Interplay of Enzyme Therapy and Dietary Management of Murine Homocystinuria



Abstract
:1. Introduction
2. Materials and Methods
2.1. Test Compounds
2.2. Diets
2.3. Animals and Study Design
2.4. Blood Collection and Analysis
2.5. Statistical Analysis
3. Results
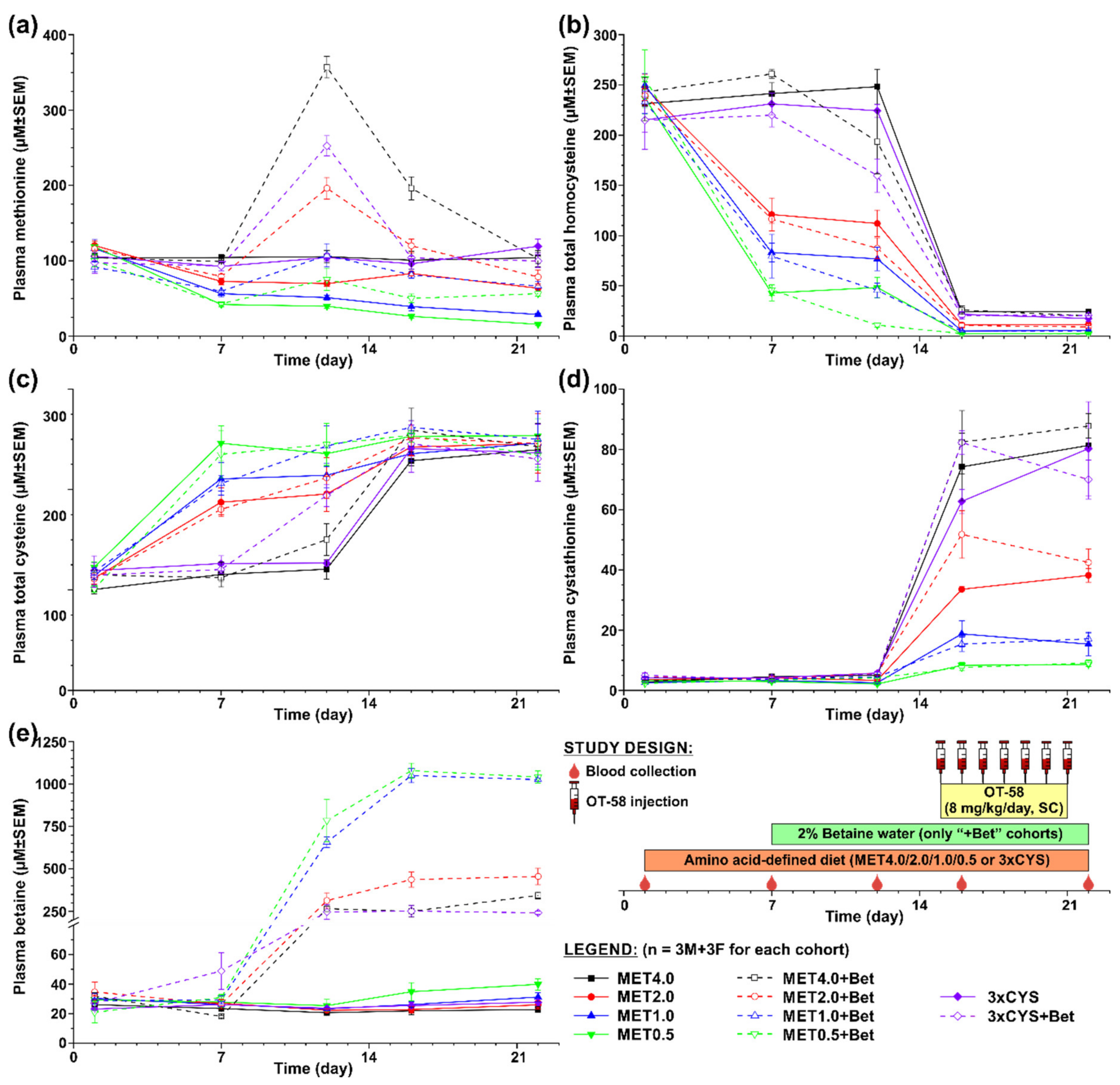
3.1. Short-Term Evaluation of OT-58 on the Background of Current Standard of Care for HCU
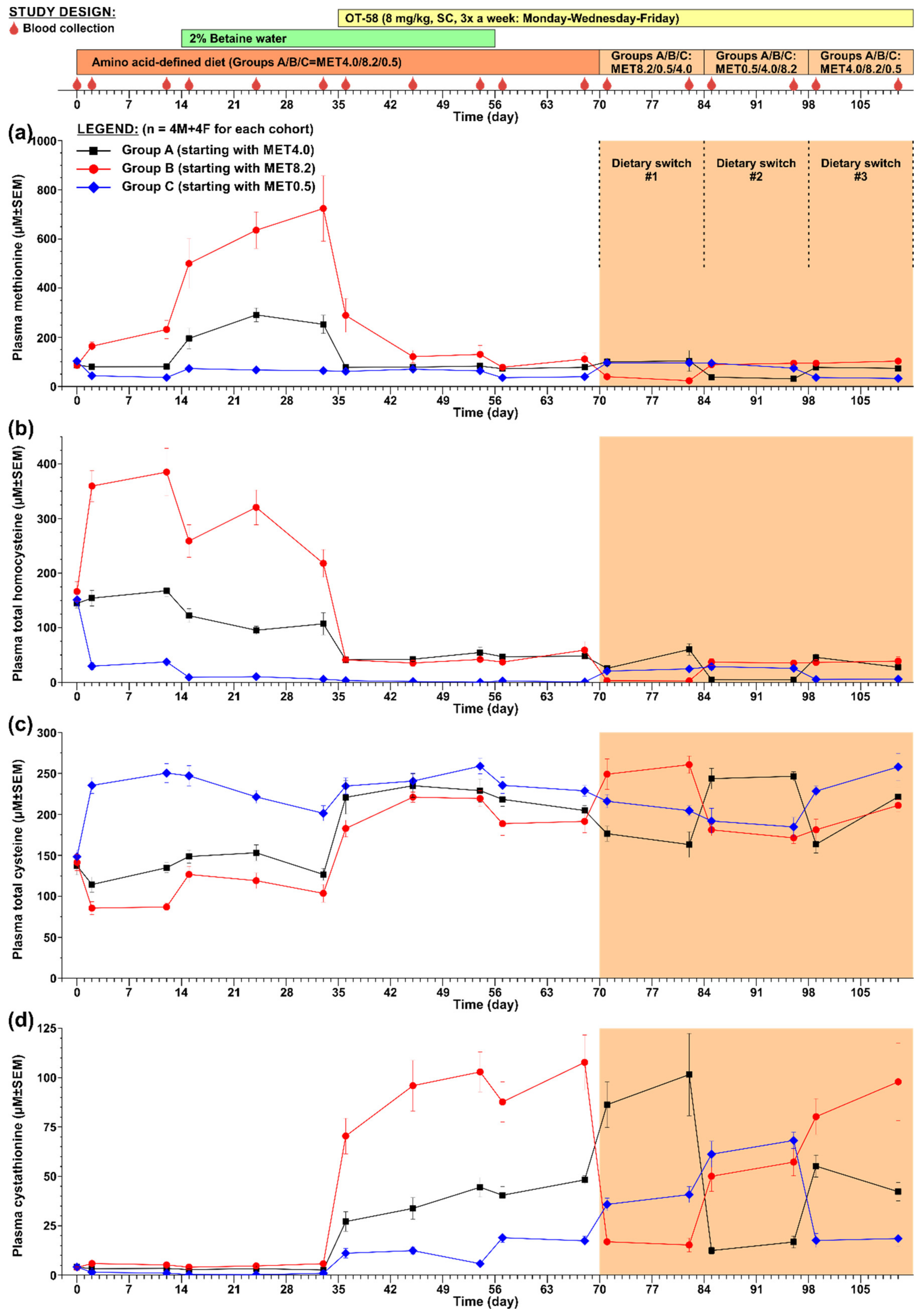
3.2. Long-Term Evaluation of OT-58
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mudd, S.H.; Levy, H.L.; Kraus, J.P. Disorders of Transsulfuration. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Childs, B., Kinzler, K., Vogelstein, B., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2007–2056. [Google Scholar]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-beta-Synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef]
- Morris, A.A.; Kozich, V.; Santra, S.; Andria, G.; Ben-Omran, T.I.; Chakrapani, A.B.; Crushell, E.; Henderson, M.J.; Hochuli, M.; Huemer, M.; et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J. Inherit. Metab. Dis. 2017, 40, 49–74. [Google Scholar] [CrossRef] [Green Version]
- Walter, J.H.; Wraith, J.E.; White, F.J.; Bridge, C.; Till, J. Strategies for the treatment of cystathionine β-synthase deficiency: The experience of the Willink Biochemical Genetics Unit over the past 30 years. Eur. J. Pediatrics 1998, 157 (Suppl. 2), S71–S76. [Google Scholar] [CrossRef]
- Majtan, T.; Park, I.; Carrillo, R.S.; Bublil, E.M.; Kraus, J.P. Engineering and Characterization of an Enzyme Replacement Therapy for Classical Homocystinuria. Biomacromolecules 2017, 18, 1747–1761. [Google Scholar] [CrossRef]
- Majtan, T.; Hulkova, H.; Park, I.; Krijt, J.; Kozich, V.; Bublil, E.M.; Kraus, J.P. Enzyme replacement prevents neonatal death, liver damage, and osteoporosis in murine homocystinuria. FASEB J. 2017, 31, 5495–5506. [Google Scholar] [CrossRef] [Green Version]
- Majtan, T.; Jones, W., Jr.; Krijt, J.; Park, I.; Kruger, W.D.; Kozich, V.; Bassnett, S.; Bublil, E.M.; Kraus, J.P. Enzyme Replacement Therapy Ameliorates Multiple Symptoms of Murine Homocystinuria. Mol. Ther. 2018, 26, 834–844. [Google Scholar] [CrossRef]
