
Graptopetalum paraguayense Extract Ameliorates Proteotoxicity in Aging and Age-Related Diseases in Model Systems

 , , and
, , and
Abstract
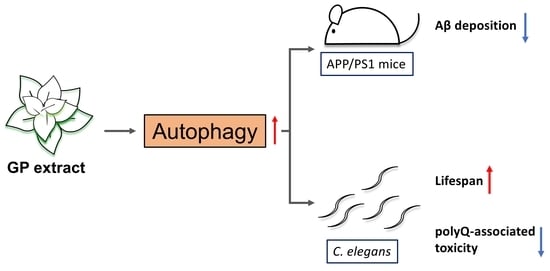
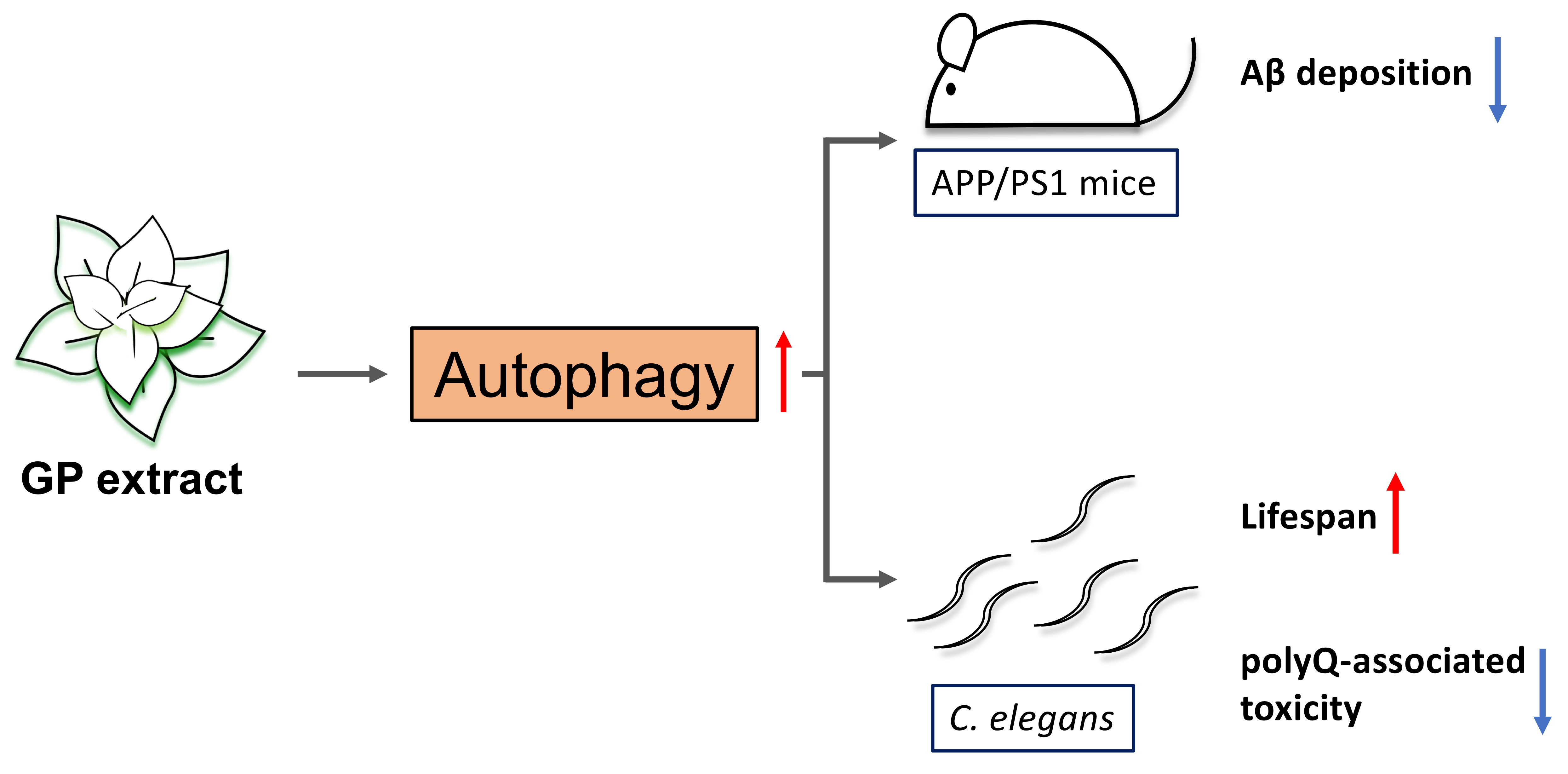
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Preparation of GP Extract
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. Extracellular Aβ1-40 and Aβ1-42 Detection
2.5. Western Blot Assay
2.6. Mouse Studies
2.7. Feeding Protocol
2.8. Thioflavin-S (ThS) Fluorescent Staining in Brain Sections
2.9. Detection of Soluble and Insoluble Aβ
2.10. L1000 Expression Profiling
2.11. Gene Set Enrichment Analysis (GSEA)
2.12. C. elegans Strains
2.13. GFP::LGG-1 Puncta Quantification
2.14. HLH-30::GFP Nuclear-Cytoplasmic Ratio (N/C Ratio) Quantification
2.15. Mobility Analysis in C. elegans
2.16. Lifespan Analysis in C. elegans
3. Results
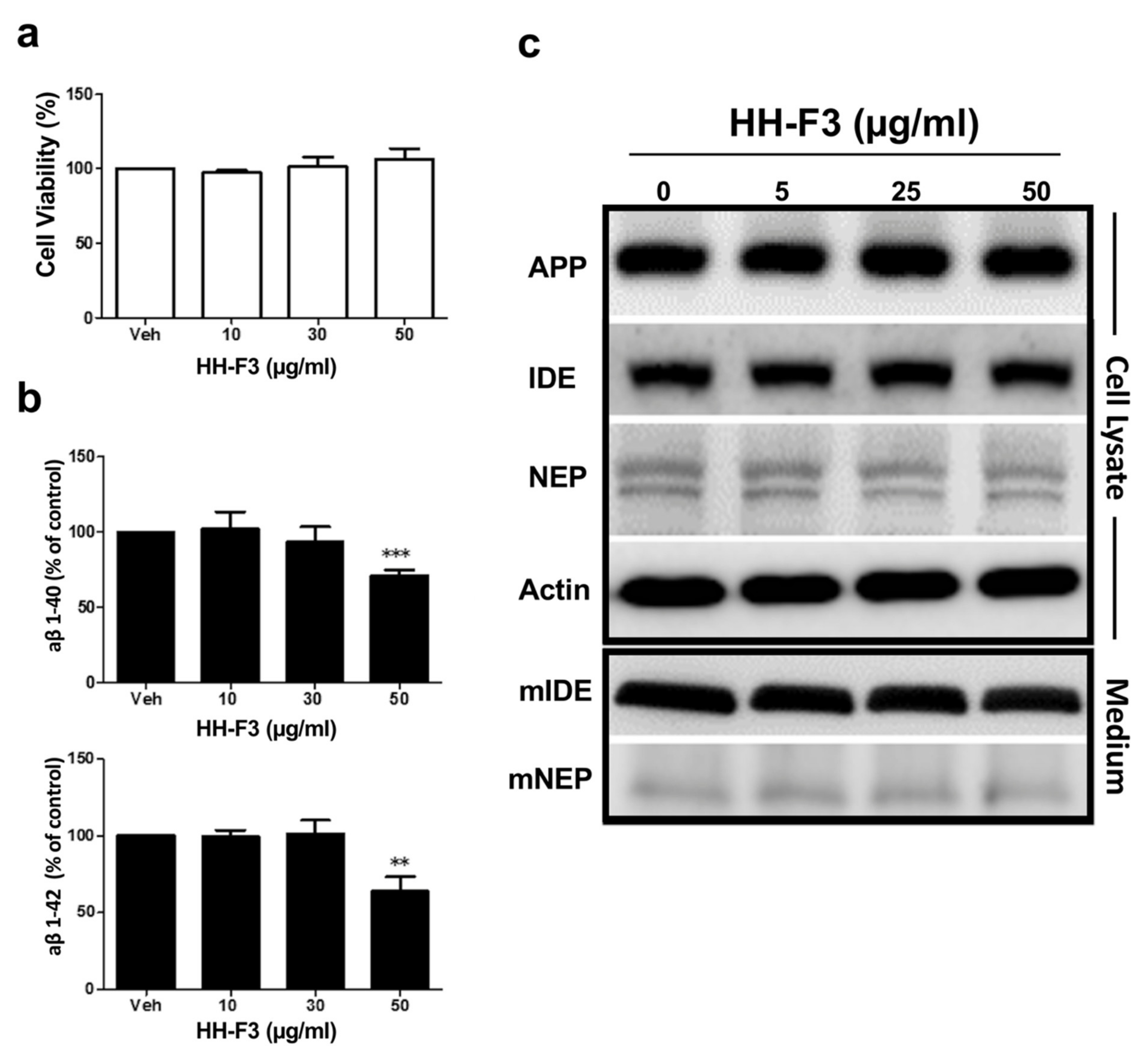
3.1. GP Extract Inhibits the Secretion of Amyloid-Aβs in the Human Neuroblastoma SH-SY5Y-APP695 Cells
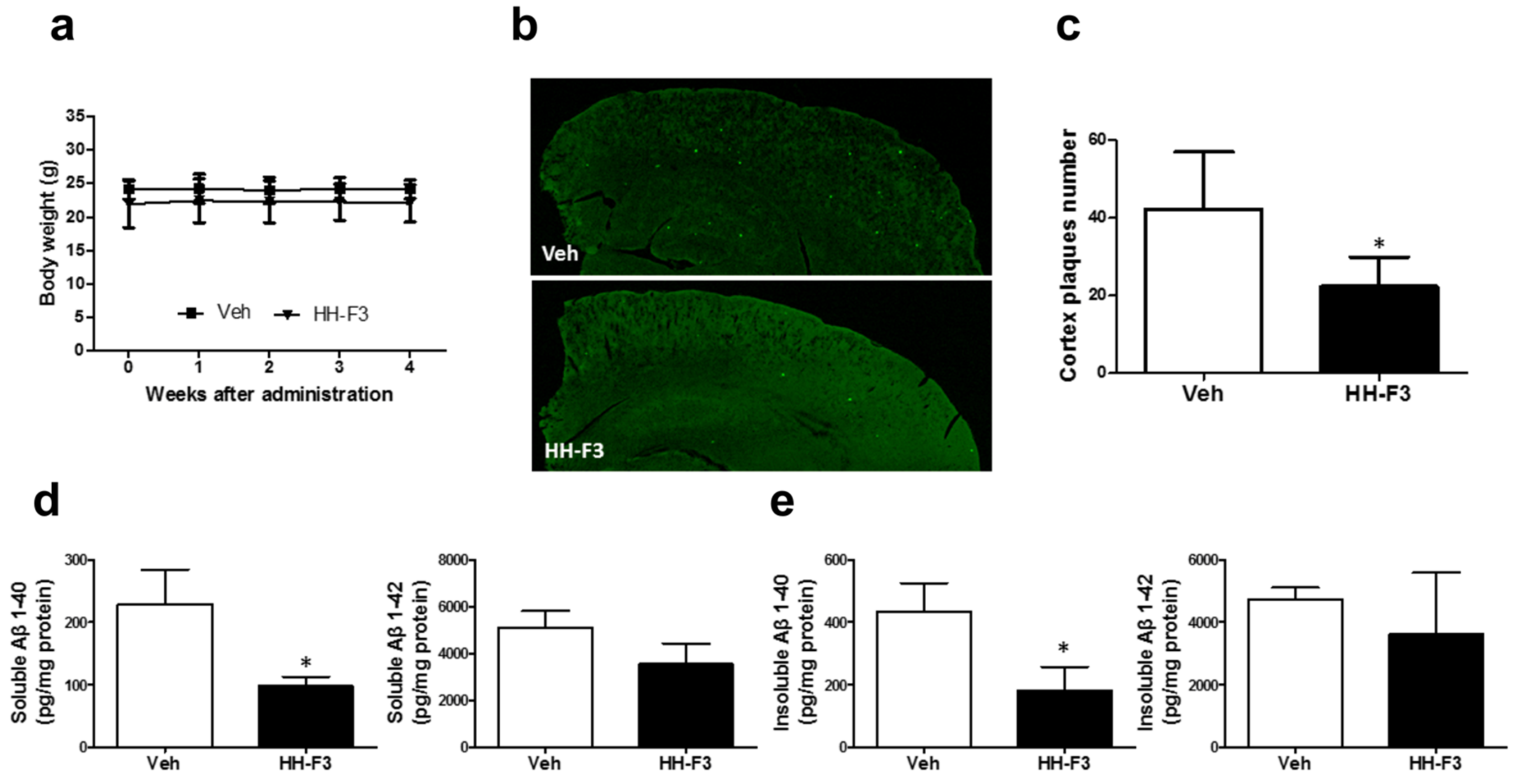
3.2. GP Extract Reduces the Plaque Formation in the Cerebral Cortex of APP/PS1 Transgenic Mice
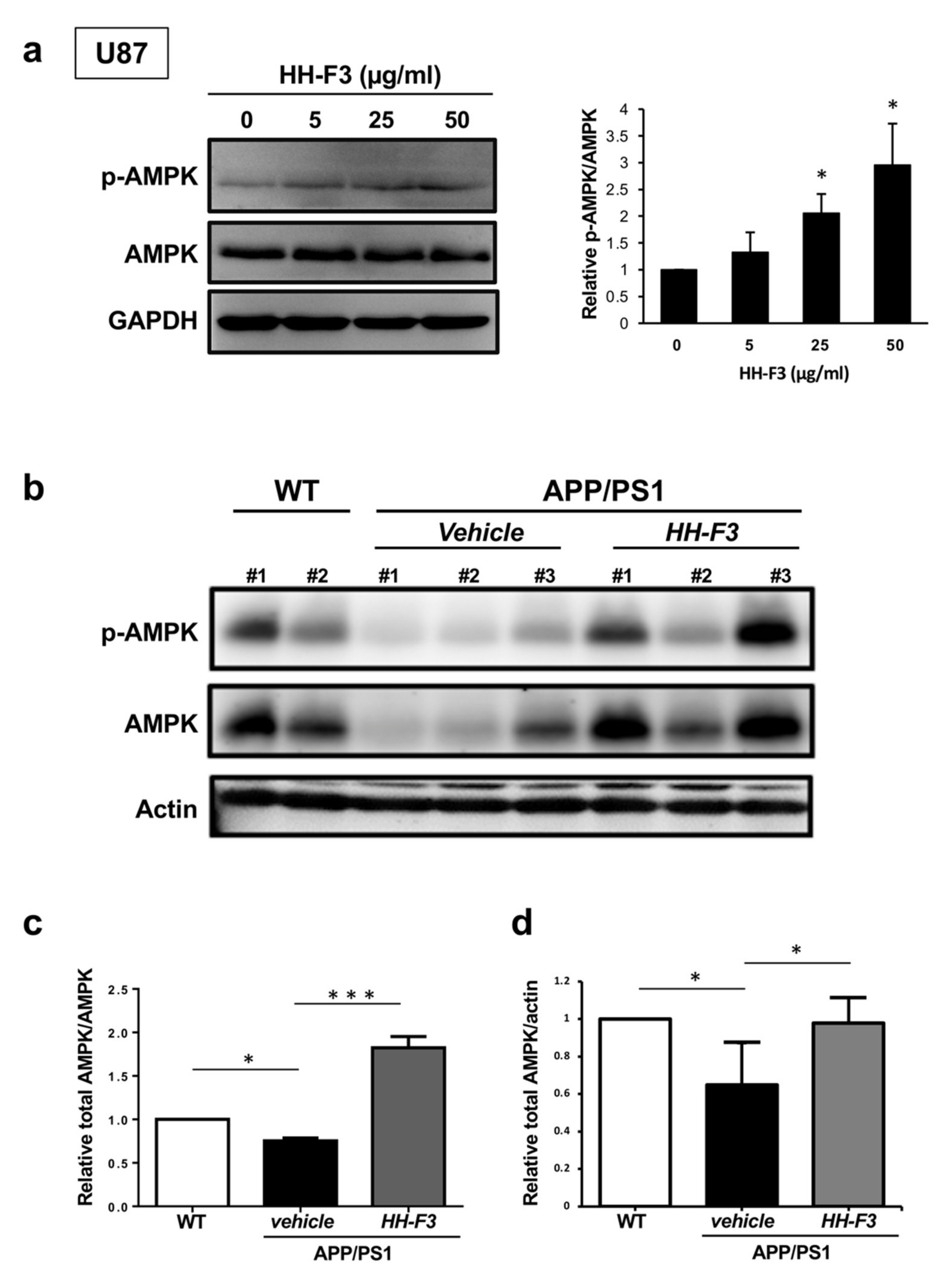
3.3. GP Extract Activates AMPK in U87 Cells and the Cerebral Cortex of APP/PS1 Mice
