
Albumin Substitution in Decompensated Liver Cirrhosis: Don’t Forget Zinc


Abstract
:1. Introduction
2. Hallmarks of Decompensated Liver Cirrhosis
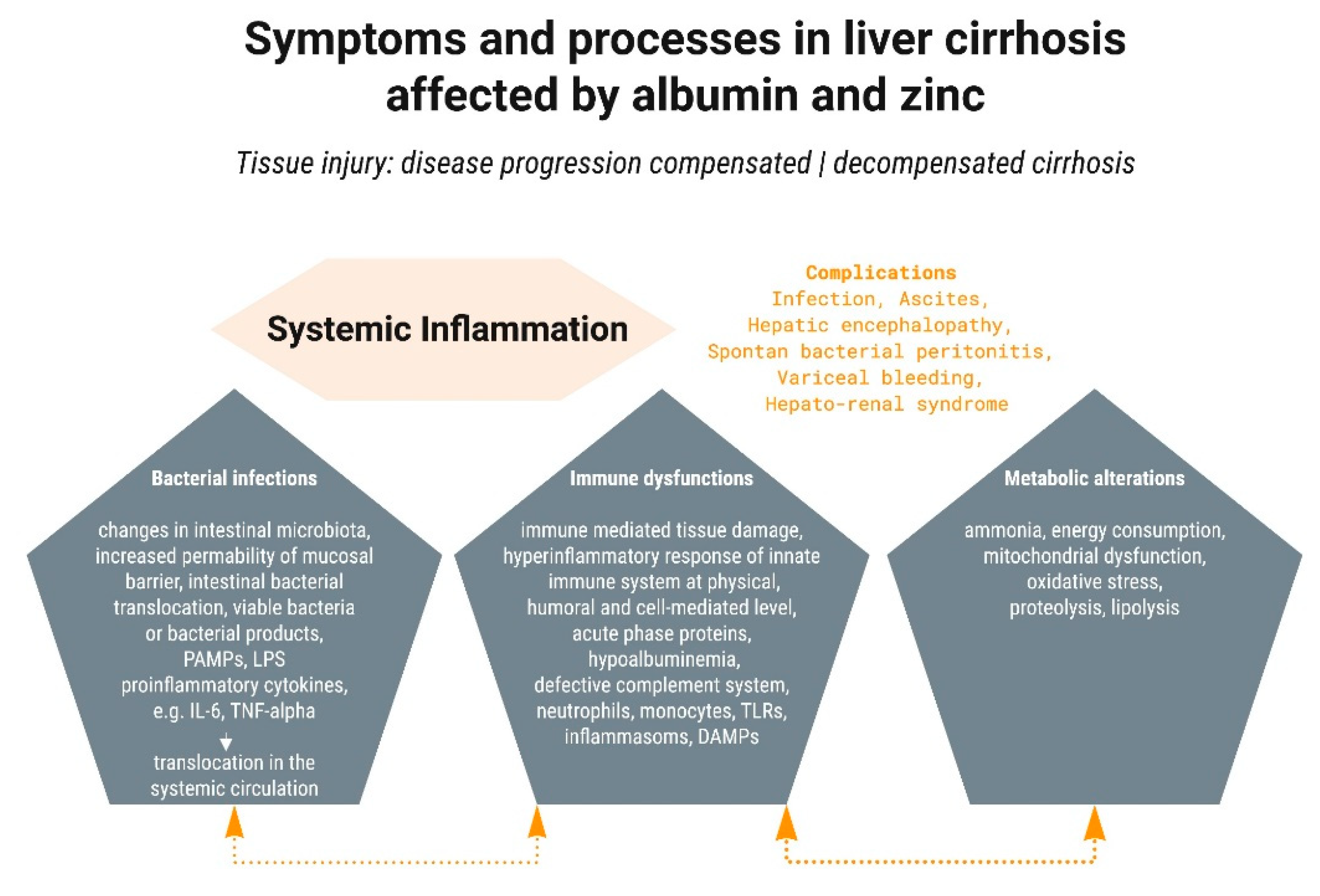
2.1. Systemic Inflammation
2.2. Bacterial Infections
2.3. Hyperammonemia
3. Albumin: Physiological Functions and Changes in Liver Disease
4. Albumin Substitution in Decompensated Liver Cirrhosis: Latest Insights
Immunomodulatory and Anti-Inflammatory Effects
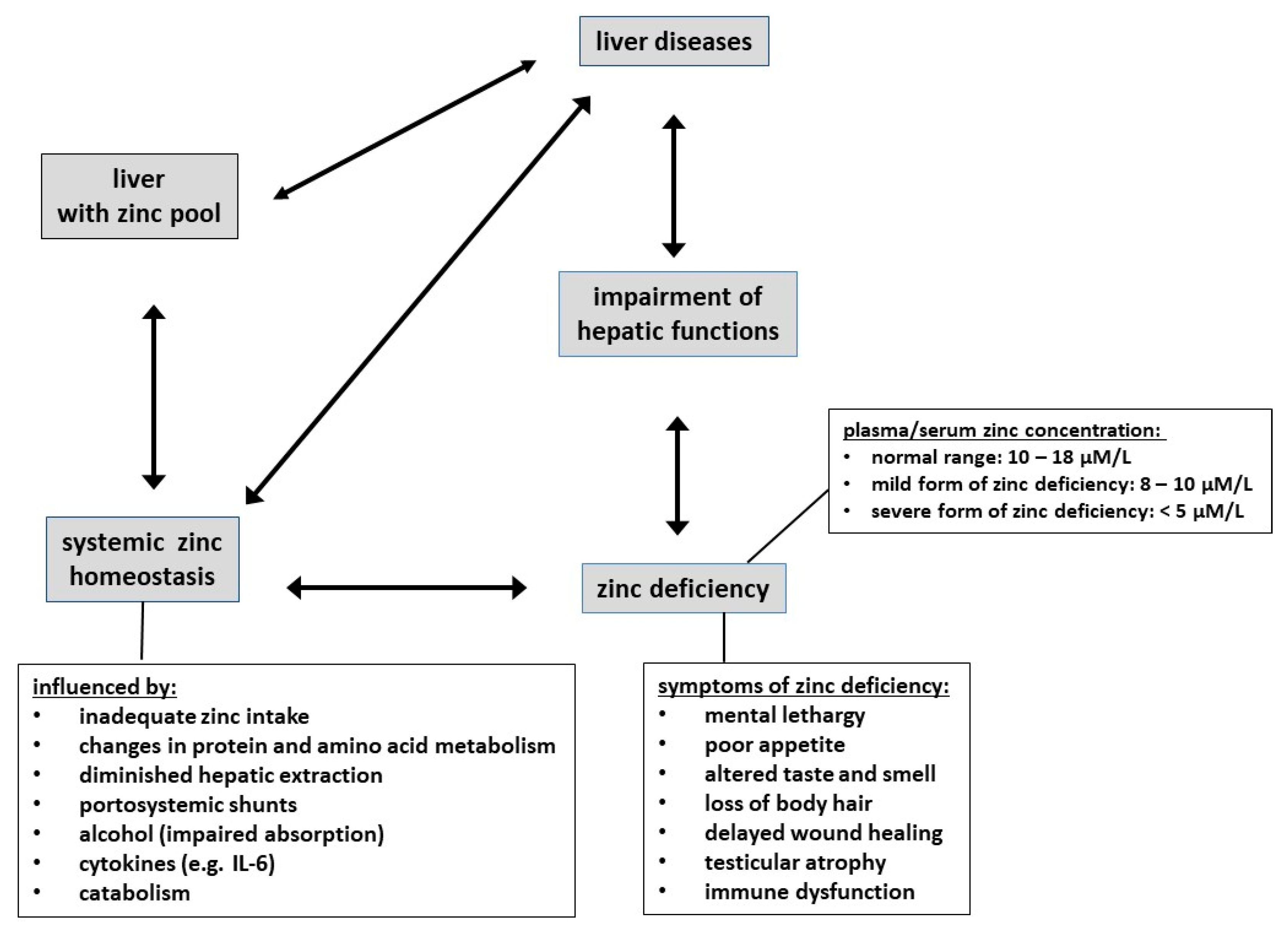
5. Zinc: Physiological Functions, Deficiency, Homeostasis, and Changes in Liver Disease
5.1. Physiological Functions
5.2. Zinc Deficiency: Prevalence and Symptoms
5.3. The Central Role of Albumin in Zinc Homeostasis
5.3.1. General Considerations
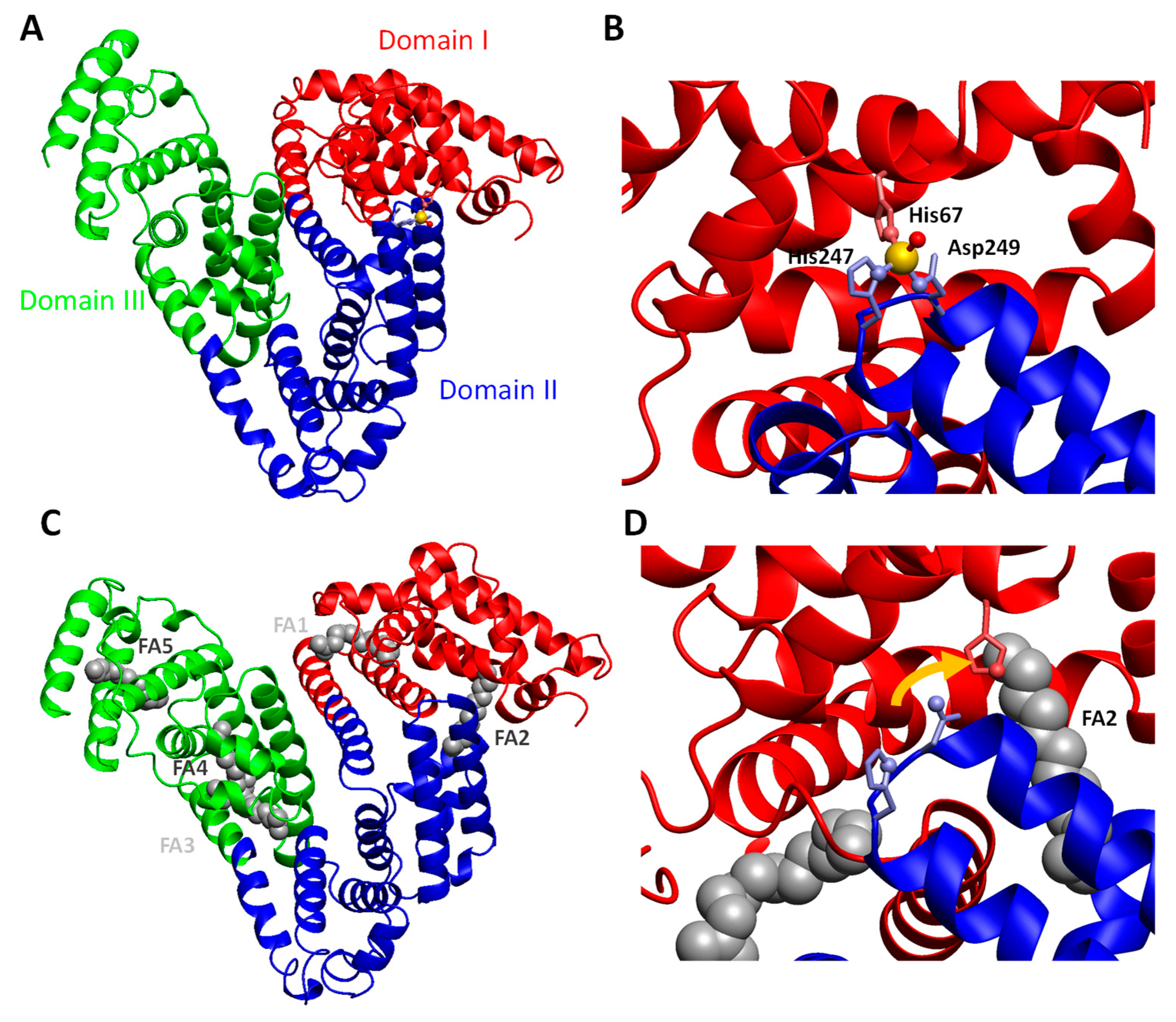
5.3.2. Molecular Details of Zinc-Albumin Interactions
5.4. Zinc, the Liver, and Changes in Plasma Zinc of Patients with Cirrhosis
6. Zinc Supplementation in Liver Disease
6.1. What Are the Conceivable Effects of Zinc Supplementation in HE?
6.2. Practical Recommendations
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Garcia-Martinez, R.; Caraceni, P.; Bernardi, M.; Gines, P.; Arroyo, V.; Jalan, R. Albumin: Pathophysiologic basis of its role in the treatment of cirrhosis and its complications. Hepatology 2013, 58, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.R.; Machado, M.V. New insights about albumin and liver disease. Ann. Hepatol. 2018, 17, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Domenicali, M.; Baldassarre, M.; Giannone, F.A.; Naldi, M.; Mastroroberto, M.; Biselli, M.; Laggetta, M.; Patrono, D.; Bertucci, C.; Bernardi, M.; et al. Posttranscriptional changes of serum albumin: Clinical and prognostic significance in hospitalized patients with cirrhosis. Hepatology 2014, 60, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Sauerbruch, T. (D)ILC–Entwicklungen in der Hepatologie. Z. Gastroenterol. 2020, 58, 83–86. [Google Scholar]
- Bernardi, M.; Angeli, P.; Claria, J.; Moreau, R.; Gines, P.; Jalan, R.; Caraceni, P.; Fernandez, J.; Gerbes, A.L.; O’Brien, A.J.; et al. Albumin in decompensated cirrhosis: New concepts and perspectives. Gut 2020, 69, 1127–1138. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, V.; Garcia-Martinez, R.; Salvatella, X. Human serum albumin, systemic inflammation, and cirrhosis. J. Hepatol. 2014, 61, 396–407. [Google Scholar] [CrossRef] [Green Version]
- Losowsky, M.S.; Atkinson, M. Intravenous albumin in the treatment of diuretic-resistant ascites in portal cirrhosis. Lancet 1961, 2, 386–389. [Google Scholar] [CrossRef]
- Fernández, J.; Clària, J.; Amorós, A.; Aguilar, F.; Castro, M.; Casulleras, M.; Acevedo, J.; Duran-Güell, M.; Nuñez, L.; Costa, M.; et al. Effects of Albumin Treatment on Systemic and Portal Hemodynamics and Systemic Inflammation in Patients with Decompensated Cirrhosis. Gastroenterology 2019, 157, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Caraceni, P.; Tufoni, M.; Zaccherini, G.; Riggio, O.; Angeli, P.; Alessandria, C.; Neri, S.; Foschi, F.G.; Levantesi, F.; Airoldi, A.; et al. ANSWER Study Investigators. On-treatment serum albumin level can guide long-term treatment in patients with cirrhosis and uncomplicated ascites. J. Hepatol. 2021, 74, 340–349. [Google Scholar] [CrossRef]
- Bal, W.; Sokołowska, M.; Kurowska, E.; Faller, P. Binding of transition metal ions to albumin: Sites, affinities and rates. Biochim. Biophys. Acta 2013, 1830, 5444–5455. [Google Scholar] [CrossRef]
- Ito, T.; Ishigami, M.; Ishizu, Y.; Kuzuya, T.; Honda, T.; Ishikawa, T.; Toyoda, H.; Kumada, T.; Fujishiro, M. Correlation of serum zinc levels with pathological and laboratory findings in patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2020, 32, 748–753. [Google Scholar] [CrossRef]
- Katayama, K. Zinc and protein metabolism in chronic liver diseases. Nutr. Res. 2020, 74, 1–9. [Google Scholar] [CrossRef]
