
Advances in NAD-Lowering Agents for Cancer Treatment

Abstract
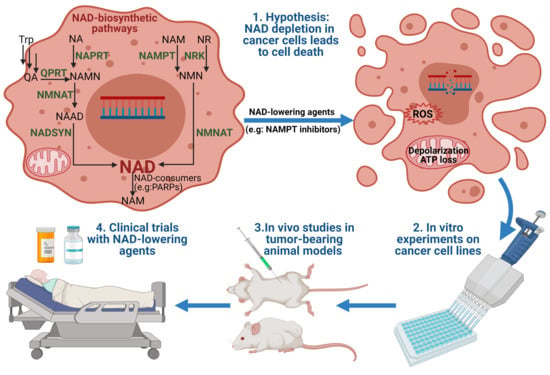
:
1. Introduction
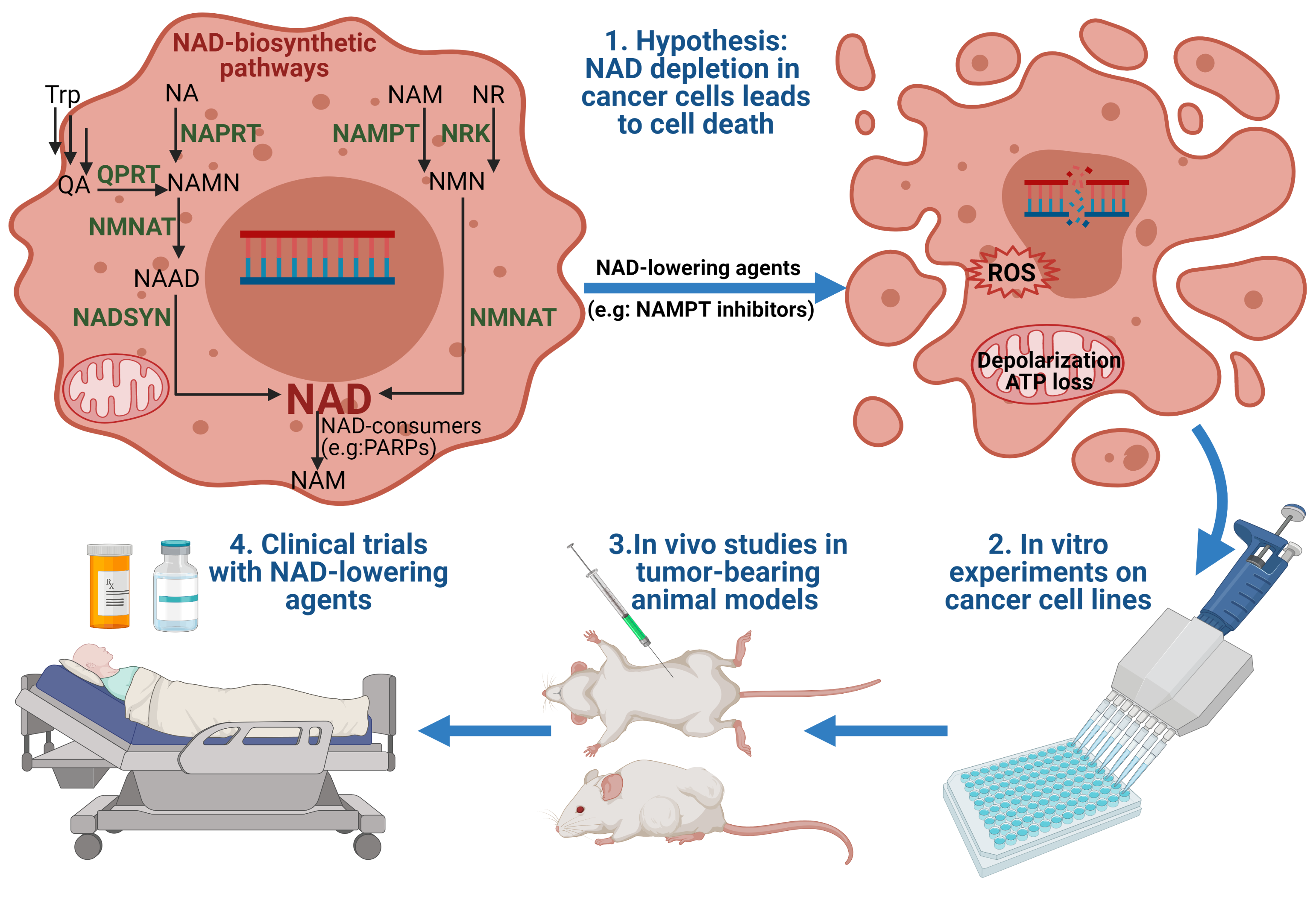
2. NAD Biosynthesis in Mammals
3. Regulation of NAD Production in Cancer Cells
3.1. NAMPT Regulation
3.2. NAPRT Regulation
3.3. QAPRT Regulation
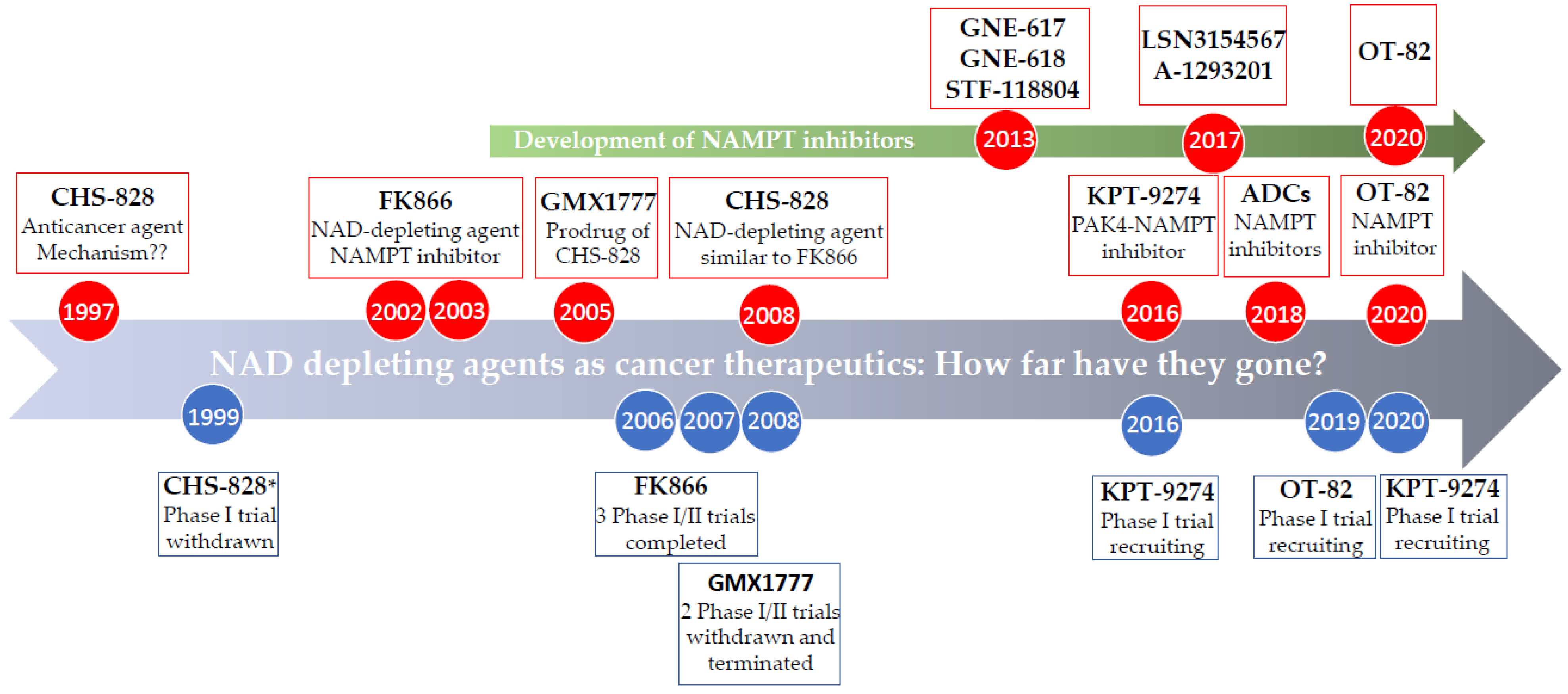
4. Chemical Inhibitors of NAD Biosynthesis
4.1. Specific NAMPT Inhibitors
4.1.1. FK866 (also known as APO866, (E)-Daporinad, and WK175)
4.1.2. CHS-828 (GMX1778)
4.1.3. GMX1777 (EB1627)
4.1.4. OT-82
4.2. Dual NAMPT Inhibitors
4.2.1. KPT-9274 (ATG-019)
4.2.2. STF-31
4.2.3. Chidamide
4.3. Inhibitors of Other NAD-Producing Enzymes
4.3.1. Vacor
4.3.2. 2-Hydroxy Nicotinic Acid (2-HNA)
4.3.3. N-(3,4-dichlorophenyl)-4-{[(4-nitrophenyl)carbamoyl]amino}benzenesulfonamide (Compound 5824)
5. Effects of NAD Production Inhibition in Cancer
5.1. NAD Depletion and Cancer Cell Death
5.2. NAD Depletion and Oxidative Stress
5.3. NAD Depletion and DNA Damage and Repair
5.4. NAD Depletion and Targeted Therapy
6. In Vivo Studies of NAD Production Inhibitors in Mice
6.1. Efficacy of NAMPT Inhibitors In Vivo
6.2. Impact of NA on the Efficacy of NAMPT Inhibitors In Vivo
6.3. Toxicity of NAMPT Inhibitors In Vivo
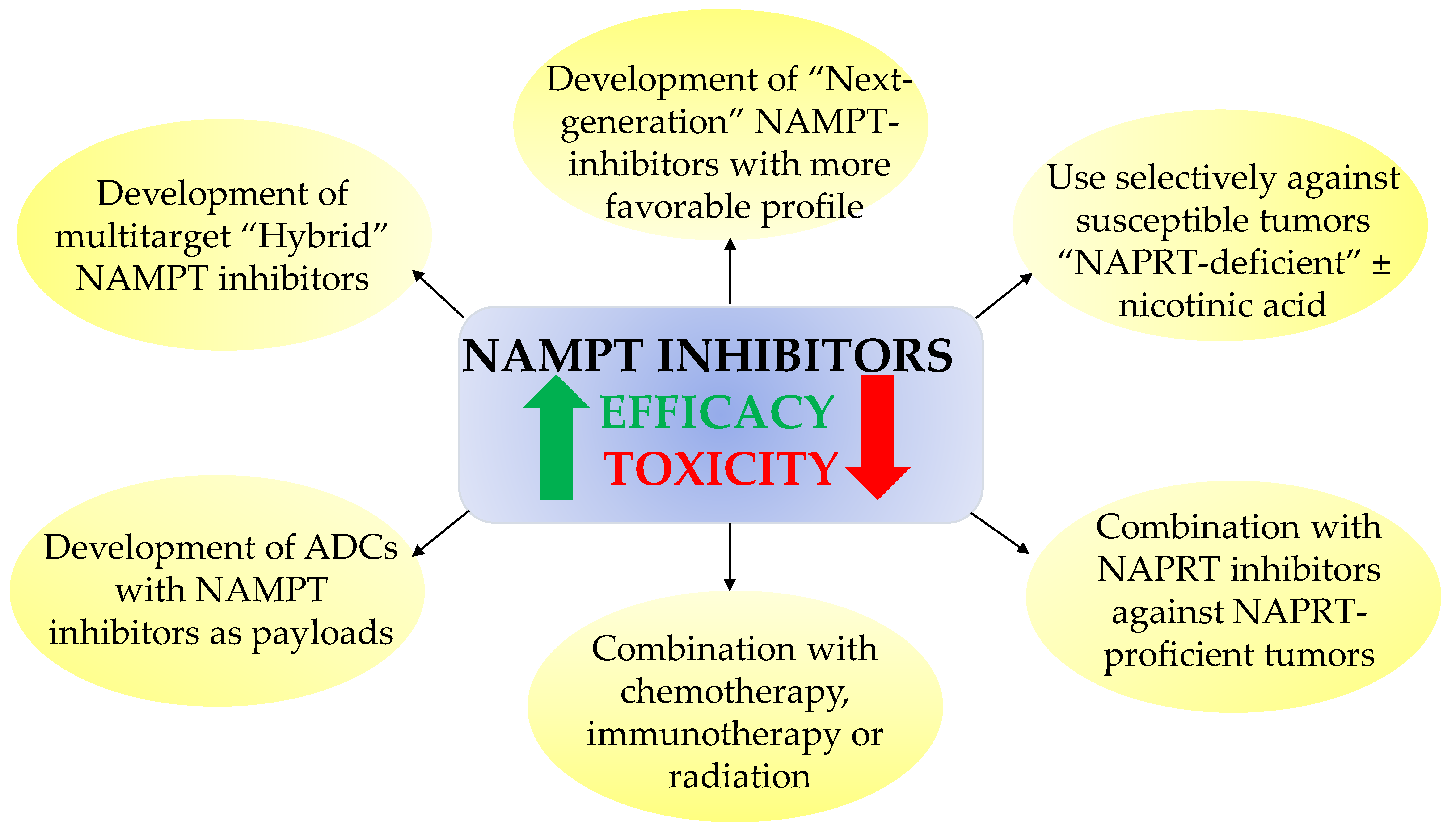
7. Perspectives and Obstacles for Clinical Uses of NAD Biosynthesis Inhibitors
- (i)
- Developing safer and more effective “next-generation” NAMPT inhibitors;
- (ii)
- Using NAMPT inhibitors against a subset of cancers that show unique sensitivity to NAMPT inhibitors, such as hematological cancers or IDH-mutant cancers. NA might be administered simultaneously to protect normal tissues (circumventing NAMPT inhibition by the PH pathway to sustain adequate NAD stores). Although NA might alleviate the systemic toxicity and widen the therapeutic index of NAMPT inhibitors, abrogation of antitumor efficacy could represent a caveat to this approach;
- (iii)
- Combining NAMPT inhibitors with chemotherapy, immunotherapy, or radiation to achieve a synergistic effect;
- (iv)
- Combining NAMPT inhibitors with NAPRT inhibitors against NAPRT-positive cancer subtypes [62];
- (v)
- Development of “broad-spectrum” NAMPT inhibitors such as the dual NAMPT-PAK inhibitors, NAMPT-HDAC inhibitors, NAMPT-GLUT1 inhibitors, and recently the NAMPT-EGFR inhibitors;
- (vi)
- Development of NAMPTi-ADCs that selectively target NAMPT in cancer cells through antibody binding to cancer-specific cell surface markers, thereby sparing the normal cells from systemic NAD depletion.
8. Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On Respiratory Impairment in Cancer Cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Garten, A.; Schuster, S.; Penke, M.; Gorski, T.; de Giorgis, T.; Kiess, W. Physiological and Pathophysiological Roles of NAMPT and NAD Metabolism. Nat. Rev. Endocrinol. 2015, 11, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [Green Version]
- Yaku, K.; Okabe, K.; Nakagawa, T. NAD Metabolism: Implications in Aging and Longevity. Ageing Res. Rev. 2018, 47, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Cantó, C.; Wanders, R.J.; Auwerx, J. The Secret Life of NAD+: An Old Metabolite Controlling New Metabolic Signaling Pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [Green Version]
- Chiarugi, A.; Dölle, C.; Felici, R.; Ziegler, M. The NAD Metabolome—A Key Determinant of Cancer Cell Biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD+ in Aging, Metabolism, and Neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ Metabolism: Pathophysiologic Mechanisms and Therapeutic Potential. Signal Transduct. Target. Ther. 2020, 5, 1–37. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic Acid, Nicotinamide, and Nicotinamide Riboside: A Molecular Evaluation of NAD+ Precursor Vitamins in Human Nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Preiss, J.; Handler, P. Biosynthesis of Diphosphopyridine Nucleotide. I. Identification of Intermediates. J. Biol. Chem. 1958, 233, 488–492. [Google Scholar] [CrossRef]
