A Cecal Slurry Mouse Model of Sepsis Leads to Acute Consumption of Vitamin C in the Brain

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse CS or LPS Treatment
2.2. Evaluation of Sickness Score
2.3. Tissue Collection
2.4. Ascorbic Acid HPLC
2.5. Determination of Gene Expression
2.6. Measurement of Oxidative Stress Markers
2.7. Measurement of Cytokine Expression
2.8. Statistical Analyses
3. Results
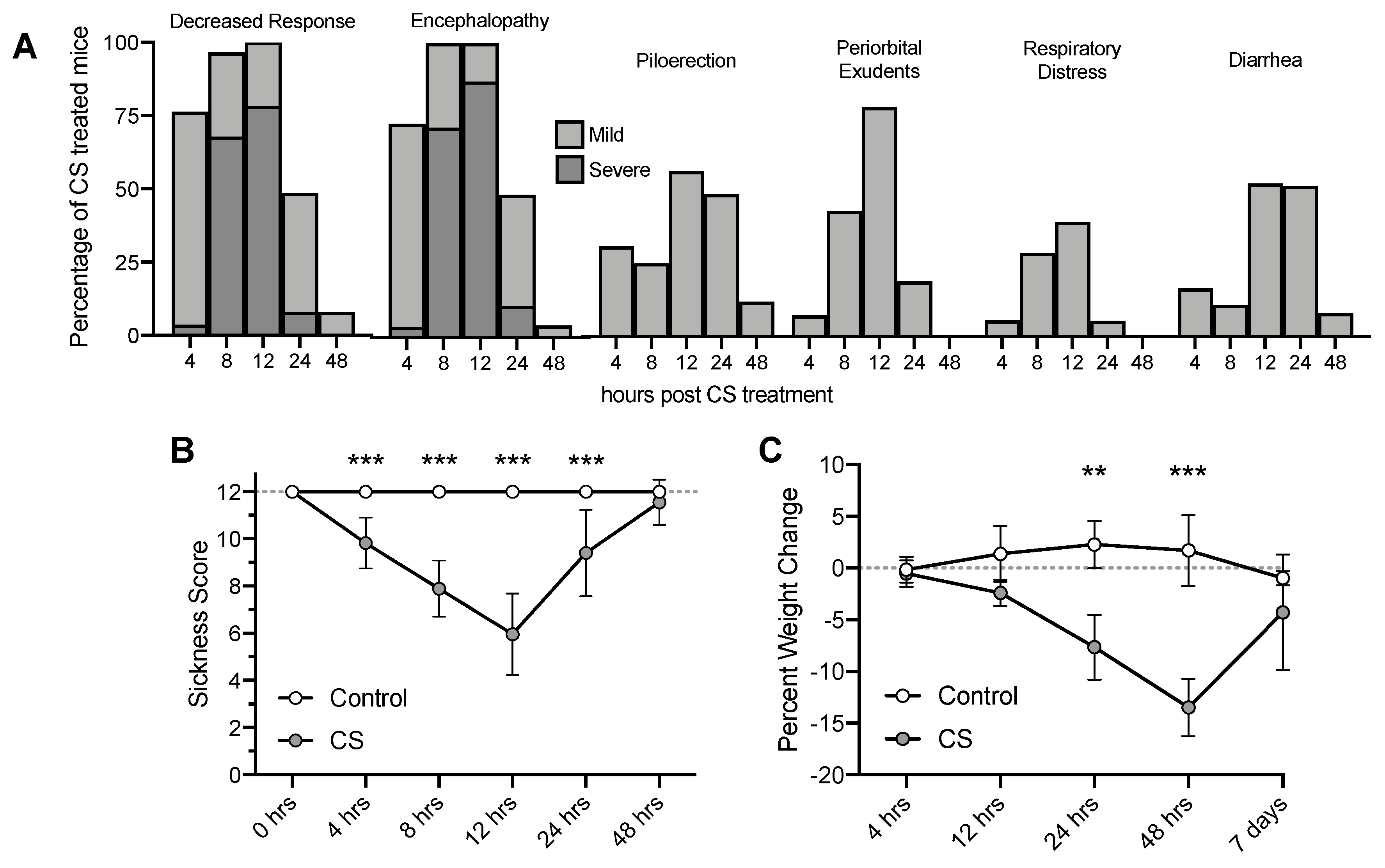
3.1. Cecal Slurry Treatment Induces Acute Peritonitis and Weight Loss
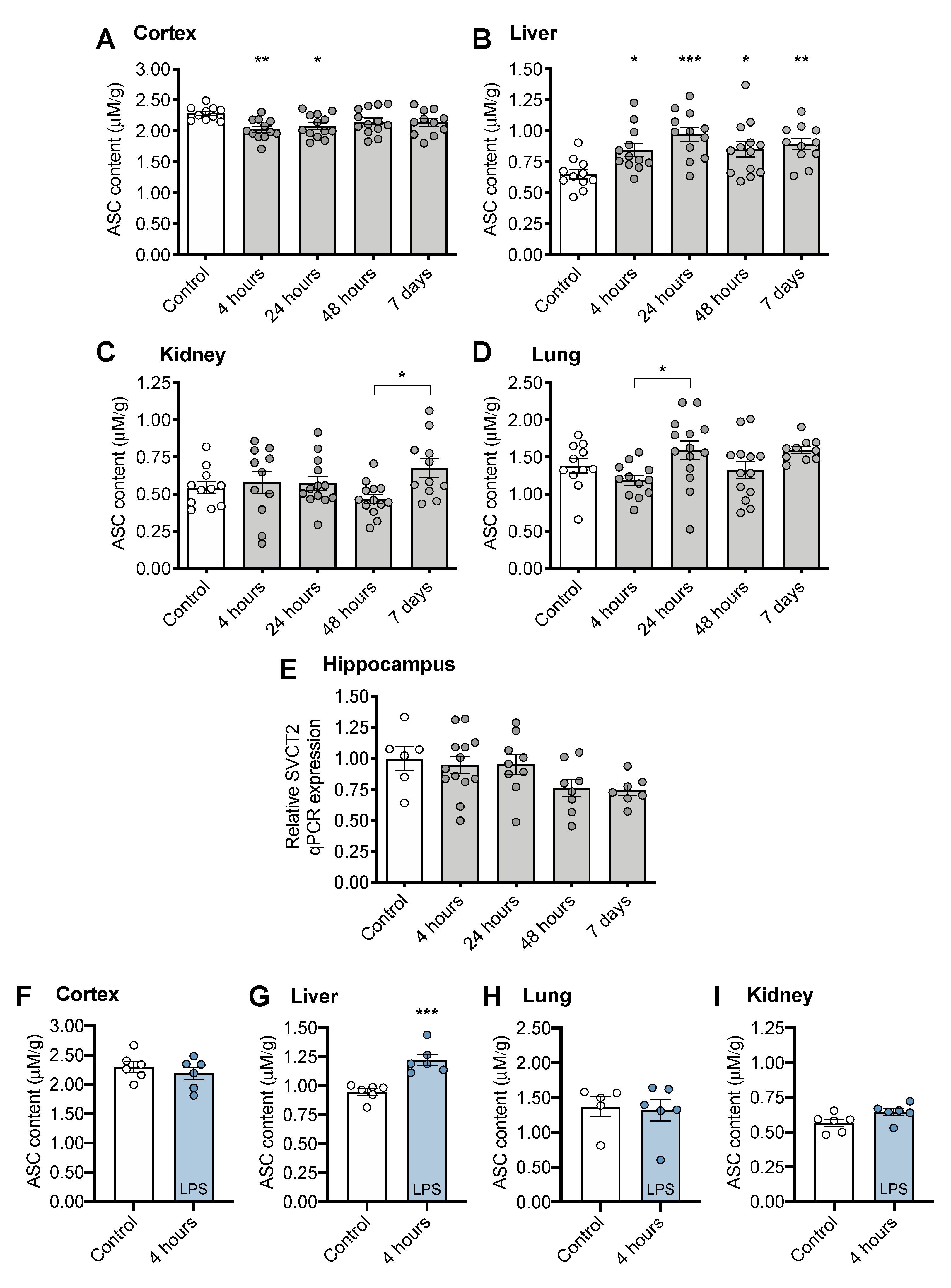
3.2. Tissue ASC Concentrations Following CS Treatment
3.3. CS Treatment Does Not Induce Changes in Oxidative Stress Measurements
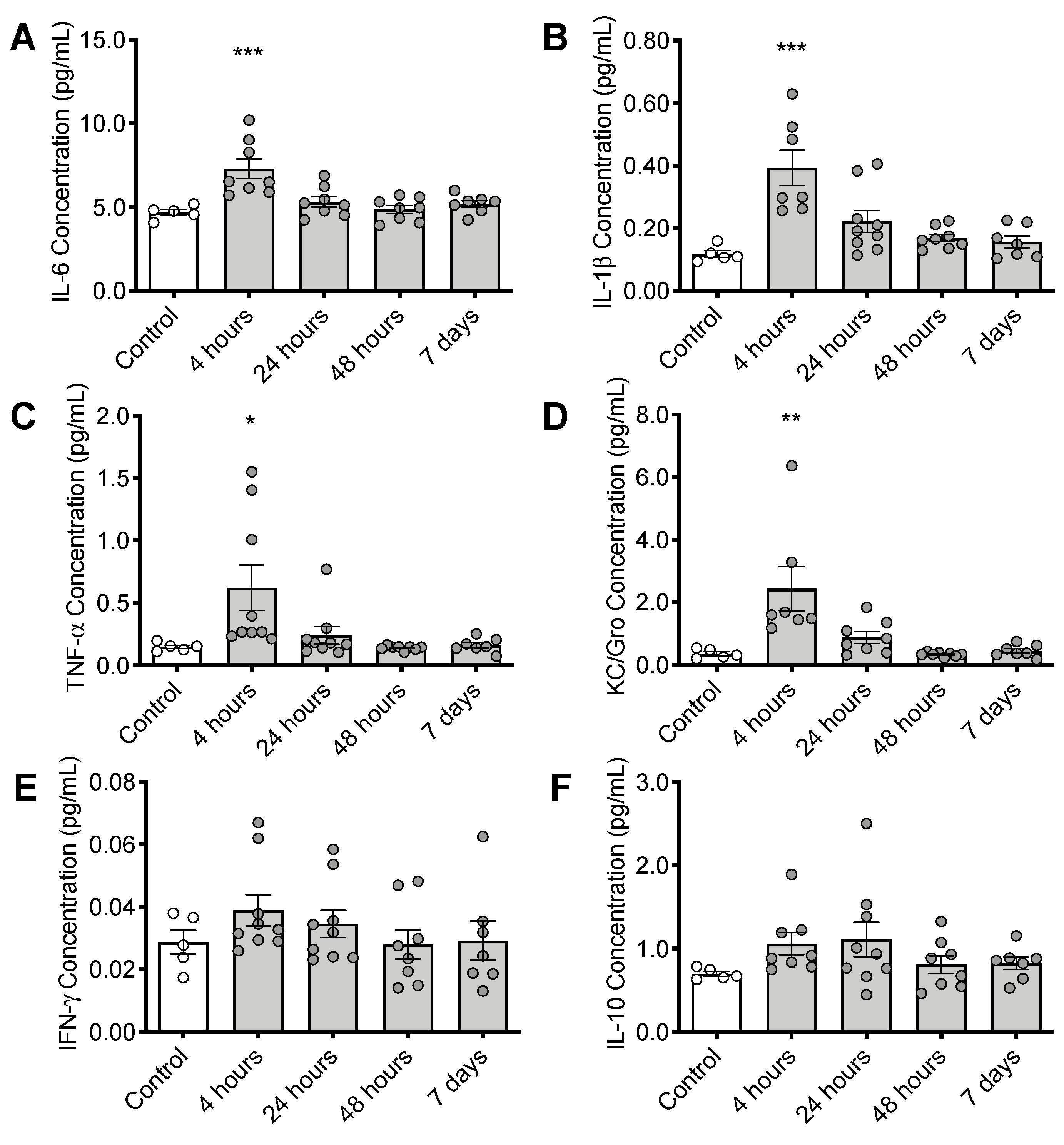
3.4. CS Treatment Initiates an Inflammatory Response in the Brain
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fleischmann, C.; Scherag, A.; Adhikari, N.K.J.; Hartog, C.S.; Tsaganos, T.; Schlattmann, P.; Angus, D.C.; Reinhart, K. Assessment of global incidence and mortality of hospital-treated sepsis current estimates and limitations. Am. J. Respir. Crit. Care Med. 2016, 193, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.O.; Meissner, K.; Mayes, L.M.; Bartels, K. Vitamin C in sepsis. Curr. Opin. Anaesthesiol. 2018, 31, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Ince, C.; Mayeux, P.R.; Nguyen, T.; Gomez, H.; Kellum, J.A.; Ospina-Tascón, G.A.; Hernandez, G.; Murray, P.; De Backer, D. The endothelium in sepsis. Shock 2016, 45, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biesalski, H.K.; McGregor, G.P. Antioxidant therapy in critical care–Is the microcirculation the primary target? Proc. Crit. Care Med. 2007, 35, S577–S583. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, F.G.; Park, M.; Machado, F.S.; Azevedo, L.C.P. Sepsis-associated encephalopathy: Not just delirium. Clinics 2011, 66, 1825–1831. [Google Scholar] [CrossRef] [Green Version]