- Majtan, T.; Park, I.; Cox, A.; Branchford, B.R.; di Paola, J.; Bublil, E.M.; Kraus, J.P. Behavior, body composition, and vascular phenotype of homocystinuric mice on methionine-restricted diet or enzyme replacement therapy. FASEB J. 2019, 33, 12477–12486. [Google Scholar] [CrossRef] [Green Version]
- Maclean, K.N.; Sikora, J.; Kozich, V.; Jiang, H.; Greiner, L.S.; Kraus, E.; Krijt, J.; Overdier, K.H.; Collard, R.; Brodsky, G.L.; et al. A novel transgenic mouse model of CBS-deficient homocystinuria does not incur hepatic steatosis or fibrosis and exhibits a hypercoagulative phenotype that is ameliorated by betaine treatment. Mol. Genet. Metab. 2010, 101, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Bublil, E.M.; Majtan, T.; Park, I.; Carrillo, R.S.; Hulkova, H.; Krijt, J.; Kozich, V.; Kraus, J.P. Enzyme replacement with PEGylated cystathionine beta-synthase ameliorates homocystinuria in murine model. J. Clin. Investig. 2016, 126, 2372–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arning, E.; Bottiglieri, T. Quantitation of S-Adenosylmethionine and S-Adenosylhomocysteine in Plasma Using Liquid Chromatography-Electrospray Tandem Mass Spectrometry. Methods Mol. Biol. 2016, 1378, 255–262. [Google Scholar] [CrossRef]
- Majtan, T.; Park, I.; Bublil, E.M.; Kraus, J.P. Enzyme replacement therapy prevents loss of bone and fat mass in murine homocystinuria. Hum. Mutat. 2018, 39, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Komrower, G.M.; Lambert, A.M.; Cusworth, D.C.; Westall, R.G. Dietary treatment of homocystinuria. Arch. Dis. Child. 1966, 41, 666–671. [Google Scholar] [CrossRef] [Green Version]
- Carson, N.A.J.; Cusworth, D.C.; Dent, C.E.; Field, C.M.B.; Neill, D.W.; Westall, R.G. Homocystinuria: A new inborn error of metabolism associated with mental deficiency. Arch. Dis. Child. 1963, 38, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Smolin, L.A.; Benevenga, N.J.; Berlow, S. The use of betaine for the treatment of homocystinuria. J. Pediatrics 1981, 99, 467–472. [Google Scholar] [CrossRef]
- Wilcken, D.E.L.; Wilcken, B.; Dudman, N.P.B.; Tyrrell, P.A. Homocystinuria-The effects of betaine in the treatment of patients not responsive to pyridoxine. N. Engl. J. Med. 1983, 309, 448–453. [Google Scholar] [CrossRef]