- Preiss, J.; Handler, P. Biosynthesis of Diphosphopyridine Nucleotide. II. Enzymatic Aspects. J. Biol. Chem. 1958, 233, 493–500. [Google Scholar] [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of Nicotinamide Riboside as a Nutrient and Conserved NRK Genes Establish a Preiss-Handler Independent Route to NAD+ in Fungi and Humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Tempel, W.; Rabeh, W.M.; Bogan, K.L.; Belenky, P.; Wojcik, M.; Seidle, H.F.; Nedyalkova, L.; Yang, T.; Sauve, A.A.; Park, H.-W.; et al. Nicotinamide Riboside Kinase Structures Reveal New Pathways to NAD+. PLoS Biol. 2007, 5, e263. [Google Scholar] [CrossRef]
- Berger, F.; Lau, C.; Dahlmann, M.; Ziegler, M. Subcellular Compartmentation and Differential Catalytic Properties of the Three Human Nicotinamide Mononucleotide Adenylyltransferase Isoforms. J. Biol. Chem. 2005, 280, 36334–36341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, C.; Niere, M.; Ziegler, M. The NMN/NaMN Adenylyltransferase (NMNAT) Protein Family. Front. Biosci. J. Virtual Libr. 2009, 14, 410–431. [Google Scholar] [CrossRef]
- Trammell, S.A.; Yu, L.; Redpath, P.; Migaud, M.E.; Brenner, C. Nicotinamide Riboside Is a Major NAD+ Precursor Vitamin in Cow Milk. J. Nutr. 2016, 146, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Long, D.J.; Jaiswal, A.K. NRH:Quinone Oxidoreductase2 (NQO2). Chem. Biol. Interact. 2000, 129, 99–112. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, N.; Zhang, G.; Sauve, A.A. NRH Salvage and Conversion to NAD+ Requires NRH Kinase Activity by Adenosine Kinase. Nat. Metab. 2020, 2, 364–379. [Google Scholar] [CrossRef]
- Kulikova, V.A.; Nikiforov, A.A. Role of NUDIX Hydrolases in NAD and ADP-Ribose Metabolism in Mammals. Biochem. Mosc. 2020, 85, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Pérez, R.; Tammaro, A.; Schomakers, B.V.; Scantlebery, A.M.L.; Denis, S.; Elfrink, H.L.; Giroud-Gerbetant, J.; Cantó, C.; López-Leonardo, C.; McIntyre, R.L.; et al. Reduced Nicotinamide Mononucleotide Is a New and Potent NAD+ Precursor in Mammalian Cells and Mice. FASEB J. 2021, 35, e21456. [Google Scholar] [CrossRef] [PubMed]
- Giroud-Gerbetant, J.; Joffraud, M.; Giner, M.P.; Cercillieux, A.; Bartova, S.; Makarov, M.V.; Zapata-Pérez, R.; Sánchez-García, J.L.; Houtkooper, R.H.; Migaud, M.E.; et al. A Reduced Form of Nicotinamide Riboside Defines a New Path for NAD+ Biosynthesis and Acts as an Orally Bioavailable NAD+ Precursor. Mol. Metab. 2019, 30, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mohammed, F.S.; Zhang, N.; Sauve, A.A. Dihydronicotinamide Riboside Is a Potent NAD+ Concentration Enhancer in Vitro and in Vivo. J. Biol. Chem. 2019, 294, 9295–9307. [Google Scholar] [CrossRef]
- Shats, I.; Williams, J.G.; Liu, J.; Makarov, M.V.; Wu, X.; Lih, F.B.; Deterding, L.J.; Lim, C.; Xu, X.; Randall, T.A.; et al. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metab. 2020, 31, 564–579.e7. [Google Scholar] [CrossRef]
- Liu, L.; Su, X.; Quinn, W.J.; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ Homeostasis in Health and Disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef]