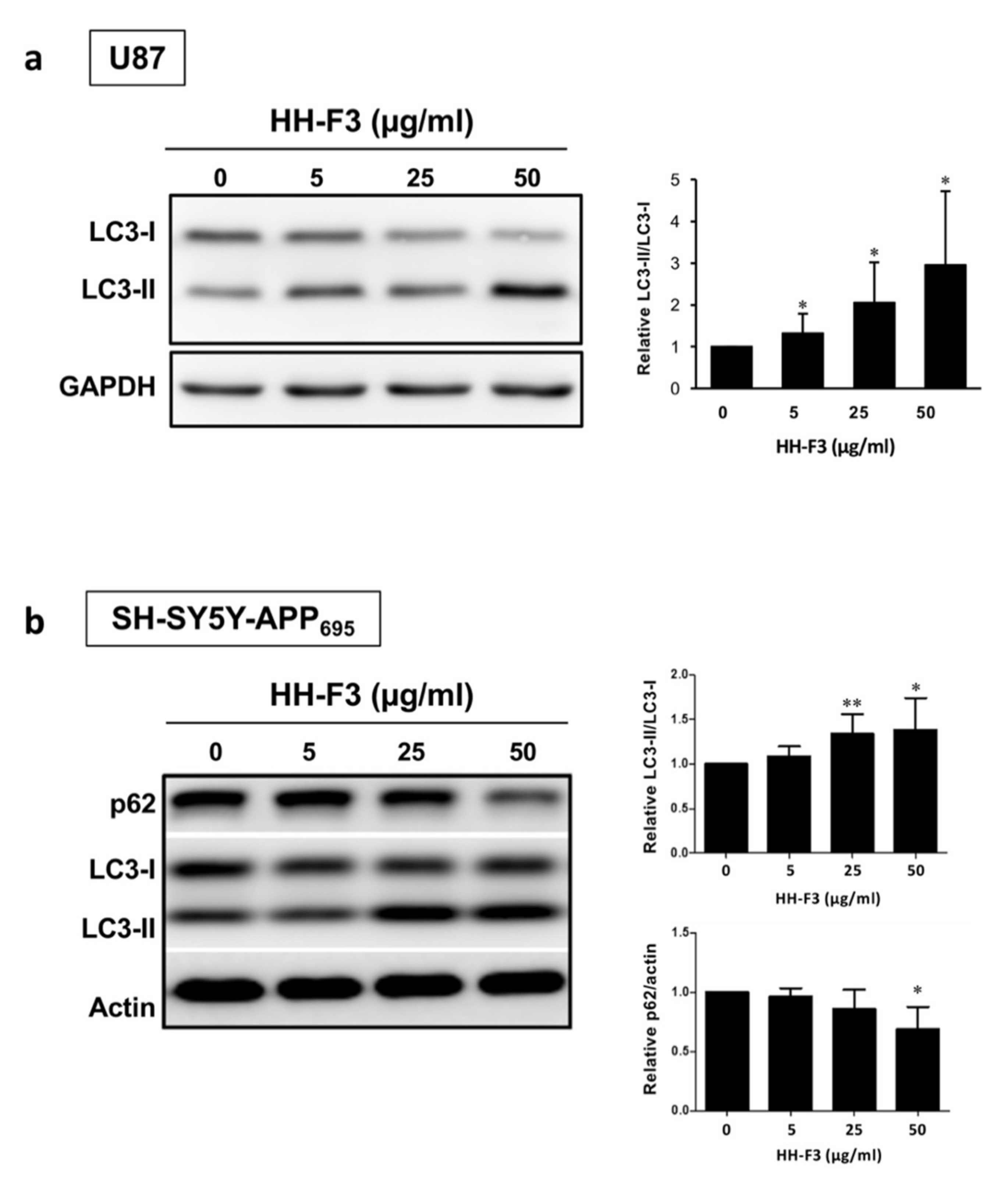
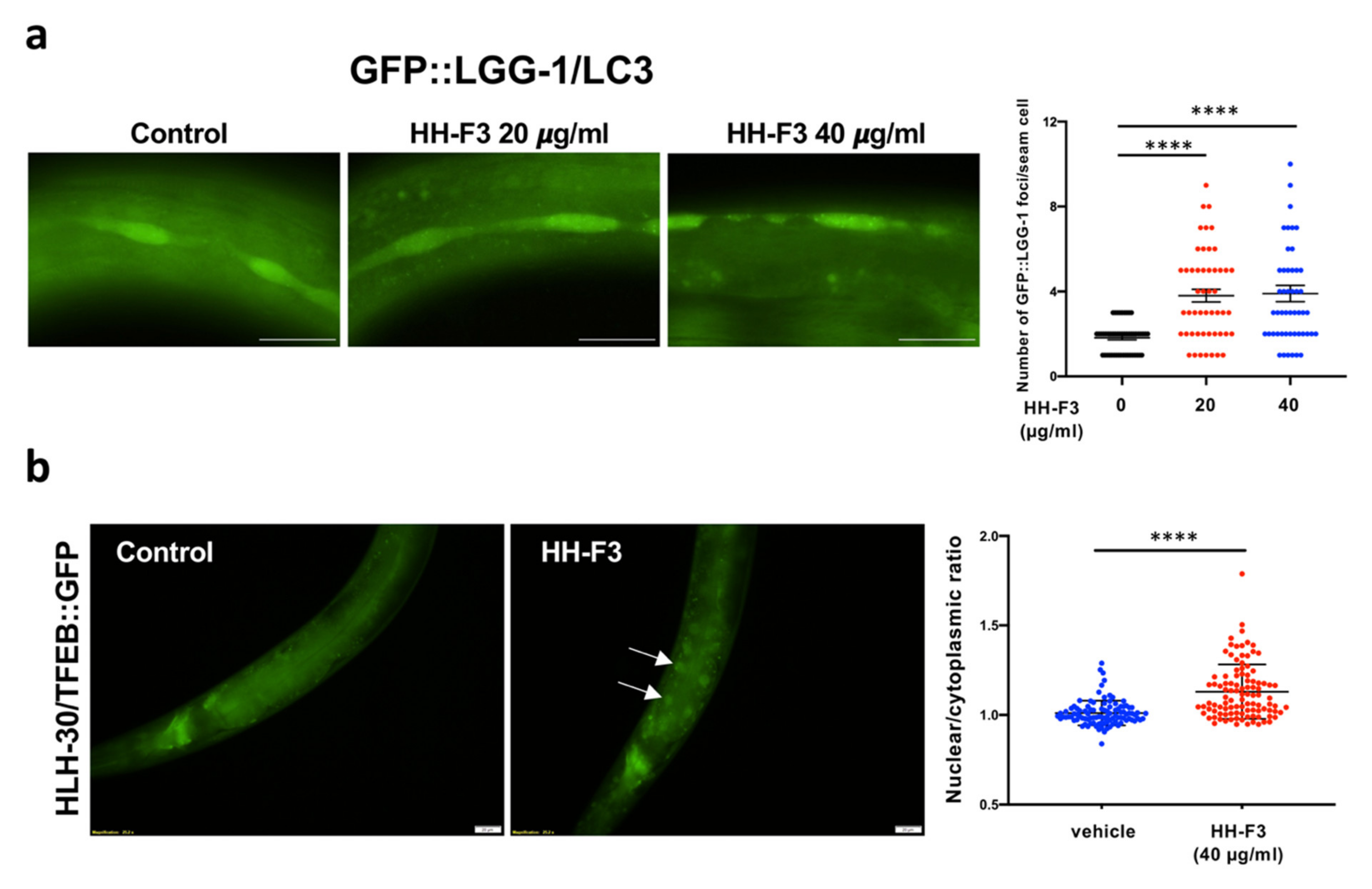
3.4. Autophagy Is Elevated by GP Extract Both in Cells and C. elegans
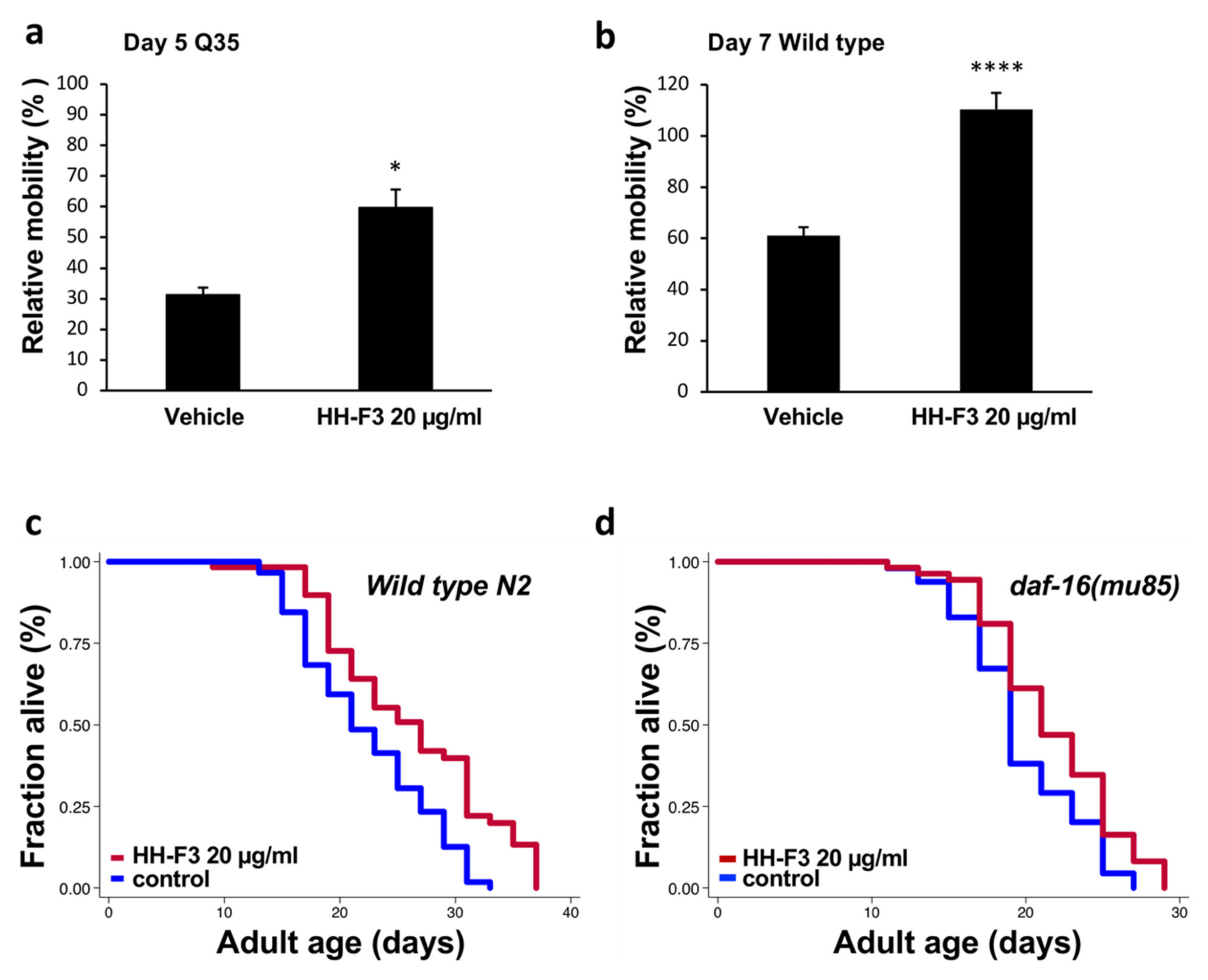
3.5. GP Extract Extended Lifespan in C. elegans in a Daf-16-Independent Manner
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khan, S.S.; Singer, B.D.; Vaughan, D.E. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell 2017, 16, 624–633. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [PubMed] [Green Version]
- Halama, A.; Kulinski, M.; Dib, S.S.; Zaghlool, S.B.; Siveen, K.S.; Iskandarani, A.; Zierer, J.; Prabhu, K.S.; Satheesh, N.J.; Bhagwat, A.M.; et al. Accelerated lipid catabolism and autophagy are cancer survival mechanisms under inhibited glutaminolysis. Cancer Lett. 2018, 430, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Caballero, B.; Wang, Y.; Diaz, A.; Tasset, I.; Juste, Y.R.; Stiller, B.; Mandelkow, E.M.; Mandelkow, E.; Cuervo, A.M. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 2018, 17, e12692. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Sheehan, P.; Chen, S.; Yue, Z. Is amyotrophic lateral sclerosis/frontotemporal dementia an autophagy disease? Mol. Neurodegener. 2017, 12, 90. [Google Scholar]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar]
