1. Introduction
In the last century, great advances were made in modern medicine to control diseases. However, many diseases, such as cancers, are not yet completely curable. To find out new and alternative therapies, researchers are working with traditional medicines (bioactive molecules), in parallel with modern medicine, to create new combinations for better treatment and management of diseases.
Nigella sativa (
N. sativa), has been used for medicinal purposes for centuries [
1].
N. sativa belongs to the botanical family of Ranunculaceae. It is a small shrub with tapering green leaves and rosaceous white and purplish flowers [
2]. The most important bioactive elements found in
N. sativa are; thymoquinone, thymohydroquinone, dithymoquinone, thymol, nigellimine-N-oxide, nigellicine, nigellidine, arvacrol, and alpha-hederin [
2]. Among these, Thymoquinone (TQ) is an important bioactive ingredient primarily found in black seed oil. Recent scientific investigations on TQ indicate a number of bioactivities, which include anti-carcinogenetic, anti-inflammatory, antiulcer, antihypertensive, antibacterial and antifungal, hepatoprotective, antipyretic and analgesic, as well as antioxidant activities such as reducing reactive oxygen species, inhibition of rheumatoid arthritis in rat models, and antihyperlipidemic [
3]. Treatment of cancer cells with TQ can result in inhibition of tumor cell proliferation within modulation of apoptosis signaling, inhibition of angiogenesis, and cell cycle arrest [
4].
TQ has been shown to negatively modulate pyruvate kinase M2 (PKM2), an enzyme related to cancer cell energy pathways [
5]. Similarly, TQ treatment has been shown to modulate various TCA cycle metabolites and lipids in cancer cells, which are critical for their survival. Further, TQ represses many signaling pathways directly involved in controlling the metabolic pathways of cancer cells, like PI3K, AKT, JNK and STAT3 [
6].
System-wide analyses of metabolites under the umbrella of metabolomics allow a unique opportunity to understand the molecular aspects of carcinogenesis and cancer biology by enabling deep investigation of targeted aspects of cancer metabolism [
7,
8]. In addition, it provides a unique opportunity to understand and quantify a global impact of anti-carcinogenic compounds affecting the metabolism of cancer cells. The major aim of the current study is to explore the metabolic impacts of TQ treatment on cancer cells (leukemia cell lines), and to obtain the differences in their metabolomic patterns, in order to identify metabolites and modified metabolic pathways.
2. Materials and Methods
2.1. Cell Culture
Acute T cell leukemia (Jurkat (clone E6-1)), acute pro-myelocytic leukemia (HL-60), and an erythroleukemia cell line derived from a chronic myeloid leukemia patient (K-562) were obtained from the American Type Culture Collection (ATCC) (Rockville, MD, USA). These cells were grown as a suspension culture. These cells were cultured in Roswell Park Memorial Institute (RPMI 1640), supplemented with 15% heat-inactivated fetal bovine serum (FBS), and 1X penicillin–streptomycin. Cells were monitored daily using a microscope to monitor confluence and general culture conditions. Every two-days, the cells were passaged at a dilution of 1:1 or 1:2. Sub-culturing was done when the cell density was more than 1 × 106 cells/mL. Frozen cell lines were stored in liquid nitrogen and thawed in a water bath for 30 to 60 s until the thawing was partially complete. Cell counting was done by using a hemocytometer.
2.2. TQ Preparation and Treatment
TQ solution was prepared in ethanol at a concentration of 100 µM. This stock was stored at −20 °C in eppendorf tubes wrapped in aluminum foil to avoid dimer formation. All cell lines were treated by TQ immediately after preparation and treated for 24 h using two different concentrations (5 µM and 10 µM) for metabolite extraction.
2.3. Measurement of Cell Viability Using Trypan Blue Exclusion Test
Trypan blue exclusion assay allows a direct identification and enumeration of live (unstained) and dead (blue) cells in a given population. however; it is not able to differentiate between apoptotic and necrotic cells. Jurkat, HL-60 and K-562 were plated in replicate (1.5 × 105 cells/well) in a 96-well micro-plate and treated with TQ (5 µM and 10 µM), followed by an incubation of 24 h at 37 °C and 5% CO2. After incubation, 20 µL from cell and 20 µL from TB was taken and mixed, and the viable and non-viable cells were counted using a hemocytometer. The negative control cell suspension was treated with ethanol of different doses (5 µM and 10 µM).