- Nishikawa, H.; Enomoto, H.; Yoh, K.; Iwata, Y.; Sakai, Y.; Kishino, K.; Ikeda, N.; Takashima, T.; Aizawa, N.; Takata, R.; et al. Serum Zinc Concentration and Sarcopenia: A Close Linkage in Chronic Liver Diseases. J. Clin. Med. 2019, 8, 336. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Stewart, A.J.; Sleep, D.; Sadler, P.J.; Pinheiro, T.J.T.; Blindauer, C.A. A molecular mechanism for modulating plasma Zn speciation by fatty acids. J. Am. Chem. Soc. 2012, 134, 1454–1457. [Google Scholar] [CrossRef] [PubMed]
- Kassaar, O.; Schwarz-Linek, U.; Blindauer, C.A.; Stewart, A.J. Plasma free fatty acid levels influence Zn2+-dependent histidine-rich glycoprotein-heparin interactions via an allosteric switch on serum albumin. J. Thromb. Haemost. 2015, 13, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coverdale, J.P.C.; Khazaipoul, S.; Arya, S.; Stewart, A.J.; Blindauer, C.A. Crosstalk between zinc and free fatty acids in plasma. Biochim. Biophys. Acta-Mol. Cell. Biol. Lipids 2019, 1864, 532–542. [Google Scholar] [CrossRef]
- Grüngreiff, K.; Reinhold, D.; Wedemeyer, H. The role of zinc in liver cirrhosis. Ann. Hepatol. 2016, 15, 7–16. [Google Scholar] [CrossRef]
- Koop, A.H.; Mousa, O.Y.; Pham, L.E.; Corral-Hurtado, J.E.; Pungpapong, S.; Keaveny, A.P. An Argument for Vitamin D, A, and Zinc Monitoring in Cirrhosis. Ann. Hepatol. 2018, 17, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, C.; Claria, J.; Szabo, G.; Bosch, J.; Bernardi, M. Pathophysiology of decompensated cirrhosis: Portal hypertension, circulatory dysfunction, inflammation, metabolism and mitochondrial dysfunction. J. Hepatol. 2021, 75, S49–S66. [Google Scholar] [CrossRef]
- Sinclair, M.; Gow, P.J.; Grossmann, M.; Angus, P.W. Review article: Sarcopenia in cirrhosis-aetiology, implications and potential therapeutic interventions. Aliment. Pharmacol. Ther. 2016, 43, 765–777. [Google Scholar] [CrossRef] [Green Version]
- Tandon, P.; Montano-Loza, A.J.; Lai, C.L.; Dasarathy, S.; Merli, M. Sarcopenia and frailty in decompensated cirrhosis. J. Hepatol. 2021, 75, S147–S162. [Google Scholar] [CrossRef] [PubMed]
- Samuel, D. Systemic inflammation and liver cirrhosis complications: Driving or secondary event? How to square the cicle? J. Hepatol. 2021, 74, 508–510. [Google Scholar] [CrossRef] [PubMed]
- Matyas, C.; Haskó, G.; Liaudet, L.; Trojnar, E.; Pacher, P. Interplay of cardiovascular mediators, oxidative stress and inflammation in liver disease and its complications. Nat. Rev. Cardiol. 2021, 18, 117–135. [Google Scholar] [CrossRef]
- Costa, D.; Simbrunner, B.; Jachs, M.; Hartl, L.; Bauer, D.; Paternostro, R.; Schwabl, P.; Scheiner, B.; Stättermayer, A.F.; Pinter, M.; et al. Systemic inflammation increases across distinct stages of advanced chronic liver disease and correlates with decompensation and mortality. J. Hepatol. 2021, 74, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Arvaniti, V.; D’Amico, G.; Fede, G.; Manousou, P.; Tsochatzis, E.; Pleguezuelo, M.; Burroughs, A.K. Infections in patients with cirrhosis increase mortality four-fold and should be used in determining prognosis. Gastroenterology 2010, 139, 1246–1256. [Google Scholar] [CrossRef]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Grüngreiff, K.; Kleine, F.D.; Lauf, H. Bakterien und Endotoxine bei der Leberzirrhose. Dt. Gesundh. Wesen. 1981, 36, 1541–1544. [Google Scholar]
- Fernández, J.; Navasa, M.; Gómez, J.; Colmenero, J.; Vila, J.; Arroyo, V.; Rodés, J. Bacterial infections in cirrhosis: Epidemiological changes with invasive procedures and norfloxacin prophylaxis. Hepatology 2002, 35, 140–148. [Google Scholar] [CrossRef]
- Borzio, M.; Salerno, F.; Piantoni, L.; Cazzaniga, M.; Angeli, P.; Bissoli, F.; Boccia, S.; Colloredo-Mels, G.; Corigliano, P.; Fornaciari, G.; et al. Bacterial infection in patients with advanced cirrhosis: A multicentre prospective study. Dig. Liver Dis. 2001, 33, 41–48. [Google Scholar] [CrossRef]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Bellot, P.; Such, J. Pathological translocation in cirrhosis. Pathophysiology, diagnosis and clinical implications. Liver Int. 2013, 33, 31–39. [Google Scholar] [CrossRef]