- Borrelli, E.; Roux-Lombard, P.; Grau, G.E.; Girardin, E.; Ricou, B.; Dayer, J.M.; Suter, P.M. Plasma concentrations of cytokines, their soluble receptors, and antioxidant vitamins can predict the development of multiple organ failure in patients at risk. Crit. Care Med. 1996, 24, 392–397. [Google Scholar] [CrossRef]
- Galley, H.F.; Davies, M.J.; Webster, N.R. Ascorbyl radical formation in patients with sepsis: Effect of ascorbate loading. Free Radic. Biol. Med. 1996, 20, 139–143. [Google Scholar] [CrossRef]
- Fujii, T.; Udy, A.A.; Deane, A.M.; Luethi, N.; Bailey, M.; Eastwood, G.M.; Frei, D.; French, C.; Orford, N.; Shehabi, Y.; et al. Vitamin C, Hydrocortisone and Thiamine in Patients with Septic Shock (VITAMINS) trial: Study protocol and statistical analysis plan. Crit. Care Resusc. 2019, 21, 119–125. [Google Scholar]
- Hudson, E.P.; Collie, J.T.; Fujii, T.; Luethi, N.; Udy, A.A.; Doherty, S.; Eastwood, G.; Yanase, F.; Naorungroj, T.; Bitker, L.; et al. Pharmacokinetic data support 6-hourly dosing of intravenous vitamin C to critically ill patients with septic shock. Crit. Care Resusc. 2019, 21, 236–242. [Google Scholar]
- Carr, A.C.; Rosengrave, P.C.; Bayer, S.; Chambers, S.; Mehrtens, J.; Shaw, G.M. Hypovitaminosis C and vitamin C deficiency in critically ill patients despite recommended enteral and parenteral intakes. Crit. Care 2017, 21, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, A.A.; Syed, A.A.; Knowlson, S.; Sculthorpe, R.; Farthing, D.; DeWilde, C.; Farthing, C.A.; Larus, T.L.; Martin, E.; Brophy, D.F.; et al. Phase I safety trial of intravenous ascorbic acid in patients with severe sepsis. J. Transl. Med. 2014, 12, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Calsavara, A.J.C.; Costa, P.A.; Nobre, V.; Teixeira, A.L. Factors Associated with Short and Long Term Cognitive Changes in Patients with Sepsis. Sci. Rep. 2018, 8, 4509. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA—J. Am. Med. Assoc. 2010, 304, 1787–1794. [Google Scholar] [CrossRef] [Green Version]
- Annane, D.; Sharshar, T. Cognitive decline after sepsis. Lancet Respir. Med. 2015, 3, 61–69. [Google Scholar] [CrossRef]