2.4. Measurement of Growth Assay and Cell Proliferation Using WST-1 Assay
Briefly, 7 × 103 cells/mL cells were seeded in a 96-well plate with a cell density of 1.5 × 105 cells/mL, and were treated with TQ (5 µM and 10 µM). The WST-1 reagent was applied to all wells, with a final dilution of 1:10 for 4 h prior to examination of the cells after 24 h treatment. The WST-1 assay is based on the cleavage of water-soluble tetrazolium salt-1, which is tetrazolium salt that is reduced extra-cellularly to formazan dye by enzymes of the plasma membrane oxidoreductase. The primary reductant is NADH, derived from the TCA of the mitochondria. WST-1 is converted by metabolically active cells and was employed to measure cell proliferation. The optical density of the formazan dye was measured to get an estimate of live cells. The absorbance was measured at a wavelength of 450 nm, using the ELISA. The value obtained from the blank (complete medium only) was subtracted manually from all wells before calculating the percentage metabolism in each well.
2.5. Apoptosis Assay
Apoptosis was assessed by using the Annexin V-PI kit. Annexin V can specifically bind to phosphatidylserine. Cells undergoing apoptosis will experience diffusion of phosphatidylserine from inner cell membrane to outer cell membrane, whereby these proteins can be quantified by targeting them with fluorescence-tagged Annexin V. The experimental procedures were carried out according to the manufacturer’s protocol. Briefly, cells were seeded (2 × 106 cells/well) in a 6-well microtiter plate followed by overnight incubation with and without treatment. After 24 h incubation, the cells were washed, and incubated with Annexin V-FITC solution for 15 min under dark conditions. After a washing step, the cells were analyzed with a flow cytometer after the adding of PI solution.
2.6. Cell Cycle Assay
Cell cycle progression of leukemia cells was examined by flow cytometry after propidium iodide (PI) staining. Cell cycle phases can be differentiated by measuring their DNA content, where G1 phase is 2n, S phase is between 2n and 4n, and G2 phase is 4n. PI can diffuse into the cell nucleus and binds to DNA proportionately to its amount; thus, by measuring the PI signal, the DNA content of each cell can be determined. Briefly, cells were seeded (2 × 106 cells/well) in a 6-well microtiter plate followed by overnight incubation. The cells were synchronized by starvation in serum-free medium for 24 h. After appropriate treatment, the cells were detached and fixed with 70% ethanol for at least 2 h, before staining with PI solution (mixed with RNase) under dark condition for 20 min. The cells were then analyzed with a flow cytometer.
2.7. Cell Morphology
Morphological study is based on the identification of the leukaemia cell line and stage of cell differentiation. Jurkat, HL-60 and K-562 cells were treated with Thymoquinone. We used Hydrogen Peroxide (H2O2) as a positive control and ethanol as a negative control. Slides of the cell suspension were prepared by Cytospins 4 (Thermo Fisher Scientific, Waltham, MA, USA), first adding Bovine Albumin 22% specific, then centrifuge at 750 rpm for 5 min. Slides were air-dried, methanol fixed. Cell were stained with Giemsa stain reagents. Slides were dried and then mounted onto a coverslip. Stained cells were observed using a light microscope and the power was 60X.
2.8. Metabolites Extraction
Metabolites were extracted from both TQ-treated and untreated cells using a combination of methanol: acetonitrile: water at a ratio of (2:2:1
v/v). Ice-cold solvent in a volume of one mL was added to the cell supernatant, and the mixture was quickly vortexed for 30s and incubated for 1 h at −20 °C, followed by spin for 15 min at 13,000 rpm at 4 °C. The supernatant was removed, and the sample was dried in a vacuum concentrator. Further, dry extracts were reconstituted in 100 μL of acetonitrile: water (1:1,
v/v), vortexed for 10 min and spin for 15 min at 13,000 rpm at 4 °C to remove insoluble debris. Finally, the resultant supernatants were taken for LC-MS/MS run [
9,
10].