- Oh, G.-S.; Kim, H.-J.; Choi, J.-H.; Shen, A.; Choe, S.-K.; Karna, A.; Lee, S.H.; Jo, H.-J.; Yang, S.-H.; Kwak, T.H.; et al. Pharmacological Activation of NQO1 Increases NAD+ Levels and Attenuates Cisplatin-Mediated Acute Kidney Injury in Mice. Kidney Int. 2014, 85, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Gaikwad, A.; Long, D.J.; Stringer, J.L.; Jaiswal, A.K. In Vivo Role of NAD(P)H:Quinone Oxidoreductase 1 (NQO1) in the Regulation of Intracellular Redox State and Accumulation of Abdominal Adipose Tissue. J. Biol. Chem. 2001, 276, 22559–22564. [Google Scholar] [CrossRef] [Green Version]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De Novo NAD+ Synthesis Enhances Mitochondrial Function and Improves Health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef]
- Palzer, L.; Bader, J.J.; Angel, F.; Witzel, M.; Blaser, S.; McNeil, A.; Wandersee, M.K.; Leu, N.A.; Lengner, C.J.; Cho, C.E.; et al. Alpha-Amino-Beta-Carboxy-Muconate-Semialdehyde Decarboxylase Controls Dietary Niacin Requirements for NAD+ Synthesis. Cell Rep. 2018, 25, 1359–1370.e4. [Google Scholar] [CrossRef] [Green Version]
- Bockwoldt, M.; Houry, D.; Niere, M.; Gossmann, T.I.; Reinartz, I.; Schug, A.; Ziegler, M.; Heiland, I. Identification of Evolutionary and Kinetic Drivers of NAD-Dependent Signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 15957–15966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.; et al. Nicotinamide N-Methyltransferase Knockdown Protects against Diet-Induced Obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT Promotes Epigenetic Remodeling in Cancer by Creating a Metabolic Methylation Sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, M.A.; Coscia, F.; Chryplewicz, A.; Chang, J.W.; Hernandez, K.M.; Pan, S.; Tienda, S.M.; Nahotko, D.A.; Li, G.; Blaženović, I.; et al. Proteomics Reveals NNMT as a Master Metabolic Regulator of Cancer Associated Fibroblasts. Nature 2019, 569, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kim, L.J.; Wang, X.; Wu, Q.; Sanvoranart, T.; Hubert, C.G.; Prager, B.C.; Wallace, L.C.; Jin, X.; Mack, S.C.; et al. Nicotinamide Metabolism Regulates Glioblastoma Stem Cell Maintenance. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Zanca, C.; Rajkumar, U.; Koga, T.; Diao, Y.; Raviram, R.; Liu, F.; Turner, K.; Yang, H.; Brunk, E.; et al. NAD Metabolic Dependency in Cancer Is Shaped by Gene Amplification and Enhancer Remodelling. Nature 2019, 569, 570–575. [Google Scholar] [CrossRef]
- Menssen, A.; Hydbring, P.; Kapelle, K.; Vervoorts, J.; Diebold, J.; Luscher, B.; Larsson, L.-G.; Hermeking, H. The C-MYC Oncoprotein, the NAMPT Enzyme, the SIRT1-Inhibitor DBC1, and the SIRT1 Deacetylase Form a Positive Feedback Loop. Proc. Natl. Acad. Sci. USA 2012, 109, E187–E196. [Google Scholar] [CrossRef] [Green Version]
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD+ Metabolism Governs the Proinflammatory Senescence-Associated Secretome. Nat. Cell Biol. 2019, 21, 397–407. [Google Scholar] [CrossRef]
- Sociali, G.; Grozio, A.; Caffa, I.; Schuster, S.; Becherini, P.; Damonte, P.; Sturla, L.; Fresia, C.; Passalacqua, M.; Mazzola, F.; et al. SIRT6 Deacetylase Activity Regulates NAMPT Activity and NAD(P)(H) Pools in Cancer Cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 3704–3717. [Google Scholar] [CrossRef]
- Yoon, M.J.; Yoshida, M.; Johnson, S.; Takikawa, A.; Usui, I.; Tobe, K.; Nakagawa, T.; Yoshino, J.; Imai, S. SIRT1-Mediated ENAMPT Secretion from Adipose Tissue Regulates Hypothalamic NAD+ and Function in Mice. Cell Metab. 2015, 21, 706–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, B.; Park, J.W.; Kim, J.G.; Lee, B.J. FOXO1 Functions in the Regulation of Nicotinamide Phosphoribosyltransferase (Nampt) Expression. Biochem. Biophys. Res. Commun. 2019, 511, 398–403. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, N.; Liu, Y.; Su, P.; Liang, Y.; Li, Y.; Wang, X.; Chen, T.; Song, X.; Sang, Y.; et al. Epigenetic Regulation of NAMPT by NAMPT-AS Drives Metastatic Progression in Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 3347–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhang, M.; Lu, W. Long Noncoding RNA GACAT3 Promotes Glioma Progression by Sponging MiR-135a. J. Cell. Physiol. 2019, 234, 10877–10887. [Google Scholar] [CrossRef] [PubMed]