- Anderson, S.T.; Commins, S.; Moynagh, P.N.; Coogan, A.N. Lipopolysaccharide-induced sepsis induces long-lasting affective changes in the mouse. Brain. Behav. Immun. 2015, 43, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Zaghloul, N.; Addorisio, M.E.; Silverman, H.A.; Patel, H.L.; Valdés-Ferrer, S.I.; Ayasolla, K.R.; Lehner, K.R.; Olofsson, P.S.; Nasim, M.; Metz, C.N.; et al. Forebrain Cholinergic Dysfunction and Systemic and Brain Inflammation in Murine Sepsis Survivors. Front. Immunol. 2017, 8, 1673. [Google Scholar] [CrossRef] [Green Version]
- Hippensteel, J.A.; Anderson, B.J.; Orfila, J.E.; McMurtry, S.A.; Dietz, R.M.; Su, G.; Ford, J.A.; Oshima, K.; Yang, Y.; Zhang, F.; et al. Circulating heparan sulfate fragments mediate septic cognitive dysfunction. J. Clin. Investig. 2019, 129, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Carr, A.C.; Maggini, S. Vitamin C and immune function. Nutrients 2017, 9, 1211. [Google Scholar] [CrossRef] [Green Version]
- Armour, J.; Tyml, K.; Lidington, D.; Wilson, J.X. Ascorbate prevents microvascular dysfunction in the skeletal muscle of the septic rat. J. Appl. Physiol. 2001, 90, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Wilson, J.X.; Tyml, K. Ascorbate protects against impaired arteriolar constriction in sepsis by inhibiting inducible nitric oxide synthase expression. Free Radic. Biol. Med. 2004, 37, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Mckinnon, R.L.; Lidington, D.; Tyml, K. Ascorbate inhibits reduced arteriolar conducted vasoconstriction in septic mouse cremaster muscle. Microcirculation 2007, 14, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Pendem, S.; Teh, S.L.; Sukumaran, D.K.; Wu, F.; Wilson, J.X. Ascorbate protects endothelial barrier function during septic insult: Role of protein phosphatase type 2A. Free Radic. Biol. Med. 2010, 48, 128–135. [Google Scholar] [CrossRef] [Green Version]
