.; Kessler, S.P.; Amin, R.; Homer, C.R.; McDonald, C.; Cowman, M.K.; de la Motte, C.A. Human milk hyaluronan enhances innate defense of the intestinal epithelium. J. Biol. Chem. 2013, 288, 29090–29104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Plaen, I.G. Inflammatory signaling in necrotizing enterocolitis. Clin. Perinatol. 2013, 40, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Riehl, T.E.; Stenson, W.F. Regulation of colonic epithelial repair in mice by Toll-like receptors and hyaluronic acid. Gastroenterology 2009, 137, 2041–2051. [Google Scholar] [CrossRef] [Green Version]
- Riehl, T.E.; Foster, L.; Stenson, W.F. Hyaluronic acid is radioprotective in the intestine through a TLR4 and COX-2-mediated mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G309–G316. [Google Scholar] [CrossRef] [Green Version]
- Asari, A.; Kanemitsu, T.; Kurihara, H. Oral administration of high molecular weight hyaluronan (900 kDa) controls immune system via Toll-like receptor 4 in the intestinal epithelium. J. Biol. Chem. 2010, 285, 24751–24758. [Google Scholar] [CrossRef] [Green Version]
- Hill, D.R.; Kessler, S.P.; Rho, H.K.; Cowman, M.K.; de la Motte, C.A. Specific-sized hyaluronan fragments promote expression of human beta-defensin 2 in intestinal epithelium. J. Biol. Chem. 2012, 287, 30610–30624. [Google Scholar] [CrossRef] [Green Version]
- Gunasekaran, A.; Eckert, J.; Burge, K.; Zheng, W.; Yu, Z.; Kessler, S.; de la Motte, C.; Chaaban, H. Hyaluronan 35 kDa enhances epithelial barrier function and protects against the development of murine necrotizing enterocolitis. Pediatr. Res. 2019. [Google Scholar] [CrossRef]
- Stabler, T.V.; Huang, Z.; Montell, E.; Verges, J.; Kraus, V.B. Chondroitin sulphate inhibits NF-kappaB activity induced by interaction of pathogenic and damage associated molecules. Osteoarthr. Cartil. 2017, 25, 166–174. [Google Scholar] [CrossRef] [Green Version]
- du Souich, P.; Garcia, A.G.; Verges, J.; Montell, E. Immunomodulatory and anti-inflammatory effects of chondroitin sulphate. J. Cell Mol. Med. 2009, 13, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Garcia, A.G.; Verges, J.; Montell, E.; Lopez, M.G. Antioxidant, antiinflammatory and neuroprotective actions of chondroitin sulfate and proteoglycans. Osteoarthr. Cartil. 2010, 18 (Suppl. 1), S24–S27. [Google Scholar] [CrossRef] [Green Version]
- Linares, P.M.; Chaparro, M.; Algaba, A.; Roman, M.; Moreno Arza, I.; Abad Santos, F.; Ochoa, D.; Guerra, I.; Bermejo, F.; Gisbert, J.P. Effect of Chondroitin Sulphate on Pro-Inflammatory Mediators and Disease Activity in Patients with Inflammatory Bowel Disease. Digestion 2015, 92, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Hoshino, J.; Yamazaki, C.; Sekiguchi, T.; Miyauchi, S.; Horie, K. Effects of chondroitin sulfate on colitis induced by dextran sulfate sodium in rats. Jpn. J. Pharm. 2001, 85, 155–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segarra, S.; Martinez-Subiela, S.; Cerda-Cuellar, M.; Martinez-Puig, D.; Munoz-Prieto, A.; Rodriguez-Franco, F.; Rodriguez-Bertos, A.; Allenspach, K.; Velasco, A.; Ceron, J. Oral chondroitin sulfate and prebiotics for the treatment of canine Inflammatory Bowel Disease: A randomized, controlled clinical trial. BMC Vet. Res 2016, 12, 49. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Cao, J.; Jiang, X.; Cui, H. Effect of low molecular weight heparin rectal suppository on experimental ulcerative colitis in mice. Biomed. Pharm. 2010, 64, 441–445. [Google Scholar] [CrossRef]