- Bolandghamat Pour, Z.; Nourbakhsh, M.; Mousavizadeh, K.; Madjd, Z.; Ghorbanhosseini, S.S.; Abdolvahabi, Z.; Hesari, Z.; Mobaser, S.E. Up-Regulation of MiR-381 Inhibits NAD+ Salvage Pathway and Promotes Apoptosis in Breast Cancer Cells. EXCLI J. 2019, 18, 683–696. [Google Scholar] [CrossRef]
- Hesari, Z.; Nourbakhsh, M.; Hosseinkhani, S.; Abdolvahabi, Z.; Alipour, M.; Tavakoli-Yaraki, M.; Ghorbanhosseini, S.S.; Yousefi, Z.; Jafarzadeh, M.; Yarahmadi, S. Down-Regulation of NAMPT Expression by Mir-206 Reduces Cell Survival of Breast Cancer Cells. Gene 2018, 673, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Ghorbanhosseini, S.S.; Nourbakhsh, M.; Zangooei, M.; Abdolvahabi, Z.; Bolandghamtpour, Z.; Hesari, Z.; Yousefi, Z.; Panahi, G.; Meshkani, R. MicroRNA-494 Induces Breast Cancer Cell Apoptosis and Reduces Cell Viability by Inhibition of Nicotinamide Phosphoribosyltransferase Expression and Activity. EXCLI J. 2019, 18, 838–851. [Google Scholar] [CrossRef]
- Bolandghamat Pour, Z.; Nourbakhsh, M.; Mousavizadeh, K.; Madjd, Z.; Ghorbanhosseini, S.S.; Abdolvahabi, Z.; Hesari, Z.; Ezzati Mobasser, S. Suppression of Nicotinamide Phosphoribosyltransferase Expression by MiR-154 Reduces the Viability of Breast Cancer Cells and Increases Their Susceptibility to Doxorubicin. BMC Cancer 2019, 19, 1027. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Tong, J.; Huang, G. Nicotinamide Phosphoribosyl Transferase (Nampt) Is a Target of MicroRNA-26b in Colorectal Cancer Cells. PLoS ONE 2013, 8, e69963. [Google Scholar] [CrossRef] [Green Version]
- Ju, H.-Q.; Zhuang, Z.-N.; Li, H.; Tian, T.; Lu, Y.-X.; Fan, X.-Q.; Zhou, H.-J.; Mo, H.-Y.; Sheng, H.; Chiao, P.J.; et al. Regulation of the Nampt-Mediated NAD Salvage Pathway and Its Therapeutic Implications in Pancreatic Cancer. Cancer Lett. 2016, 379, 1–11. [Google Scholar] [CrossRef]
- Lv, R.; Yu, J.; Sun, Q. Anti-Angiogenic Role of MicroRNA-23b in Melanoma by Disturbing NF-ΚB Signaling Pathway via Targeted Inhibition of NAMPT. Future Oncol. Lond. Engl. 2020, 16, 541-458. [Google Scholar] [CrossRef]
- Shames, D.S.; Elkins, K.; Walter, K.; Holcomb, T.; Du, P.; Mohl, D.; Xiao, Y.; Pham, T.; Haverty, P.M.; Liederer, B.; et al. Loss of NAPRT1 Expression by Tumor-Specific Promoter Methylation Provides a Novel Predictive Biomarker for NAMPT Inhibitors. Clin. Cancer Res. 2013, 19, 6912–6923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fons, N.R.; Sundaram, R.K.; Breuer, G.A.; Peng, S.; McLean, R.L.; Kalathil, A.N.; Schmidt, M.S.; Carvalho, D.M.; Mackay, A.; Jones, C.; et al. PPM1D Mutations Silence NAPRT Gene Expression and Confer NAMPT Inhibitor Sensitivity in Glioma. Nat. Commun. 2019, 10, 3790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LI, H.; FENG, Z.; WU, W.; LI, J.; ZHANG, J.; XIA, T. SIRT3 Regulates Cell Proliferation and Apoptosis Related to Energy Metabolism in Non-Small Cell Lung Cancer Cells through Deacetylation of NMNAT2. Int. J. Oncol. 2013, 43, 1420–1430. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.-C.; Tong, J.; Shi, X.-F.; Zhang, T. MicroRNA-654-3p Enhances Cisplatin Sensitivity by Targeting QPRT and Inhibiting the PI3K/AKT Signaling Pathway in Ovarian Cancer Cells. Exp. Ther. Med. 2020, 20, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Shusheng, J.; Hongtao, S.; Shu, Z.; Lan, H.; Qingyuan, Z.; Shaoqiang, C.; Yuanxi, H. Silencing DSCAM-AS1 Suppresses the Growth and Invasion of ER-Positive Breast Cancer Cells by Downregulating Both DCTPP1 and QPRT. Aging 2020, 12, 14754–14774. [Google Scholar] [CrossRef]