- Tyml, K.; Li, F.; Wilson, J.X. Septic impairment of capillary blood flow requires nicotinamide adenine dinucleotide phosphate oxidase but not nitric oxide synthase and is rapidly reversed by ascorbate through an endothelial nitric oxide synthase-dependent mechanism. Crit. Care Med. 2008, 36, 2355–2362. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Kamenos, G.; Pendem, S.; Wilson, J.X.; Wu, F. Ascorbate protects against vascular leakage in cecal ligation and puncture-induced septic peritonitis. Am. J. Physiol.—Regul. Integr. Comp. Physiol. 2012, 302, R409–R416. [Google Scholar] [CrossRef]
- Fisher, B.J.; Seropian, I.M.; Kraskauskas, D.; Thakkar, J.N.; Voelkel, N.F.; Fowler, A.A.; Natarajan, R. Ascorbic acid attenuates lipopolysaccharide-induced acute lung injury. Crit. Care Med. 2011, 39, 1454–1460. [Google Scholar] [CrossRef]
- Fisher, B.J.; Kraskauskas, D.; Martin, E.J.; Farkas, D.; Puri, P.; Massey, H.D.; Idowu, M.O.; Brophy, D.F.; Voelkel, N.F.; Fowler, A.A.; et al. Attenuation of sepsis-induced organ injury in mice by vitamin C. J. Parenter. Enter. Nutr. 2014, 38, 825–839. [Google Scholar] [CrossRef]
- Gao, Y.-L.; Lu, B.; Zhai, J.-H.; Liu, Y.-C.; Qi, H.-X.; Yao, Y.; Chai, Y.-F.; Shou, S.-T. The Parenteral Vitamin C Improves Sepsis and Sepsis-Induced Multiple Organ Dysfunction Syndrome via Preventing Cellular Immunosuppression. Mediat. Inflamm. 2017, 2017, 4024672. [Google Scholar] [CrossRef]
- Fujii, T.; Luethi, N.; Young, P.J.; Frei, D.R.; Eastwood, G.M.; French, C.J.; Deane, A.M.; Shehabi, Y.; Hajjar, L.A.; Oliveira, G.; et al. Effect of Vitamin C, Hydrocortisone, and Thiamine vs Hydrocortisone Alone on Time Alive and Free of Vasopressor Support among Patients with Septic Shock: The VITAMINS Randomized Clinical Trial. JAMA—J. Am. Med. Assoc. 2020, 323, 423–431. [Google Scholar] [CrossRef]
- Fowler, A.A.; Truwit, J.D.; Hite, R.D.; Morris, P.E.; Dewilde, C.; Priday, A.; Fisher, B.; Thacker, L.R.; Natarajan, R.; Brophy, D.F.; et al. Effect of Vitamin C Infusion on Organ Failure and Biomarkers of Inflammation and Vascular Injury in Patients with Sepsis and Severe Acute Respiratory Failure: The CITRIS-ALI Randomized Clinical Trial. JAMA—J. Am. Med. Assoc. 