- Schaeffer, V.; Lavenir, I.; Ozcelik, S.; Tolnay, M.; Winkler, D.T.; Goedert, M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 2012, 135, 2169–2177. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.L.; Jahrling, J.B.; Zhang, W.; DeRosa, N.; Bakshi, V.; Romero, P.; Galvan, V.; Richardson, A. Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2017, 37, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congdon, E.E.; Wu, J.W.; Myeku, N.; Figueroa, Y.H.; Herman, M.; Marinec, P.S.; Gestwicki, J.E.; Dickey, C.A.; Yu, W.H.; Duff, K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 2012, 8, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.D.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. Autophagy and Longevity. Mol. Cells 2018, 41, 65–72. [Google Scholar]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, A.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef]
- Hsu, W.H.; Liao, S.C.; Chyan, Y.J.; Huang, K.W.; Hsu, S.L.; Chen, Y.C.; Siu, M.L.; Chang, C.C.; Chung, Y.S.; Huang, C.F. Graptopetalum paraguayense Inhibits Liver Fibrosis by Blocking TGF-beta Signaling In Vivo and In Vitro. Int. J. Mol. Sci. 2019, 20, 2592. [Google Scholar] [CrossRef] [Green Version]
- Duh, P.D.; Lin, S.L.; Wu, S.C. Hepatoprotection of Graptopetalum paraguayense E. Walther on CCl(4)-induced liver damage and inflammation. J. Ethnopharmacol. 2011, 134, 379–385. [Google Scholar] [CrossRef]
- Su, L.J.; Chang, C.C.; Yang, C.H.; Hsieh, S.J.; Wu, Y.C.; Lai, J.M.; Tseng, T.L.; Huang, C.Y.; Hsu, S.L. Graptopetalum paraguayense ameliorates chemical-induced rat hepatic fibrosis in vivo and inactivates stellate cells and Kupffer cells in vitro. PLoS ONE 2013, 8, e53988. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.C.; Fann, M.J.; Tran, T.T.; Chen, S.C.; Devina, T.; Cheng, I.H.; Lien, C.C.; Kao, L.S.; Wang, S.J.; Fuh, J.L.; et al. Assessing the therapeutic potential of Graptopetalum paraguayense on Alzheimer’s disease using patient iPSC-derived neurons. Sci. Rep. 2019, 9, 19301. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.H.; Chang, C.C.; Huang, K.W.; Chen, Y.C.; Hsu, S.L.; Wu, L.C.; Tsou, A.P.; Lai, J.M.; Huang, C.Y. Evaluation of the medicinal herb Graptopetalum paraguayense as a treatment for liver cancer. PLoS ONE 2015, 10, e0121298. [Google Scholar]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.L.; Murphy, C.T.; Kenyon, C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 2003, 300, 1142–1145. [Google Scholar] [CrossRef] [Green Version]