- Yap, S.; Naughten, E. Homocystinuria due to cystathionine beta-synthase deficiency in Ireland-25 years experience of a newborn screened and treated population with reference to clinical outcome and biochemical control. J. Inherit. Metab. Dis. 1998, 21, 738–747. [Google Scholar] [CrossRef]
- Gupta, S.; Wang, L.; Kruger, W.D. Betaine supplementation is less effective than methionine restriction in correcting phenotypes of CBS deficient mice. J. Inherit. Metab. Dis. 2016, 39, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, R.M. Development of recombinant methioninase to target the general cancer-specific metabolic defect of methionine dependence: A 40-year odyssey. Expert Opin. Biol. Ther. 2015, 15, 21–31. [Google Scholar] [CrossRef]
- Gupta, S.; Kruger, W.D. Cystathionine beta-synthase deficiency causes fat loss in mice. PLoS ONE 2011, 6, e27598. [Google Scholar] [CrossRef]
- Jiang, H.; Stabler, S.P.; Allen, R.H.; Abman, S.H.; Maclean, K.N. Altered hepatic sulfur metabolism in cystathionine beta-synthase-deficient homocystinuria: Regulatory role of taurine on competing cysteine oxidation pathways. FASEB J. 2014, 28, 4044–4054. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.J.; Briddon, A. A rationale for cystine supplementation in severe homocystinuria. J. Inherit. Metab. Dis. 2007, 30, 35–38. [Google Scholar] [CrossRef]
- Lawson-Yuen, A.; Levy, H.L. The use of betaine in the treatment of elevated homocysteine. Mol. Genet. Metab. 2006, 88, 201–207. [Google Scholar] [CrossRef]
- Yaghmai, R.; Kashani, A.H.; Geraghty, M.T.; Okoh, J.; Pomper, M.; Tangerman, A.; Wagner, C.; Stabler, S.P.; Allen, R.H.; Mudd, S.H.; et al. Progressive cerebral edema associated with high methionine levels and betaine therapy in a patient with cystathionine beta-synthase (CBS) deficiency. Am. J. Med. Genet. 2002, 108, 57–63. [Google Scholar] [CrossRef]
- Devlin, A.M.; Hajipour, L.; Gholkar, A.; Fernandes, H.; Ramesh, V.; Morris, A.A. Cerebral edema associated with betaine treatment in classical homocystinuria. J. Pediatrics 2004, 144, 545–548. [Google Scholar] [CrossRef]