2019, 322, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Harrison, F.E.; Bowman, G.L.; Polidori, M.C. Ascorbic acid and the brain: Rationale for the use against cognitive decline. Nutrients 2014, 6, 1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballaz, S.J.; Rebec, G.V. Neurobiology of vitamin C: Expanding the focus from antioxidant to endogenous neuromodulator. Pharm. Res. 2019, 146, 104321. [Google Scholar] [CrossRef] [PubMed]
- Kuck, J.L.; Bastarache, J.A.; Shaver, C.M.; Fessel, J.P.; Dikalov, S.I.; May, J.M.; Ware, L.B. Ascorbic acid attenuates endothelial permeability triggered by cell-free hemoglobin. Biochem. Biophys. Res. Commun. 2018, 495, 433–437. [Google Scholar] [CrossRef]
- Lin, J.L.; Huang, Y.H.; Shen, Y.C.; Huang, H.C.; Liu, P.H. Ascorbic acid prevents blood-brain barrier disruption and sensory deficit caused by sustained compression of primary somatosensory cortex. J. Cereb. Blood Flow Metab. 2010, 30, 1121–1136. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.X.; Wu, F. Vitamin C in sepsis. Subcell. Biochem. 2012, 56, 67–83. [Google Scholar]
- Young, J.I.; Züchner, S.; Wang, G. Regulation of the Epigenome by Vitamin C. Annu. Rev. Nutr. 2015, 35, 545–564. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.X.; Peters, C.E.; Sitar, S.M.; Daoust, P.; Gelb, A.W. Glutamate stimulates ascorbate transport by astrocytes. Brain Res. 2000, 858, 61–66. [Google Scholar] [CrossRef]
- May, J.M. Vitamin C transport and its role in the central nervous system. Subcell. Biochem. 2012, 56, 85–103. [Google Scholar]
- Mi, D.J.; Dixit, S.; Warner, T.A.; Kennard, J.A.; Scharf, D.A.; Kessler, E.S.; Moore, L.M.; Consoli, D.C.; Bown, C.W.; Eugene, A.J.; et al. Altered glutamate clearance in ascorbate deficient mice increases seizure susceptibility and contributes to cognitive impairment in APP/PSEN1 mice. Neurobiol. Aging 2018, 71, 241–254. [Google Scholar] [CrossRef]
- Shaver, C.M.; Paul, M.G.; Putz, N.D.; Landstreet, S.R.; Kuck, J.L.; Scarfe, L.; Skrypnyk, N.; Yang, H.; Harrison, F.E.; de Caestecker, M.P.; et al. Cell-free hemoglobin augments acute kidney injury during experimental sepsis. Am. J. Physiol. Physiol. 2019, 317, F922–F929. [Google Scholar] [CrossRef]