- Apfeld, J.; Kenyon, C. Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature 1999, 402, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Lopez Sanchez, M.I.G.; Waugh, H.S.; Tsatsanis, A.; Wong, B.X.; Crowston, J.G.; Duce, J.A.; Trounce, I.A. Amyloid precursor protein drives down-regulation of mitochondrial oxidative phosphorylation independent of amyloid beta. Sci. Rep. 2017, 7, 9835. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Chen, Y.; Vingtdeux, V.; Zhao, H.; Viollet, B.; Marambaud, P.; Klann, E. Inhibition of AMP-activated protein kinase signaling alleviates impairments in hippocampal synaptic plasticity induced by amyloid beta. J. Neurosci. 2014, 34, 12230–12238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Sato, Y.; Nixon, R.A. Primary lysosomal dysfunction causes cargo-specific deficits of axonal transport leading to Alzheimer-like neuritic dystrophy. Autophagy 2011, 7, 1562–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.F.; Huang, Y.; Chen, S.D.; Halliday, G. Immunohistochemical evidence for macroautophagy in neurones and endothelial cells in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2010, 36, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Park, H.J.; Kim, H.N.; Oh, S.H.; Bae, J.S.; Ha, H.J.; Lee, P.H. Mesenchymal stem cells enhance autophagy and increase beta-amyloid clearance in Alzheimer disease models. Autophagy 2014, 10, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Rocchi, A.; Yamamoto, S.; Ting, T.; Fan, Y.; Sadleir, K.; Wang, Y.; Zhang, W.; Huang, S.; Levine, B.; Vassar, R.; et al. A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer’s disease. PLoS Genet. 2017, 13, e1006962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, A.; De Pinto, V.; Messina, A.; Branca, C.; Oddo, S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J. Neurosci. 2014, 34, 7988–7998. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzamba, D.; Harantova, L.; Butenko, O.; Anderova, M. Glial Cells—The Key Elements of Alzheimer s Disease. Curr. Alzheimer Res. 2016, 13, 894–911. [Google Scholar] [CrossRef]
- Simonovitch, S.; Schmukler, E.; Bespalko, A.; Iram, T.; Frenkel, D.; Holtzman, D.M.; Masliah, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Impaired Autophagy in APOE4 Astrocytes. J. Alzheimers Dis. 2016, 51, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzales, E.; Burchett, J.M.; Schuler, D.R.; Cirrito, J.R.; et al. Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 2014, 34, 9607–9620. [Google Scholar] [CrossRef] [Green Version]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apfeld, J.; O’Connor, G.; McDonagh, T.; DiStefano, P.S.; Curtis, R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004, 18, 3004–3009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapierre, L.R.; De Magalhaes Filho, C.D.; McQuary, P.R.; Chu, C.C.; Visvikis, O.; Chang, J.T.; Gelino, S.; Ong, B.; Davis, A.E.; Irazoqui, J.E.; et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 2013, 4, 2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, H.J.; Yao, P.; Huynh, F.K.; Escoubas, C.C.; Goncalves, R.L.; Burkewitz, K.; Laboy, R.; Hirschey, M.D.; Mair, W.B. Dietary Restriction and AMPK Increase Lifespan via Mitochondrial Network and Peroxisome Remodeling. Cell Metab. 2017, 26, 884–896.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.T.; You, G.T.; Jian, Y.J.; Chen, T.S.; Siao, Y.C.; Hsu, A.L.; Ching, T.T. AMPK-mediated formation of stress granules is required for dietary restriction-induced longevity in Caenorhabditis elegans. Aging Cell 2020, 19, e13157. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-X.; Le, P.T.N.; Tzeng, T.-T.; Tran, T.-H.; Nguyen, A.T.; Cheng, I.H.-J.; Huang, C.-Y.F.; Shiao, Y.-J.; Ching, T.-T. Graptopetalum paraguayense Extract Ameliorates Proteotoxicity in Aging and Age-Related Diseases in Model Systems. Nutrients 2021, 13, 4317. https://doi.org/10.3390/nu13124317
Chen Y-X, Le PTN, Tzeng T-T, Tran T-H, Nguyen AT, Cheng IH-J, Huang C-YF, Shiao Y-J, Ching T-T. Graptopetalum paraguayense Extract Ameliorates Proteotoxicity in Aging and Age-Related Diseases in Model Systems. Nutrients. 2021; 13(12):4317. https://doi.org/10.3390/nu13124317
Chicago/Turabian StyleChen, Yan-Xi, Phuong Thu Nguyen Le, Tsai-Teng Tzeng, Thu-Ha Tran, Anh Thuc Nguyen, Irene Han-Juo Cheng, Chi-Ying F. Huang, Young-Ji Shiao, and Tsui-Ting Ching. 2021. "Graptopetalum paraguayense Extract Ameliorates Proteotoxicity in Aging and Age-Related Diseases in Model Systems" Nutrients 13, no. 12: 4317. https://doi.org/10.3390/nu13124317
APA StyleChen, Y. -X., Le, P. T. N., Tzeng, T. -T., Tran, T. -H., Nguyen, A. T., Cheng, I. H. -J., Huang, C. -Y. F., Shiao, Y. -J., & Ching, T. -T. (2021). Graptopetalum paraguayense Extract Ameliorates Proteotoxicity in Aging and Age-Related Diseases in Model Systems. Nutrients, 13(12), 4317. https://doi.org/10.3390/nu13124317