- Ullmark, T.; Montano, G.; Järvstråt, L.; Jernmark Nilsson, H.; Håkansson, E.; Drott, K.; Nilsson, B.; Vidovic, K.; Gullberg, U. Anti-Apoptotic Quinolinate Phosphoribosyltransferase (QPRT) Is a Target Gene of Wilms’ Tumor Gene 1 (WT1) Protein in Leukemic Cells. Biochem. Biophys. Res. Commun. 2017, 482, 802–807. [Google Scholar] [CrossRef] [Green Version]
- Brandl, L.; Kirstein, N.; Neumann, J.; Sendelhofert, A.; Vieth, M.; Kirchner, T.; Menssen, A. The C-MYC/NAMPT/SIRT1 Feedback Loop Is Activated in Early Classical and Serrated Route Colorectal Cancer and Represents a Therapeutic Target. Med. Oncol. 2018, 36, 5. [Google Scholar] [CrossRef]
- Brandl, L.; Zhang, Y.; Kirstein, N.; Sendelhofert, A.; Boos, S.L.; Jung, P.; Greten, F.; Rad, R.; Menssen, A. Targeting C-MYC through Interference with NAMPT and SIRT1 and Their Association to Oncogenic Drivers in Murine Serrated Intestinal Tumorigenesis. Neoplasia 2019, 21, 974–988. [Google Scholar] [CrossRef]
- Sumter, T.F.; Xian, L.; Huso, T.; Koo, M.; Chang, Y.-T.; Almasri, T.N.; Chia, L.; Inglis, C.; Reid, D.; Resar, L.M.S. The High Mobility Group A1 (HMGA1) Transcriptome in Cancer and Development. Curr. Mol. Med. 2016, 16, 353–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piacente, F.; Caffa, I.; Ravera, S.; Sociali, G.; Passalacqua, M.; Vellone, V.G.; Becherini, P.; Reverberi, D.; Monacelli, F.; Ballestrero, A.; et al. Nicotinic Acid Phosphoribosyltransferase Regulates Cancer Cell Metabolism, Susceptibility to NAMPT Inhibitors, and DNA Repair. Cancer Res. 2017, 77, 3857–3869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte-Pereira, S.; Pereira-Castro, I.; Silva, S.S.; Correia, M.G.; Neto, C.; da Costa, L.T.; Amorim, A.; Silva, R.M. Extensive Regulation of Nicotinate Phosphoribosyltransferase (NAPRT) Expression in Human Tissues and Tumors. Oncotarget 2016, 7, 1973–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, H.; Lee, J.E.; Shin, S.-J.; Oh, S.; Kwon, G.; Kim, H.; Choi, Y.Y.; White, M.A.; Paik, S.; et al. Selective Cytotoxicity of the NAMPT Inhibitor FK866 Toward Gastric Cancer Cells With Markers of the Epithelial-Mesenchymal Transition, Due to Loss of NAPRT. Gastroenterology 2018, 155, 799–814.e13. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Elkins, K.; Durieux, J.K.; Lee, L.; Oeh, J.; Yang, L.X.; Liang, X.; DelNagro, C.; Tremayne, J.; Kwong, M.; et al. Dependence of Tumor Cell Lines and Patient-Derived Tumors on the NAD Salvage Pathway Renders Them Sensitive to NAMPT Inhibition with GNE-618. Neoplasia 2013, 15, 1151-IN23. [Google Scholar] [CrossRef]
- Thongon, N.; Zucal, C.; D’Agostino, V.G.; Tebaldi, T.; Ravera, S.; Zamporlini, F.; Piacente, F.; Moschoi, R.; Raffaelli, N.; Quattrone, A.; et al. Cancer Cell Metabolic Plasticity Allows Resistance to NAMPT Inhibition but Invariably Induces Dependence on LDHA. Cancer Metab. 2018, 6, 1. [Google Scholar] [CrossRef]
- Guo, J.; Lam, L.T.; Longenecker, K.L.; Bui, M.H.; Idler, K.B.; Glaser, K.B.; Wilsbacher, J.L.; Tse, C.; Pappano, W.N.; Huang, T.-H. Identification of Novel Resistance Mechanisms to NAMPT Inhibition via the de Novo NAD+ Biosynthesis Pathway and NAMPT Mutation. Biochem. Biophys. Res. Commun. 2017, 491, 681–686. [Google Scholar] [CrossRef]
- Jane, E.P.; Premkumar, D.R.; Thambireddy, S.; Golbourn, B.; Agnihotri, S.; Bertrand, K.C.; Mack, S.C.; Myers, M.I.; Chattopadhyay, A.; Taylor, D.L.; et al. Targeting NAD+ Biosynthesis Overcomes Panobinostat and Bortezomib-Induced Malignant Glioma Resistance. Mol. Cancer Res. MCR 2020, 18, 1004–1017. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.-T.; Yeh, K.-C.; Lee, C.-H.; Chen, Y.-Y.; Ho, P.-J.; Chang, K.-Y.; Chen, C.-H.; Lai, Y.-K.; Chen, C.-T. Molecular Profiling of Afatinib-Resistant Non-Small Cell Lung Cancer Cells in Vivo Derived from Mice. Pharmacol. Res. 2020, 161, 105183. [Google Scholar] [CrossRef]