- Liehr, H.; Grün, M. Endotoxine und RES-Funktion in der Pathogenese von Lebererkrankungen. Internist 1976, 36, 122–128. [Google Scholar]
- Triger, D.R. The liver as an immunological organ. Gastroenterology 1976, 71, 162–164. [Google Scholar] [CrossRef]
- Nolan, J.P. The role of endotoxin in liver injury. Gastroenterology 1975, 69, 1346–1356. [Google Scholar] [CrossRef]
- Rose, C.F. Ammonia-lowering strategies for the treatment of hepatic encephalopathy. Clin. Pharmacol. Ther. 2012, 92, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Sawhney, R.; Holland-Fischer, P.; Rosselli, M.; Mookerjee, R.P.; Agarwal, B.; Jalan, R. Role of ammonia, inflammation, and cerebral oxygenation in brain dysfunction of acute-on-chronic liver failure patients. Liver Transpl. 2016, 22, 732–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, D.; O’Grady, J.; Patel, A.; Shawcross, D.; Connor, S.; Deasy, N.; Willars, C.; Bernal, W.; Wendon, J.; Auzinger, G. Cerebral oedema is rare in acute-on-chronic liver failure patients presenting with high-grade hepatic encephalopathy. Liver Int. 2014, 34, 362–366. [Google Scholar] [CrossRef]
- Strauss, A.W.; Donohue, A.M.; Bennett, C.D.; Rodkey, J.A.; Alberts, A.W. Rat liver preproalbumin: In vitro synthesis and partial amino acid sequence. Proc. Natl. Acad. Sci. USA 1977, 74, 1358–1362. [Google Scholar] [CrossRef] [Green Version]
- Peters, T. Serum albumin. Adv. Chem. 1985, 37, 161–245. [Google Scholar]
- Ghashut, R.A.; McMillan, D.C.; Kinsella, J.; Vasilaki, A.T.; Talwar, D.; Duncan, A. The effect of the systemic inflammatory response on plasma zinc and selenium adjusted for albumin. Clin. Nutr. 2016, 35, 381–387. [Google Scholar] [CrossRef]
- Simard, J.R.; Zunszain, P.A.; Hamilton, J.A.; Curry, S. Location of high and low affinity fatty acid binding sites on human serum albumin revealed by NMR drug-competition analysis. J. Mol. Biol. 2006, 361, 336–351. [Google Scholar] [CrossRef] [PubMed]
- Jalan, R.; Schnurr, K.; Mookerjee, R.P.; Sen, S.; Cheshire, L.; Hodges, S.; Muravsky, V.; Williams, R.; Matthes, G.; Davies, N.A. Alterations in the functional capacity of albumin in patients with decompensated cirrhosis is associated with increased mortality. Hepatology 2009, 50, 555–564. [Google Scholar] [CrossRef]
- Solà, E.; Solé, C.; Simón-Talero, M.; Martín-Llahí, M.; Castellote, J.; Garcia-Martínez, R.; Moreira, R.; Torrens, M.; Márquez, F.; Fabrellas, N.; et al. Midodrine and albumin for prevention of complications in patients with cirrhosis awaiting liver transplantation. A randomized placebo-controlled trial. J. Hepatol. 2018, 69, 1250–1259. [Google Scholar] [CrossRef]
- China, L.; Freemantle, N.; Forrest, E.; Kallis, Y.; Ryder, S.D.; Wright, G.; Portal, A.J.; Becares Salles, N.; Gilroy, D.W.; O’Brien, A.; et al. A Randomized Trial of Albumin Infusions in Hospitalized Patients with Cirrhosis. N. Engl. J. Med. 2021, 384, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Caraceni, P.; Riggio, O.; Angeli, P.; Alessandria, C.; Neri, S.; Foschi, F.G.; Levantesi, F.; Airoldi, A.; Boccia, S.; Svegliati-Baroni, G.; et al. Long-term albumin administration in decompensated cirrhosis (ANSWER): An open-label randomised trial. Lancet 2018, 391, 2417–2429. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Aldini, G.; Ito, S.; Morishita, N.; Shibata, T.; Vistoli, G.; Carini, M.; Uchida, K. Delta12-prostaglandin j2 as a product and ligand of human serum albumin: Formation of an unusual covalent adduct at His146. J. Am. Chem. Soc. 2010, 132, 824–832. [Google Scholar] [CrossRef]
- Casulleras, M.; Flores-Costa, R.; Duran-Güell, M.; Alcaraz-Quiles, J.; Sanz, S.; Titos, E.; López-Vicario, C.; Fernández, J.; Horrillo, R.; Costa, M.; et al. Albumin internalizes and inhibits endosomal TLR signaling in leukocytes from patients with decompensated cirrhosis. Sci. Transl. Med. 2020, 12, 5135. [Google Scholar] [CrossRef]