- Maclean, K.N.; Jiang, H.; Greiner, L.S.; Allen, R.H.; Stabler, S.P. Long-term betaine therapy in a murine model of cystathionine beta-synthase deficient homocystinuria: Decreased efficacy over time reveals a significant threshold effect between elevated homocysteine and thrombotic risk. Mol. Genet Metab. 2012, 105, 395–403. [Google Scholar] [CrossRef]
- Maclean, K.N.; Jiang, H.; Phinney, W.N.; Keating, A.K.; Hurt, K.J.; Stabler, S.P. Taurine alleviates repression of betaine-homocysteine S-methyltransferase and significantly improves the efficacy of long-term betaine treatment in a mouse model of cystathionine beta-synthase-deficient homocystinuria. FASEB J. 2019, 33, 6339–6353. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Schiff, M.; Guffon, N.; Nadjar, Y.; Garcia-Cazorla, A.; Martinez-Pardo Casanova, M.; Cano, A.; Couce, M.L.; Dalmau, J.; Pena-Quintana, L.; et al. Betaine anhydrous in homocystinuria: Results from the RoCH registry. Orphanet J. Rare Dis. 2019, 14, 66. [Google Scholar] [CrossRef] [Green Version]
- Bublil, E.M.; Majtan, T. Classical homocystinuria: From cystathionine beta-synthase deficiency to novel enzyme therapies. Biochimie 2020, 173, 48–56. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Component (g/kg) | MET4.0 (TD.170063) | MET2.0 (TD.170062) | MET1.0 (TD.170061) | MET0.5 (TD.110591) | 3xCYS (TD.170065) | MET8.2 (TD.01084) |
|---|---|---|---|---|---|---|
| L-Alanine | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 |
| L-Arginine.HCl | 12.1 | 12.1 | 12.1 | 12.1 | 12.1 | 12.1 |
| L-Asparagine | 6.0 | 6.0 | 6.0 | 6.0 | 6.0 | 6.0 |
| L-Aspartic Acid | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 |
| L-Cystine | 3.5 | 3.5 | 3.5 | 3.5 | 10.5 | 3.5 |
| L-Glutamic Acid | 36.5 | 38.5 | 39.5 | 40.0 | 29.5 | 40.0 |
| Glycine | 23.04 | 23.04 | 23.04 | 23.04 | 23.04 | 23.3 |
| L-Histidine.HClxH2O | 4.5 | 4.5 | 4.5 | 4.5 | 4.5 | 4.5 |
| L-Isoleucine | 8.2 | 8.2 | 8.2 | 8.2 | 8.2 | 8.2 |
| L-Leucine | 11.1 | 11.1 | 11.1 | 11.1 | 11.1 | 11.1 |
| L-Lysine.HCl | 18.0 | 18.0 | 18.0 | 18.0 | 18.0 | 18.0 |
| L-Methionine | 4.0 | 2.0 | 1.0 | 0.5 | 4.0 | 8.2 |
| L-Phynylalanine | 7.5 | 7.5 | 7.5 | 7.5 | 7.5 | 7.5 |
| L-Proline | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 |