- Sahm, F.; Oezen, I.; Opitz, C.A.; Radlwimmer, B.; von Deimling, A.; Ahrendt, T.; Adams, S.; Bode, H.B.; Guillemin, G.J.; Wick, W.; et al. The Endogenous Tryptophan Metabolite and NAD+ Precursor Quinolinic Acid Confers Resistance of Gliomas to Oxidative Stress. Cancer Res. 2013, 73, 3225–3234. [Google Scholar] [CrossRef] [Green Version]
- Bi, T.-Q.; Che, X.-M.; Liao, X.-H.; Zhang, D.-J.; Long, H.-L.; Li, H.-J.; Zhao, W. Overexpression of Nampt in Gastric Cancer and Chemopotentiating Effects of the Nampt Inhibitor FK866 in Combination with Fluorouracil. Oncol. Rep. 2011, 26, 1251–1257. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Hasan, M.K.; Alvarado, E.; Yuan, H.; Wu, H.; Chen, W.Y. NAMPT Overexpression in Prostate Cancer and Its Contribution to Tumor Cell Survival and Stress Response. Oncogene 2011, 30, 907–921. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Guo, M.; Zhang, L.; Xu, T.; Wang, L.; Xu, G. Biomarker Triplet NAMPT/VEGF/HER2 as a de Novo Detection Panel for the Diagnosis and Prognosis of Human Breast Cancer. Oncol. Rep. 2016, 35, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-J.; Bi, T.-Q.; Qin, C.-X.; Yang, X.-Q.; Pang, K. Expression of NAMPT Is Associated with Breast Invasive Ductal Carcinoma Development and Prognosis. Oncol. Lett. 2018, 15, 6648–6654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Lei, J.; Mao, L.; Wang, Q.; Xu, F.; Ran, T.; Zhou, Z.; He, S. NAMPT and NAPRT, Key Enzymes in NAD Salvage Synthesis Pathway, Are of Negative Prognostic Value in Colorectal Cancer. Front. Oncol. 2019, 9, 736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, C.; Qi, L.; Li, X.; Wang, J.; Yu, J.; Zhou, B.; Guo, C.; Chen, J.; Zheng, S. Targeting the NAD+ Salvage Pathway Suppresses APC Mutation-Driven Colorectal Cancer Growth and Wnt/β-Catenin Signaling via Increasing Axin Level. Cell Commun. Signal. CCS 2020, 18, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Beijnum, J.R.; Moerkerk, P.T.M.; Gerbers, A.J.; De Bruïne, A.P.; Arends, J.-W.; Hoogenboom, H.R.; Hufton, S.E. Target Validation for Genomics Using Peptide-Specific Phage Antibodies: A Study of Five Gene Products Overexpressed in Colorectal Cancer. Int. J. Cancer 2002, 101, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jiménez-García, M.P.; Peinado-Serrano, J.; Carnero, A. NAMPT Overexpression Induces Cancer Stemness and Defines a Novel Tumor Signature for Glioma Prognosis. Oncotarget 2017, 8, 99514–99530. [Google Scholar] [CrossRef] [Green Version]
- Shackelford, R.E.; Bui, M.M.; Coppola, D.; Hakam, A. Over-Expression of Nicotinamide Phosphoribosyltransferase in Ovarian Cancers. Int. J. Clin. Exp. Pathol. 2010, 3, 522–527. [Google Scholar]
- Maldi, E.; Travelli, C.; Caldarelli, A.; Agazzone, N.; Cintura, S.; Galli, U.; Scatolini, M.; Ostano, P.; Miglino, B.; Chiorino, G.; et al. Nicotinamide Phosphoribosyltransferase (NAMPT) Is over-Expressed in Melanoma Lesions. Pigment Cell Melanoma Res. 2013, 26, 144–146. [Google Scholar] [CrossRef]
- Sawicka-Gutaj, N.; Waligórska-Stachura, J.; Andrusiewicz, M.; Biczysko, M.; Sowiński, J.; Skrobisz, J.; Ruchała, M. Nicotinamide Phosphorybosiltransferase Overexpression in Thyroid Malignancies and Its Correlation with Tumor Stage and with Survivin/Survivin DEx3 Expression. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 7859–7863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vora, M.; Ansari, J.; Shanti, R.M.; Veillon, D.; Cotelingam, J.; Coppola, D.; Shackelford, R.E. Increased Nicotinamide Phosphoribosyltransferase in Rhabdomyosarcomas and Leiomyosarcomas Compared to Skeletal and Smooth Muscle Tissue. Anticancer Res. 2016, 36, 503–507. [Google Scholar]