- Casulleras, M.; Zhang, I.W.; López-Vicario, C.; Clària, J. Leukocytes, Systemic Inflammation and Immunopathology in Acute-on-Chronic Liver Failure. Cells 2020, 9, 2632. [Google Scholar] [CrossRef]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef]
- Grüngreiff, K. Zinc and the Liver, 1st ed.; Dr. Falk Pharma GmbH: Freiburg, Germany, 2013; pp. 1–84. [Google Scholar]
- Maret, W. Human Zinc Biochemistry. In Zinc in Human Health, 1st ed.; Rink, L., Ed.; IOS Press: Amsterdam, The Netherlands, 2011; pp. 45–62. [Google Scholar]
- Maret, W. The redox biology of redox-inert ions. Free Rad. Biol. Med. 2019, 134, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.H.; Sermersheim, M.; Li, H.; Lee, P.H.U.; Steinberg, S.M.; Ma, J. Zinc in Wound Healing Modulation. Nutrients 2017, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, A.; Sada, K.K.; Ketheeswaran, S.; Dubey, A.K.; Bhat, M.S. Role of Zinc in Mucosal Health and Disease: A Review of Physiological, Biochemical, and Molecular Processes. Cureus 2020, 12, e8197. [Google Scholar] [CrossRef]
- Sarkar, P.; Saha, T.; Kazi, M.H. Zinc deficiency induces epithelial barrier dysfunction and altered intestinal ion transport in novel murine model of Shigella flexneri diarrhea. Int. J. Infect. Dis. 2020, 101, 136. [Google Scholar] [CrossRef]
- Grüngreiff, K.; Reinhold, D. Zink: Bedeutung in der Ärtzlichen Praxis, 1st ed.; Jürgen Hartmann Verlag: Heßdorf-Klebheim, Germany, 2007; pp. 1–96. [Google Scholar]
- Wessels, I.; Rolles, B.; Rink, L. The Potential Impact of Zinc Supplementation on COVID-19 Pathogenesis. Front. Immunol. 2020, 11, 1712. [Google Scholar] [CrossRef]
- Haase, H.; Schomburg, L. You’d Better Zinc-Trace Element Homeostasis in Infection and Inflammation. Nutrients 2019, 11, 2078. [Google Scholar] [CrossRef] [Green Version]
- Reinhold, D.; Guttek, K.; Reddig, A.; Voss, L.; Schubert, C.; Kahlfuss, S.; Grüngreiff, K.; Schraven, B.; Reinhold, A. Zinc Aspartate Induces IL-16 Secretion and Apoptosis in Human T Cells. Biomedicines 2021, 9, 246. [Google Scholar] [CrossRef]
- Salinas, E.; Ciminari, M.E.; Prez, C.M.V.; Gomez, N.N. Anti-Inflammatory and antioxidant effects and zinc deficiency. In Handbook of Famine, Starvation, and Nutrient Deprevitation, 1st ed.; Preedy, V.R., Patel, V.B., Eds.; Springer International Publishing AG: Cham, Switzerland, 2018; pp. 1951–1991. [Google Scholar]
- Bozym, R.A.; Chimienti, F.; Giblin, L.J.; Gross, G.W.; Korichneva, I.; Li, Y.A.; Libert, S.; Maret, W.; Parviz, M.; Frederickson, C.J.; et al. Free zinc ions outside a narrow concentration range are toxic to a variety of cells in vitro. Exp. Biol. Med. 2010, 235, 741–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, A.S. Discovery of human zinc deficiency: Its impact on human health and disease. Adv. Nutr. 2013, 4, 176–190. [Google Scholar] [CrossRef] [PubMed]
- King, J.C. Zinc: An essential but elusive nutrient. Am. J. Clin. Nutr. 2011, 94, 679S–684S. [Google Scholar] [CrossRef] [Green Version]
- Gibson, R.S.; Hess, S.Y.; Hotz, C.; Brown, K.H. Indicators of zinc status at the population level: A review of the evidence. Br. J. Nutr. 2008, 99, S14–S23. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.; Pfeiffer, C.M.; Georgieff, M.K.; Brittenham, G.; Fairweather-Tait, S.; Hurrell, R.F.; McArdle, H.J.; Raiten, D.J. Biomarkers of Nutrition for Development (BOND)-Iron. J. Nutr. 2018, 148, 1001S–1067S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rükgauer, M.; Klein, J.; Kruse-Jarres, J.D. Reference values for the trace elements copper, manganese, selenium, and zinc in the serum/plasma of children, adolescents, and adults. J. Trace Elem. Med. Biol. 1997, 11, 92–98. [Google Scholar] [CrossRef]
- King, J.C.T. Yet again, serum concentrations are unrelated to zinc intaks. J. Nutr. 2018, 148, 1399–1401. [Google Scholar] [CrossRef] [Green Version]
- Baer, M.T.; King, J.C. Tissue zinc levels and zinc excretion during experimental zinc depletion in young men. Am. J. Clin. Nutr. 1984, 39, 556–570. [Google Scholar] [CrossRef]