2.9. HPLC Workflow
Ten µL of sample was injected into a HPLC column (Hypersail gold; 150 mm × 4.6 mm, 5 μm) with a flow rate of 0.2 mL/min. The mobile phase consisted of 0.1% Formic acid and 99.9% ACN formic acid (0.1%,
v/v), using a linear gradient programme where the component of solution was changed from 5% B to 100% B over 90 min, at a constant flow rate of 0.2 mL/min (95% A from 5% to 30% over 72 min, 30% to 100% over 10 min, and kept at 100% for 5 min at a flow rate of 0.25 mL/min). The column temperature was maintained at 30 °C [
9,
10].
2.10. LC-MS/MS
A LTQ XL™ linear ion trap instrument (Thermo Fisher Scientific) was used for the quantification of metabolites. Full scan scope was chosen from 100 to 1000
m/z. The spray voltage was set at −3.0 kV. The capillary voltage was fixed at 4.0 V and the temperature was set at 270 °C. Nitrogen was used as a sheath gas and the flow rate was set at 40 arbitrary units. Further, helium was used as the buffer gas for the run [
9,
10].
2.11. Data Processing
The data obtained was processed using (XC-MS) data processing software. The raw MS data (raw files) were processed using XC-MS for feature detection, retention time correction and alignment. The parameters in XC-MS were set as follows: Cent-Wave settings for feature detection (Δ m/z = 30 ppm, minimum peak width = 10 s and maximum peak width = 120 s) and m/zwid = 0.25, min frac = 0.5, and bw = 10 for chromatogram alignment. After careful evaluation of retention time, alignment was shown not to be required. Isotopic peaks and adducts were detected using CAMERA. The precursor was matched with METLIN database at 20 ppm accuracy.
2.12. Multivariate Data Analysis
After data preprocessing, multivariate data analysis is then used to identify the metabolites that change most significantly because of treatments or over time. Different multivariate models are used in metabolomics, including principal component analysis (PCA), partial least squares discriminant analysis (PLS-DA), and orthogonal partial least squares discriminant analysis (OPLS-DA) as the most common methods.
2.13. Statistical Analysis
All the experiments were performed in triplicates. Statistical analyses were performed using the One-way ANOVA and t-test. Graphs were produced using GraphPad Prism v5.0 and the Microsoft Excel program.
4. Discussion
One of the earliest and most common methods for measuring cell viability is the trypan blue (TB) exclusion assay. Trypan blue binds to intracellular proteins, thereby rendering the cells a bluish color. The trypan blue exclusion assay allows for a direct identification and enumeration of live (unstained) and dead (blue) cells in a given population. However; it is not able to differentiate between apoptotic and necrotic cells. In the present study, Jurkat, HL-60 and K-562 were cultured in RPMI-1640 medium and treated with Thymoquinone for 24 h. TQ inhibited cell viability in leukemic cells, and the more significant cells that were affected by TQ compounds were K-562 cells.
Further analysis of leukemic cells in cell proliferation of was determined by WST-1. The WST-1 assay is based on the cleavage of water-soluble tetrazolium salt-1, which is reduced extra-cellularly to formazan dye by enzymes of the plasma membrane oxidoreductase. The primary reductant is NADH, derived from the TCA of the mitochondria. WST-1 is converted by metabolically active cells and was employed to measure cell proliferation. The optical density of the formazan dye is measured to get an estimate of live cells. In the present study, Jurkat, HL-60 and K-562 was cultured in RPMI-1640 medium and treated with Thymoquinone for 24 h. TQ treatment is significant on HL-60 and more significant on K-562 cells.
The level of cell death in cancer cells was assessed with the Annexin V-PI kit. Annexin V can specifically bind to phosphatidylserine. Cells undergoing cell death will experience diffusion of phosphatidylserine from inner cell membrane to outer cell membrane, whereby these proteins can be quantified by targeting them with fluorescence-tagged Annexin V. The necrosis as a form of cell death is almost always associated with a pathological process. We showed in (
Table S1) that cells die by necrosis more than apoptosis on Jurkat, HL-60 and K-562 after treatment by TQ.