- Meegan, J.E.; Shaver, C.M.; Putz, N.D.; Jesse, J.J.; Landstreet, S.R.; Lee, H.N.R.; Sidorova, T.N.; Brennan McNeil, J.; Wynn, J.L.; Cheung-Flynn, J.; et al. Cell-free hemoglobin increases inflammation, lung apoptosis, and microvascular permeability in murine polymicrobial sepsis. PLoS ONE 2020, 15, e0228727. [Google Scholar] [CrossRef]
- Eric Kerchberger, V.; Bastarache, J.A.; Shaver, C.M.; Nagata, H.; Brennan McNeil, J.; Landstreet, S.R.; Putz, N.D.; Kuang Yu, W.; Jesse, J.; Wickersham, N.E.; et al. Haptoglobin-2 variant increases susceptibility to acute respiratory distress syndrome during sepsis. JCI Insight 2019, 4, 21. [Google Scholar] [CrossRef]
- Su, G.; Atakilit, A.; Li, J.T.; Wu, N.; Luong, J.; Chen, R.; Bhattacharya, M.; Sheppard, D. Effective treatment of mouse sepsis with an inhibitory antibody targeting integrin αvβ5. Crit. Care Med. 2013, 41, 546–553. [Google Scholar] [CrossRef]
- Manley, M.O.; O’Riordan, M.A.; Levine, A.D.; Latifi, S.Q. Interleukin 10 extends the effectiveness of standard therapy during late sepsis with serum interleukin 6 levels predicting outcome. Shock 2005, 23, 521–526. [Google Scholar]
- Harrison, F.E.; Yu, S.S.; Van Den Bossche, K.L.; Li, L.; May, J.M.; McDonald, M.P. Elevated oxidative stress and sensorimotor deficits but normal cognition in mice that cannot synthesize ascorbic acid. J. Neurochem. 2008, 106, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- May, J.M.; Qu, Z.C.; Mendiratta, S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch. Biochem. Biophys. 1998, 349, 281–289. [Google Scholar] [CrossRef]
- Harrison, F.E.; Hosseini, A.H.; McDonald, M.P.; May, J.M. Vitamin C reduces spatial learning deficits in middle-aged and very old APP/PSEN1 transgenic and wild-type mice. Pharm. Biochem. Behav. 2009, 93, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Sgaravatti, Â.M.; Magnusson, A.S.; Oliveira, A.S.; Mescka, C.P.; Zanin, F.; Sgarbi, M.B.; Pederzolli, C.D.; Wyse, A.T.S.; Wannmacher, C.M.D.; Wajner, M.; et al. Effects of 1,4-butanediol administration on oxidative stress in rat brain: Study of the neurotoxicity of γ-hydroxybutyric acid in vivo. Metab. Brain Dis. 2009, 24, 271–282. [Google Scholar] [CrossRef]