- Foote, J.W.; Delves, H.T. Albumin bound and alpha-macroglobulin bound zinc concentrations in the sera of healthy adults. J. Clin. Pathol. 1984, 37, 1050–1054. [Google Scholar] [CrossRef] [Green Version]
- Hennigar, S.R.; Kelley, A.M.; Anderson, B.J.; Armstrong, N.J.; McClung, H.L.; Berryman, C.E.; Karl, J.P.; McClung, J.P. Sensitivity and reliability of zinc transporter and metallothionein gene expression in peripheral blood mononuclear cells as indicators of zinc status: Responses to ex vivo zinc exposure and habitual zinc intake in humans. Br. J. Nutr. 2021, 125, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.V.; Hambidge, K.M.; Naake, V.L.; Hong, Z.; Westcott, J.L.; Fennessey, P.V. Size of the zinc pools that exchange rapidly with plasma zinc in humans: Alternative techniques for measuring and relation to dietary zinc intake. J. Nutr. 1994, 124, 268–2676. [Google Scholar] [CrossRef] [PubMed]
- Grüngreiff, K.; Reinhold, D. Zinc and the Liver. In Zinc in Human Health, 1st ed.; Rink, L., Ed.; IOS Press: Amsterdam, The Netherlands, 2011; pp. 473–492. [Google Scholar]
- Grüngreiff, K.; Gottstein, T.; Reinhold, D. Zinc deficiency-An independent risk factor in the pathogenesis of haemorrhagic stroke? Nutrients 2020, 12, 3548. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, L.M.F.; Mafra, D. Don`t forget zinc. Nephrol. Dial. Transplant. 2020, 35, 1094–1098. [Google Scholar] [CrossRef]
- Haase, H.; Mocchegiani, E.; Rink, L. Correlation between zinc status and immune function in the elderly. Biogerontology 2006, 7, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Taylor, K.M.; Fu, D. Zinc transporters and their functional integration in mammalian cells. J. Biol. Chem. 2021, 296, 100320. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.P.; Blindauer, C.A.; Kassaar, O.; Khazaipoul, S.; Martin, E.M.; Sadler, P.J.; Stewart, A.J. Allosteric modulation of zinc speciation by fatty acids. Biochim. Biophys. Acta 2013, 1830, 5456–5464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blindauer, C.A.; Khazaipoul, S.; Yu, R.; Stewart, A.J. Fatty Acid-Mediated Inhibition of Metal Binding to the Multi-Metal Site on Serum Albumin: Implications for Cardiovascular Disease. Curr. Top. Med. Chem. 2016, 16, 3021–3032. [Google Scholar] [CrossRef] [Green Version]
- Sobczak, A.I.S.; Katundu, K.G.H.; Phoenix, F.A.; Khazaipoul, S.; Yu, R.; Lampiao, F.; Stefanowicz, F.; Blindauer, C.A.; Pitt, S.J.; Smith, T.K.; et al. Albumin-mediated alteration of plasma zinc speciation by fatty acids modulates blood clotting in type-2 diabetes. Chem. Sci. 2021, 12, 4079–4093. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Hebel, S.; Engelhardt, G.; Rink, L. The biochemical effects of extracellular Zn(2+) and other metal ions are severely affected by their speciation in cell culture media. Metallomics 2015, 7, 102–111. [Google Scholar] [CrossRef]
- Coverdale, J.P.C.; Barnett, J.P.; Adamu, A.H.; Griffiths, E.J.; Stewart, A.J.; Blindauer, C.A. A metalloproteomic analysis of interactions between plasma proteins and zinc: Elevated fatty acid levels affect zinc distribution. Metallomics 2019, 11, 1805–1819. [Google Scholar] [CrossRef] [Green Version]
- Cousins, R.J. Theoretical and practical aspects of zinc uptake and absorption. Adv. Exp. Med. Biol. 1989, 249, 3–12. [Google Scholar]
- Handing, K.B.; Shabalin, I.G.; Kassaar, O.; Khazaipoul, S.; Blindauer, C.A.; Stewart, A.J.; Chruszcz, M.; Minor, W. Circulatory zinc transport is controlled by distinct interdomain sites on mammalian albumins. Chem. Sci. 2016, 7, 6635–6648. [Google Scholar] [CrossRef] [Green Version]
- Curry, S.; Mandelkow, H.; Brick, P.; Franks, N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 1998, 5, 827–835. [Google Scholar] [CrossRef]