- Olesen, U.H.; Hastrup, N.; Sehested, M. Expression Patterns of Nicotinamide Phosphoribosyltransferase and Nicotinic Acid Phosphoribosyltransferase in Human Malignant Lymphomas. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2011, 119, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Messana, V.G.; Moiso, E.; Vitale, N.; Arruga, F.; Brandimarte, L.; Gaudino, F.; Pellegrino, E.; Vaisitti, T.; Riganti, C.; et al. NAMPT Over-Expression Recapitulates the BRAF Inhibitor Resistant Phenotype Plasticity in Melanoma. Cancers 2020, 12, 3855. [Google Scholar] [CrossRef]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jiménez-García, M.P.; Muñoz-Galvan, S.; Carnero, A. NAMPT Is a Potent Oncogene in Colon Cancer Progression That Modulates Cancer Stem Cell Properties and Resistance to Therapy through Sirt1 and PARP. Clin. Cancer Res. 2018, 24, 1202–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samal, B.; Sun, Y.; Stearns, G.; Xie, C.; Suggs, S.; McNiece, I. Cloning and Characterization of the CDNA Encoding a Novel Human Pre-B-Cell Colony-Enhancing Factor. Mol. Cell. Biol. 1994, 14, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Liberale, L.; Bonaventura, A.; Vecchiè, A.; Casula, M.; Cea, M.; Monacelli, F.; Caffa, I.; Bruzzone, S.; Montecucco, F.; et al. Regulation and Function of Extracellular Nicotinamide Phosphoribosyltransferase/Visfatin. Compr. Physiol. 2017, 7, 603–621. [Google Scholar] [CrossRef]
- Soncini, D.; Caffa, I.; Zoppoli, G.; Cea, M.; Cagnetta, A.; Passalacqua, M.; Mastracci, L.; Boero, S.; Montecucco, F.; Sociali, G.; et al. Nicotinamide Phosphoribosyltransferase Promotes Epithelial-to-Mesenchymal Transition as a Soluble Factor Independent of Its Enzymatic Activity. J. Biol. Chem. 2014, 289, 34189–34204. [Google Scholar] [CrossRef] [Green Version]
- Grolla, A.A.; Torretta, S.; Gnemmi, I.; Amoruso, A.; Orsomando, G.; Gatti, M.; Caldarelli, A.; Lim, D.; Penengo, L.; Brunelleschi, S.; et al. Nicotinamide Phosphoribosyltransferase (NAMPT/PBEF/Visfatin) Is a Tumoural Cytokine Released from Melanoma. Pigment Cell Melanoma Res. 2015, 28, 718–729. [Google Scholar] [CrossRef]
- Audrito, V.; Managò, A.; Zamporlini, F.; Rulli, E.; Gaudino, F.; Madonna, G.; D’Atri, S.; Antonini Cappellini, G.C.; Ascierto, P.A.; Massi, D.; et al. Extracellular Nicotinamide Phosphoribosyltransferase (ENAMPT) Is a Novel Marker for Patients with BRAF-Mutated Metastatic Melanoma. Oncotarget 2018, 9, 18997–19005. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.L.; Sun, X.; Casanova, N.; Garcia, A.N.; Oita, R.; Algotar, A.M.; Camp, S.M.; Hernon, V.R.; Gregory, T.; Cress, A.E.; et al. Role of Secreted Extracellular Nicotinamide Phosphoribosyltransferase (ENAMPT) in Prostate Cancer Progression: Novel Biomarker and Therapeutic Target. EBioMedicine 2020, 61, 103059. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an Adipocytokine with Proinflammatory and Immunomodulating Properties. J. Immunol. Baltim. Md 1950 2007, 178, 1748–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, G.; Clemente, N.; Zito, A.; Bracci, C.; Colombo, F.S.; Sangaletti, S.; Jachetti, E.; Ribaldone, D.G.; Caviglia, G.P.; Pastorelli, L.; et al. Neutralization of Extracellular NAMPT (Nicotinamide Phosphoribosyltransferase) Ameliorates Experimental Murine Colitis. J. Mol. Med. Berl. Ger. 2020, 98, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Managò, A.; Audrito, V.; Mazzola, F.; Sorci, L.; Gaudino, F.; Gizzi, K.; Vitale, N.; Incarnato, D.; Minazzato, G.; Ianniello, A.; et al. Extracellular Nicotinate Phosphoribosyltransferase Binds Toll like Receptor 4 and Mediates Inflammation. Nat. Commun. 2019, 10, 4116. [Google Scholar] [CrossRef]