Cell cycle progression of leukemia cells was examined by flow cytometry after propidium iodide (PI) staining. Cell cycle phases can be differentiated by measuring their DNA content, where G1 phase is 2n, S phase is between 2n and 4n, and G2 phase is 4n. PI can diffuse into the cell nucleus and binds to DNA proportionately to its amount. Jurkat, HL-60 and K-562 cells were stained with DNA-specific fluorochrome Propidium iodide. TQ interferes with DNA structure. It aims at cellular copper, which is current in the chromatin and is closely associated with DNA base guanine, and is the reason for oxidative breakage to DNA and consequent cancer cell death. In an earlier study, TQ was found to inhibit DNA synthesis, proliferation, and viability of cancerous cells, such as LNCaP, C4-B, DU145, and PC-3, but not noncancerous BPH-1 prostate epithelial cells [
5]. We showed in (
Table S2) that DNA content is more in G1/G0 phases on Jurkat, HL-60 and K-562 after treatment by TQ.
The morphological variation between untreated and treated leukemic cells correlated to their different types of cells and treatments. In the present study, Jurkat, HL-60 and K-562 were cultured in RPMI-1640 medium and treated with TQ for indicated time. Hydrogen Peroxide (H2O2) as a positive control and ethanol as a negative control for 24 h. On the second day, treated and untreated leukemic cell lines was prepared by Cytospins 4, cells were stained with Giemsa stain reagents and then cells were observed using a light microscope and the power was 60X. TQ treatment showed significant decrease in cell size when compared the control cells treated with H2O2 and Ethanol.
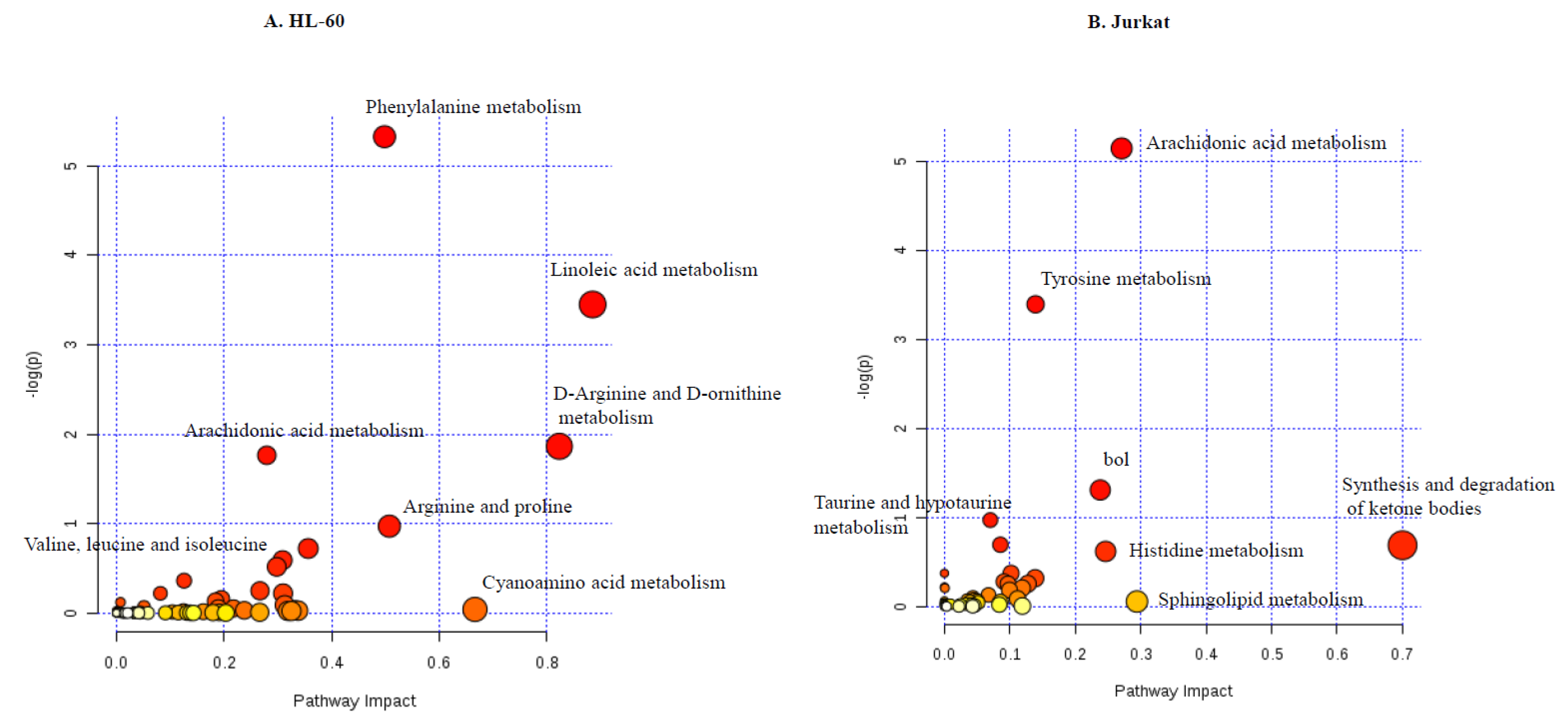
In the current work, we provided a comprehensive map of metabolites and metabolic pathways altered by TQ in cancer cells. We found that TQ alters major metabolic pathways like TCA cycle, amino acid metabolism, lipid metabolism and metabolites important for methylation-related events.
We decided to use an active ingredient extracted from natural products or traditional medicine because these medicines have been used in the human body for many generations, and any possible adverse effects/toxicities have been well identified and are likely manageable. Cancer cell lines are well established models to study specific cellular mechanisms characteristic of different types of cancer, usually by monitoring specific proteins and their actions [
11]. Thus, in this project, we focused on the anticancer activities of thymoquinone, a natural product isolated from