- Stewart, A.J.; Blindauer, C.A.; Berezenko, S.; Sleep, D.; Sadler, P.J. Interdomain zinc site on human albumin. Proc. Natl. Acad. Sci. USA 2003, 100, 3701–3706. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Stewart, A.J.; Sadler, P.J.; Pinheiro, T.J.; Blindauer, C.A. Allosteric inhibition of cobalt binding to albumin by fatty acids: Implications for the detection of myocardial ischemia. J. Med. Chem. 2012, 55, 4425–4430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coverdale, J.P.C.; Katundu, K.G.H.; Sobczak, A.I.S.; Arya, S.; Blindauer, C.A.; Stewart, A.J. Ischemia-modified albumin: Crosstalk between fatty acid and cobalt binding. Prostaglandins Leukot. Essent. Fatty Acids 2018, 135, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Bar-Or, D.; Rael, L.T.; Bar-Or, R.; Slone, D.S.; Mains, C.W.; Rao, N.K.; Curtis, C.G. The cobalt-albumin binding assay: Insights into its mode of action. Clin. Chim. Acta 2008, 387, 120–127. [Google Scholar] [CrossRef]
- Bhagavan, N.V.; Ha, J.S.; Park, J.H.; Honda, S.A.; Rios, C.N.; Sugiyama, C.; Fujitani, G.K.; Takeuchi, I.K.; Ha, C.E. Utility of serum fatty acid concentrations as a marker for acute myocardial infarction and their potential role in the formation of ischemia-modified albumin: A pilot study. Clin. Chem. 2009, 55, 1588–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, R.; Claria, J.; Aguilar, F.; Fenaille, F.; Lozano, J.J.; Junot, C.; Colsch, B.; Caraceni, P.; Trebicka, J.; Pavesi, M.; et al. Blood metabolomics uncovers inflammation-associated mitochondrial dysfunction as a potential mechanism underlying ACLF. J. Hepatol. 2020, 72, 688–701. [Google Scholar] [CrossRef]
- Iqbal, S.; Qais, F.A.; Alam, M.M.; Naseem, I. Effect of glycation on human serum albumin-zinc interaction: A biophysical study. J. Biol. Inorg. Chem. 2018, 23, 447–458. [Google Scholar] [CrossRef]
- Tuerk, M.; Fazel, N. Zinc deficiency. Curr. Opin. Gastroenterol. 2009, 25, 136–143. [Google Scholar] [CrossRef]
- Krebs, N.E.; Hambidge, K.M. Zinc metabolism and homeostasis: The application of tracer techniques to human zinc physiology. Biometals 2001, 14, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Cousins, R.J. Absorption, transport and hepatic metabolism of copper and zinc: Special reference to metallothionein and ceruloplasmin. Physiol. Rev. 1985, 65, 238–309. [Google Scholar] [CrossRef] [PubMed]
- Etzel, K.R.; Cousins, R.J. Hormonal regulation of liver metallothionein zinc: Independent and synergistic action of glucagon and glucocorticoids. Proc. Soc. Exp. Biol. Med. 1981, 167, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, S.L.; Seo, Y.A.; Lopez, V. Mammary gland zinc metabolism: Regulation and dysregulation. Genes Nutr. 2009, 4, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, J.C.; Hambidge, K.M.; Westcott, J.L.; Kern, D.L.; Marshall, G. Daily variation in plasma zinc concentrations in women fed meals at six-hour intervals. J. Nutr. 1994, 124, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Aydemir, T.B.; Cousins, R.J. The multiple faces of the metal transporter ZIP14 (SLC39A14). J. Nutr. 2018, 148, 174–184. [Google Scholar] [CrossRef]
- O’Halloran, T.V.; Kebede, M.; Philips, S.J.; Attie, A.D. Zinc, insulin, and the liver: A ménage à trois. J. Clin. Investig. 2013, 123, 4136–4139. [Google Scholar] [CrossRef] [Green Version]
- Liuzzi, J.P.; Lichten, L.A.; Rivera, S.; Blanchard, R.K.; Aydemir, T.B.; Knutson, M.D.; Ganz, T.; Cousins, R.J. Interleukin-6 regulates the zinc transporter ZIP14 in liver and contributes to the hypozincemia of the acute-phase response. Proc. Natl. Acad. Sci. USA 2005, 102, 6843–6848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieck, H.T.; Doring, F.; Roth, H.P.; Daniel, H. Changes in rat hepatic gene expression in response to zinc deficiency as assessed by DNA arrays. J. Nutr. 2003, 133, 1004–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhu, P.; Garg, M.L.; Dhawan, D.K. Protective effects of zinc on oxidative stress enzymes in liver protein deficient rats. Nutr. Hosp. 2004, 6, 341–347. [Google Scholar] [CrossRef]