- Aksenov, M.Y.; Markesbery, W.R. Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci. Lett. 2001, 302, 141–145. [Google Scholar] [CrossRef]
- Bastarache, J.A.; Koyama, T.; Wickersham, N.E.; Ware, L.B. Validation of a multiplex electrochemiluminescent immunoassay platform in human and mouse samples. J. Immunol. Methods 2014, 408, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabbay, K.H.; Bohren, K.M.; Morello, R.; Bertin, T.; Liu, J.; Vogel, P. Ascorbate synthesis pathway: Dual role of ascorbate in bone homeostasis. J. Biol. Chem. 2010, 285, 19510–19520. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.; Downing, D. New Concepts in the Biology and Biochemistry of Ascorbic Acid. J. Nutr. Med. 1992, 3, 361–362. [Google Scholar] [CrossRef]
- Ching, S.; Mahan, D.C.; Dabrowski, K. Liver L-Gulonolactone Oxidase Activity and Tissue Ascorbic Acid Concentrations in Nursing Pigs and the Effect of Various Weaning Ages. J. Nutr. 2001, 131, 2002–2006. [Google Scholar] [CrossRef]
- Ching, S.; Mahan, D.C.; Ottobre, J.S.; Dabrowski, K. Ascorbic Acid Synthesis in Fetal and Neonatal Pigs and in Pregnant and Postpartum Sows. J. Nutr. 2001, 131, 1997–2001. [Google Scholar] [CrossRef]
- Harrison, F.E.; Dawes, S.M.; Meredith, M.E.; Babaev, V.R.; Li, L.; May, J.M. Low vitamin C and increased oxidative stress and cell death in mice that lack the sodium-dependent vitamin C transporter SVCT2. Free Radic. Biol. Med. 2010, 49, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Jackson, T.S.; Xu, A.; Vita, J.A.; Keaney, J.F. Ascorbate prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations. Circ. Res. 1998, 83, 916–922. [Google Scholar] [CrossRef] [Green Version]
- Kuo, S.-M.; Tan, C.-H.; Dragan, M.; Wilson, J.X. Endotoxin Increases Ascorbate Recycling and Concentration in Mouse Liver. J. Nutr. 2005, 135, 2411–2416. [Google Scholar] [CrossRef] [Green Version]