- Zezos, P.; Kouklakis, G.; Saibil, F. Inflammatory bowel disease and thromboembolism. World J. Gastroenterol. 2014, 20, 13863–13878. [Google Scholar] [CrossRef]
- Mousavi, S.; Moradi, M.; Khorshidahmad, T.; Motamedi, M. Anti-Inflammatory Effects of Heparin and Its Derivatives: A Systematic Review. Adv. Pharm. Sci. 2015, 2015, 507151. [Google Scholar] [CrossRef] [Green Version]
- Lean, Q.Y.; Gueven, N.; Eri, R.D.; Bhatia, R.; Sohal, S.S.; Stewart, N.; Peterson, G.M.; Patel, R.P. Heparins in ulcerative colitis: Proposed mechanisms of action and potential reasons for inconsistent clinical outcomes. Exp. Rev. Clin. Pharm. 2015, 8, 795–811. [Google Scholar] [CrossRef]
- Remon, J.I.; Amin, S.C.; Mehendale, S.R.; Rao, R.; Luciano, A.A.; Garzon, S.A.; Maheshwari, A. Depth of bacterial invasion in resected intestinal tissue predicts mortality in surgical necrotizing enterocolitis. J. Perinatol. 2015, 35, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Heida, F.H.; Harmsen, H.J.; Timmer, A.; Kooi, E.M.; Bos, A.F.; Hulscher, J.B. Identification of bacterial invasion in necrotizing enterocolitis specimens using fluorescent in situ hybridization. J. Perinatol. 2017, 37, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.; Dymock, D.; Corfield, A.P.; Weaver, G.; Woodward, M.; Berry, M. Bacterial invasion of HT29-MTX-E12 monolayers: Effects of human breast milk. J. Pediatr. Surg. 2013, 48, 353–357, discussion 357–358. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kessler, S.P.; Obery, D.R.; Homer, C.R.; McDonald, C.; de la Motte, C.A. Hyaluronan 35 kDa treatment protects mice from Citrobacter rodentium infection and induces epithelial tight junction protein ZO-1 in vivo. Matrix Biol. 2017, 62, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Burge, K.Y.; Hannah, L.; Eckert, J.V.; Gunasekaran, A.; Chaaban, H. The Protective Influence of Chondroitin Sulfate, a Component of Human Milk, on Intestinal Bacterial Invasion and Translocation. J. Hum. Lact. 2019, 35, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Bueno, S.; Day, M.W.; Toby, I.T.; Akins, D.R.; Dyer, D.W. Genome Sequence of SCB34, a Sequence Type 131 Multidrug-Resistant Escherichia coli Isolate Causing Neonatal Early-Onset Sepsis. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burge, K.; Bergner, E.; Gunasekaran, A.; Eckert, J.; Chaaban, H. The Role of Glycosaminoglycans in Protection from Neonatal Necrotizing Enterocolitis: A Narrative Review. Nutrients 2020, 12, 546. https://doi.org/10.3390/nu12020546
Burge K, Bergner E, Gunasekaran A, Eckert J, Chaaban H. The Role of Glycosaminoglycans in Protection from Neonatal Necrotizing Enterocolitis: A Narrative Review. Nutrients. 2020; 12(2):546. https://doi.org/10.3390/nu12020546
Chicago/Turabian StyleBurge, Kathryn, Erynn Bergner, Aarthi Gunasekaran, Jeffrey Eckert, and Hala Chaaban. 2020. "The Role of Glycosaminoglycans in Protection from Neonatal Necrotizing Enterocolitis: A Narrative Review" Nutrients 12, no. 2: 546. https://doi.org/10.3390/nu12020546
APA StyleBurge, K., Bergner, E., Gunasekaran, A., Eckert, J., & Chaaban, H. (2020). The Role of Glycosaminoglycans in Protection from Neonatal Necrotizing Enterocolitis: A Narrative Review. Nutrients, 12(2), 546. https://doi.org/10.3390/nu12020546