- Dieck, H.T.; Döring, F.; Fuchs, D.; Roth, H.P.; Daniel, H. Transcriptome and proteome analysis identifies the pathways that increase hepatic lipid accumulation in zinc-deficient rats. J. Nutr. 2005, 135, 199–205. [Google Scholar] [CrossRef]
- Giroux, E.; Schechter, P.J.; Schoun, J.; Sjoerdsma, A. Reduced binding of added zinc in serum of patients with decompensated hepatic cirrhosis. Eur. J. Clin. Investig. 1977, 7, 71–73. [Google Scholar] [CrossRef]
- Cuthbertson, D.P.; Fell, G.S.; Smith, C.M.; Tilstone, W.J. Metabolism after injury. I. Effects of severity, nutrition, and environmental temperature on protein potassium, zinc, and creatine. Br. J. Surg. 1972, 59, 926–931. [Google Scholar] [CrossRef]
- Barry, M.; Keeling, P.W.; Feely, J. Tissue zinc status and drug elimination in patients with chronic liver disease. Clin. Sci. 1990, 78, 547–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diglio, D.C.; Fernandes, S.A.; Stein, J.; Azeredo-da-Silva, A.; de Mattos, A.A.; Tovo, C.V. Role of zinc supplementation in the management of chronic liver diseases: A systematic review and meta-analysis. Ann. Hepatol. 2020, 19, 190–196. [Google Scholar] [CrossRef]
- Tan, H.K.; Streeter, A.; Cramp, M.E.; Dhanda, A.D. Effect of zinc treatment on clinical outcomes in patients with liver cirrhosis: A systematic review and meta-analysis. World J. Hepatol. 2020, 12, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Himoto, T.; Masaki, T. Current Trends of Essential Trace Elements in Patients with Chronic Liver Diseases. Nutrients 2020, 12, 2084. [Google Scholar] [CrossRef] [PubMed]
- Kozeniecki, M.; Ludke, R.; Kerner, J.; Patterson, B. Micronutrients in Liver Disease: Roles, Risk Factors for Deficiency, and Recommendations for Supplementation. Nutr. Clin. Pract. 2020, 35, 50–62. [Google Scholar] [CrossRef] [Green Version]
- Grüngreiff, K.; Grüngreiff, S.; Reinhold, D. Zinc deficiency and hepatic encephalopathy: Results of a long-term follow-up on zinc supplementation. J. Trace Elem. Exp. Med. 2000, 13, 21–31. [Google Scholar] [CrossRef]
- Fathi, M.; Alavinejad, P.; Haidari, Z.; Amani, R. The effects of zinc supplementation on metabolic profile and oxidative stress in overweight/obese patients with non-alcoholic fatty liver disease: A randomized, double-blind, placebo-controlled trial. J. Trace Elem. Med. Biol. 2020, 62, 126635. [Google Scholar] [CrossRef]
- Vilar Gomez, E.; Sanchez Rodriguez, Y.; Torres Gonzalez, A.; Calzadilla Bertot, L.; Arus Soler, E.; Martinez Perez, Y.; Yasells Garcia, A.; Abreu Vazquez Mdel, R. Viusid, a nutritional supplement, increases survival and reduces disease progression in HCV-related decompensated cirrhosis: A randomised and controlled trial. BMJ Open 2011, 1, e000140. [Google Scholar] [CrossRef]
- Takuma, Y.; Nouso, K.; Makino, Y.; Hayashi, M.; Takahashi, H. Clinical trial: Oral zinc in hepatic encephalopathy. Aliment. Pharmacol. Ther. 2010, 32, 1080–1090. [Google Scholar] [CrossRef]
- Reding, P.; Duchateau, J.; Bataille, C. Oral zinc supplementation improves hepatic encephalopathy. Results of a randomised controlled trial. Lancet 1984, 2, 493–495. [Google Scholar] [CrossRef]
- Schölmerich, J. Zinc and vitamin A in liver cirrhosis. In Liver Cirrhosis, 1st ed.; Boyer, J.L., Bianchi, L., Eds.; MTP Press Limited: Lancaster, UK, 1987; pp. 421–432. [Google Scholar]
- Van der Rijt, C.C.; Schalm, S.W.; Schat, H.; Foeken, K.; De Jong, G. Overt hepatic encephalopathy precipitated by zinc deficiency. Gastroenterology 1991, 100, 1114–1118. [Google Scholar] [CrossRef]
- Horiguchi, S.; Naganuma, A.; Tateyama, Y.; Suzuki, Y.; Hoshino, T.; Saito, N.; Hatanaka, T.; Takakusagi, S.; Kosone, T.; Takagi, H.; et al. Efficacy of Zinc Acetate Treatment for Patients with Decompensated Liver Cirrhosis Complicated by Hypozincemia. Biol. Trace. Elem. Res. 2021, 1–8. [Google Scholar] [CrossRef]
- Miwa, T.; Hanai, T.; Toshihide, M.; Ogiso, Y.; Imai, K.; Suetsugu, A.; Takai, K.; Shiraki, M.; Katsumura, N.; Shimizu, M. Zinc deficiency predicts overt hepatic encephalopathy and mortality in liver cirrhosis patients with minimal hepatic encephalopathy. Hepatol. Res. 2021, 51, 662–673. [Google Scholar] [CrossRef]