| L-Serine | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 |
| L-Threonine | 8.2 | 8.2 | 8.2 | 8.2 | 8.2 | 8.2 |
| L-Tryptophan | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 |
| L-Tyrosine | 5.0 | 5.0 | 5.0 | 5.0 | 5.0 | 5.0 |
| L-Valine | 8.2 | 8.2 | 8.2 | 8.2 | 8.2 | 8.2 |
| Sucrose | 353.14 | 353.14 | 353.14 | 353.14 | 353.14 | 344.98 |
| Corn Starch | 150.0 | 150.0 | 150.0 | 150.0 | 150.0 | 150.0 |
| Maltodextrin | 150.0 | 150.0 | 150.0 | 150.0 | 150.0 | 150.0 |
| Soybean Oil | 80.0 | 80.0 | 80.0 | 80.0 | 80.0 | 80.0 |
| Cellulose | 30.0 | 30.0 | 30.0 | 30.0 | 30.0 | 30.0 |
| Mineral Mix (AIN-93M-MX) | 35.0 | 35.0 | 35.0 | 35.0 | 35.0 | 35.0 |
| Ca(H2PO4)2xH2O | 8.2 | 8.2 | 8.2 | 8.2 | 8.2 | 8.2 |
| Vitamin Mix (AIN-93-VX) | 19.5 | 19.5 | 19.5 | 19.5 | 19.5 | 19.5 |
| Choline bitartarate | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | 2.7 |
| TBHQ (antioxidant) | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 |
| Summary Nutrient Information (% by weight/% kcal from) | ||||||
| Proteins | 14.9/15.1 | 14.9/15.1 | 14.9/15.1 | 16.5/16.4 | 15.0/15.2 | 17.3/17.2 |
| Carbohydrates | 65.7/66.7 | 65.7/66.7 | 65.7/66.6 | 65.7/65.6 | 65.7/66.6 | 64.9/64.8 |
| Fat | 8.0/18.3 | 8.0/18.3 | 8.0/18.3 | 8.0/18.0 | 8.0/18.3 | 8.0/18.0 |
| Energy density (Kcal/g) | 3.9 | 3.9 | 3.9 | 4.0 | 3.9 | 4.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, I.; Bublil, E.M.; Glavin, F.; Majtan, T. Interplay of Enzyme Therapy and Dietary Management of Murine Homocystinuria. Nutrients 2020, 12, 2895. https://doi.org/10.3390/nu12092895
Park I, Bublil EM, Glavin F, Majtan T. Interplay of Enzyme Therapy and Dietary Management of Murine Homocystinuria. Nutrients. 2020; 12(9):2895. https://doi.org/10.3390/nu12092895
Chicago/Turabian StylePark, Insun, Erez M. Bublil, Frank Glavin, and Tomas Majtan. 2020. "Interplay of Enzyme Therapy and Dietary Management of Murine Homocystinuria" Nutrients 12, no. 9: 2895. https://doi.org/10.3390/nu12092895
APA StylePark, I., Bublil, E. M., Glavin, F., & Majtan, T. (2020). Interplay of Enzyme Therapy and Dietary Management of Murine Homocystinuria. Nutrients, 12(9), 2895. https://doi.org/10.3390/nu12092895