- Maeda, N.; Hagihara, H.; Nakata, Y.; Hiller, S.; Wilder, J.; Reddick, R. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc. Natl. Acad. Sci. USA 2000, 97, 841–846. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.X. Evaluation of Vitamin C for Adjuvant Sepsis Therapy. Antioxid. Redox Signal 2013, 19, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Prauchner, C.A. Oxidative stress in sepsis: Pathophysiological implications justifying antioxidant co-therapy. Burns 2017, 43, 471–485. [Google Scholar] [CrossRef]
- Ahn, J.H.; Oh, D.K.; Huh, J.W.; Lim, C.M.; Koh, Y.; Hong, S.B. Vitamin C alone does not improve treatment outcomes in mechanically ventilated patients with severe sepsis or septic shock: A retrospective cohort study. J. Thorac. Dis. 2019, 11, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Crit. Care Med. 2017, 45, 486–552. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Consoli, D.C.; Jesse, J.J.; Klimo, K.R.; Tienda, A.A.; Putz, N.D.; Bastarache, J.A.; Harrison, F.E. A Cecal Slurry Mouse Model of Sepsis Leads to Acute Consumption of Vitamin C in the Brain. Nutrients 2020, 12, 911. https://doi.org/10.3390/nu12040911
Consoli DC, Jesse JJ, Klimo KR, Tienda AA, Putz ND, Bastarache JA, Harrison FE. A Cecal Slurry Mouse Model of Sepsis Leads to Acute Consumption of Vitamin C in the Brain. Nutrients. 2020; 12(4):911. https://doi.org/10.3390/nu12040911
Chicago/Turabian StyleConsoli, David C., Jordan J. Jesse, Kelly R. Klimo, Adriana A. Tienda, Nathan D. Putz, Julie A. Bastarache, and Fiona E. Harrison. 2020. "A Cecal Slurry Mouse Model of Sepsis Leads to Acute Consumption of Vitamin C in the Brain" Nutrients 12, no. 4: 911. https://doi.org/10.3390/nu12040911
APA StyleConsoli, D. C., Jesse, J. J., Klimo, K. R., Tienda, A. A., Putz, N. D., Bastarache, J. A., & Harrison, F. E. (2020). A Cecal Slurry Mouse Model of Sepsis Leads to Acute Consumption of Vitamin C in the Brain. Nutrients, 12(4), 911. https://doi.org/10.3390/nu12040911