- Riggio, O.; Ariosto, F.; Merli, M.; Caschera, M.; Zullo, A.; Balducci, G.; Ziparo, V.; Pedretti, G.; Fiaccadori, F.; Bottari, E.; et al. Short-term oral zinc supplementation does not improve chronic hepatic encephalopathy. Results of a double-blind crossover trial. Dig. Dis. Sci. 1991, 36, 1204–1208. [Google Scholar] [CrossRef]
- Riggio, O.; Ridola, L.; Pasquale, C. Hepatic encephalopathy therapy: An overview. World J. Gastrointest. Pharmacol. Ther. 2010, 1, 54–63. [Google Scholar] [CrossRef]
- Schliess, F.; Görg, B.; Häussinger, D. RNA oxidation and zinc in hepatic encephalopathy and hyperammonemia. Metab. Brain Dis. 2009, 24, 119–134. [Google Scholar] [CrossRef]
- Shawcross, D.L.; Shabbir, S.S.; Taylor, N.J.; Hughes, R.D. Ammonia and the neutrophil in the pathogenesis of hepatic encephalopathy in cirrhosis. Hepatology 2010, 51, 1062–1069. [Google Scholar] [CrossRef]
- Prasad, A.S.; Rabbani, P.; Abbasii, A.; Bowersox, E.; Fox, M.R. Experimental zinc deficiency in humans. Ann. Intern. Med. 1978, 89, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, P.; Prasad, A.S. Plasma ammonia and liver ornithine transcarbamoylase activity in zinc-deficient rats. Am. J. Physiol. 1978, 235, E203–E206. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Fabbri, A.; Bianchi, G.; Brizi, M.; Zoli, M. Zinc supplementation and amino acid-nitrogen metabolism in patients with advanced cirrhosis. Hepatology 1996, 23, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Bogden, J.D.; Oleske, J.M.; Lavenhar, M.A.; Munves, E.M.; Kemp, F.W.; Bruening, K.S.; Holding, K.J.; Denny, T.N.; Guarino, M.A.; Holland, B.K. Effects of one year of supplementation with zinc and other micronutrients on cellular immunity in the elderly. J. Am. Coll. Nutr. 1990, 9, 214–225. [Google Scholar] [CrossRef]
- Chandra, R.K. Excessive intake of zinc impairs immune responses. JAMA 1984, 252, 1443–1446. [Google Scholar] [CrossRef]
- Reinhold, D.; Ansorge, S.; Grüngreiff, K. Zinc regulates DNA synthesis and IL-2, IL-6 and IL-10 production of PWM-stimulated PBMC and normalises the periphere cytokine concentration in chronic liver disease. J. Trace Elem. Exp. Med. 1997, 10, 19–27. [Google Scholar] [CrossRef]
- Prasad, A.S. Essentiality and toxicity of zinc. Scand. J. Work Environ. Health 1993, 19, 134–136. [Google Scholar]
- Nath, S.; Baidya, D.K. Mucormycosis in COVID-19: Is Zinc a Silent Killer in India? Indian J. Crit. Care Med. 2021, 25, 1079–1080. [Google Scholar]
- Wilson, D. An evolutionary perspective on zinc uptake by human fungal pathogens. Metallomics 2015, 7, 979–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staats, C.C.; Kmetzsch, L.; Schrank, A.; Vainstein, M.H. Fungal zinc metabolism and its connections to virulence. Front. Cell Infect. Microbiol. 2013, 3, 65. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| ZC (µmol L−1) | Recommended Regime (Elemental Zn, Daily over 6 Weeks) |
|---|---|
| 11–9.5 | 10–15 mg |
| below 9.0 | 30 mg |
| below 6.0 | up to 45 mg |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grüngreiff, K.; Gottstein, T.; Reinhold, D.; Blindauer, C.A. Albumin Substitution in Decompensated Liver Cirrhosis: Don’t Forget Zinc. Nutrients 2021, 13, 4011. https://doi.org/10.3390/nu13114011
Grüngreiff K, Gottstein T, Reinhold D, Blindauer CA. Albumin Substitution in Decompensated Liver Cirrhosis: Don’t Forget Zinc. Nutrients. 2021; 13(11):4011. https://doi.org/10.3390/nu13114011
Chicago/Turabian StyleGrüngreiff, Kurt, Thomas Gottstein, Dirk Reinhold, and Claudia A. Blindauer. 2021. "Albumin Substitution in Decompensated Liver Cirrhosis: Don’t Forget Zinc" Nutrients 13, no. 11: 4011. https://doi.org/10.3390/nu13114011
APA StyleGrüngreiff, K., Gottstein, T., Reinhold, D., & Blindauer, C. A. (2021). Albumin Substitution in Decompensated Liver Cirrhosis: Don’t Forget Zinc. Nutrients, 13(11), 4011. https://doi.org/10.3390/nu13114011