Nigella sativa, in the treatment of leukemia, rather than chemotherapy and radiotherapy, which targets cancerous and healthy cells, and tried to elucidate its mechanism of action in multiple signaling pathways. Resent advances in omics technologies allow for more systematic investigation to characterize parameters like gene expression, protein and metabolite profiles. For the application of omics technologies to the understanding of compound action, it is essential that the effects of compounds produce a sensitive response, which can be reproduced with sufficiently high quality. Here, we have shown that the metabolic profiling of cancer cells can not only identify phenotypic differences between cell lines, but can also sensitively detect and distinguish changes in metabolite concentrations induced by relatively short exposure to differing compounds. It is important to mention that all treatments caused a distinct reproducible profile, which is observed in unsupervised PCA. TQ alters the metabolic signature of leukemic cells, and earlier it showed that TQ activates ROS production and p38 phosphorylation in breast cancer cells. This results in TQ’s effects in anti-proliferative and pro-apoptotic effects. Metabolic pathways play a key role in cellular physiology. TQ alters major metabolic pathways, which results in induction of apoptosis and disturbances in cellular homeostasis. Even though there is heterogeneity between the two cell lines, TQ activates some similar metabolite changes, which results in apoptosis and epigenetic variation.
It has been shown that TQ can induce DNA damage in glioblastoma cells [
12]. Here, we found significant accumulation of thymine glycol, an intermediary metabolite known to induce DNA damage and apoptosis in cancer cells [
13]. Therefore, we believe that TQ promotes the accumulation of thymine glycol to promote DNA damage of leukemic cells, to induce apoptosis. However, this assumption needs further scientific evidence. Further, we observed a strong reduction in cellular guanine pools during TQ treatment. High guanine levels are maintained by cancer cells to facilitate nucleotide metabolism and their sufficient pools [
14,
15,
16].
We found that α-KG is acceleration of cell proliferation, which shows decreases in its fold variation irrespective of two cell lines and doses. It was also shown to be a reduced accumulation of onco-metabolites like Fumarate. The palmitic acid pathway is a key regulatory element in cell survival apoptosis. Palmitic acid converts into ceramide, and then to sphingosine. Two major metabolites involved in palmitic acid pathways are ceramide and sphingosine, which are involved in cell toxicity and cell survival. Significantly, ceramide shows an increase in its accumulation, which supports the TQ-induced apoptosis. TQ also has major effects on DNA methylation metabolites.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