- Audrito, V.; Messana, V.G.; Deaglio, S. NAMPT and NAPRT: Two Metabolic Enzymes With Key Roles in Inflammation. Front. Oncol. 2020, 10, 358. [Google Scholar] [CrossRef] [Green Version]
- Dakroub, A.; Nasser, S.A.; Younis, N.; Bhagani, H.; Al-Dhaheri, Y.; Pintus, G.; Eid, A.A.; El-Yazbi, A.F.; Eid, A.H. Visfatin: A Possible Role in Cardiovasculo-Metabolic Disorders. Cells 2020, 9, 2444. [Google Scholar] [CrossRef]
- Sampath, D.; Zabka, T.S.; Misner, D.L.; O’Brien, T.; Dragovich, P.S. Inhibition of Nicotinamide Phosphoribosyltransferase (NAMPT) as a Therapeutic Strategy in Cancer. Pharmacol. Ther. 2015, 151, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Galli, U.; Colombo, G.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Grolla, A.A. Recent Advances in NAMPT Inhibitors: A Novel Immunotherapic Strategy. Front. Pharmacol. 2020, 11, 656. [Google Scholar] [CrossRef] [PubMed]
- Hasmann, M.; Schemainda, I. FK866, a Highly Specific Noncompetitive Inhibitor of Nicotinamide Phosphoribosyltransferase, Represents a Novel Mechanism for Induction of Tumor Cell Apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar] [PubMed]
- Wosikowski, K.; Mattern, K.; Schemainda, I.; Hasmann, M.; Rattel, B.; Löser, R. WK175, a Novel Antitumor Agent, Decreases the Intracellular Nicotinamide Adenine Dinucleotide Concentration and Induces the Apoptotic Cascade in Human Leukemia Cells. Cancer Res. 2002, 62, 1057–1062. [Google Scholar]
- Kim, M.-K.; Lee, J.H.; Kim, H.; Park, S.J.; Kim, S.H.; Kang, G.B.; Lee, Y.S.; Kim, J.B.; Kim, K.K.; Suh, S.W.; et al. Crystal Structure of Visfatin/Pre-B Cell Colony-Enhancing Factor 1/Nicotinamide Phosphoribosyltransferase, Free and in Complex with the Anti-Cancer Agent FK-866. J. Mol. Biol. 2006, 362, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.B.; Bae, M.-H.; Kim, M.-K.; Im, I.; Kim, Y.-C.; Eom, S.H. Crystal Structure of Rattus Norvegicus Visfatin/PBEF/Nampt in Complex with an FK866-Based Inhibitor. Mol. Cells 2009, 27, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.A.; Tao, X.; Tong, L. Molecular Basis for the Inhibition of Human NMPRTase, a Novel Target for Anticancer Agents. Nat. Struct. Mol. Biol. 2006, 13, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Hjarnaa, P.J.; Jonsson, E.; Latini, S.; Dhar, S.; Larsson, R.; Bramm, E.; Skov, T.; Binderup, L. CHS 828, a Novel Pyridyl Cyanoguanidine with Potent Antitumor Activity in Vitro and in Vivo. Cancer Res. 1999, 59, 5751–5757. [Google Scholar]
- Schou, C.; Ottosen, E.R.; Petersen, H.J.; Björkling, F.; Latini, S.; Hjarnaa, P.V.; Bramm, E.; Binderup, L. Novel Cyanoguanidines with Potent Oral Antitumour Activity. Bioorg. Med. Chem. Lett. 1997, 7, 3095–3100. [Google Scholar] [CrossRef]
- Hovstadius, P.; Larsson, R.; Jonsson, E.; Skov, T.; Kissmeyer, A.-M.; Krasilnikoff, K.; Bergh, J.; Karlsson, M.O.; Lönnebo, A.; Ahlgren, J. A Phase I Study of CHS 828 in Patients with Solid Tumor Malignancy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 2843–2850. [Google Scholar]
- Olesen, U.H.; Christensen, M.K.; Björkling, F.; Jäättelä, M.; Jensen, P.B.; Sehested, M.; Nielsen, S.J. Anticancer Agent CHS-828 Inhibits Cellular Synthesis of NAD. Biochem. Biophys. Res. Commun. 2008, 367, 799–804. [Google Scholar] [CrossRef]
- Binderup, E.; Björkling, F.; Hjarnaa, P.V.; Latini, S.; Baltzer, B.; Carlsen, M.; Binderup, L. EB1627: A Soluble Prodrug of the Potent Anticancer Cyanoguanidine CHS828. Bioorg. Med. Chem. Lett. 2005, 15, 2491–2494. [Google Scholar] [CrossRef]
- Korotchkina, L.; Kazyulkin, D.; Komarov, P.G.; Polinsky, A.; Andrianova, E.L.; Joshi, S.; Gupta, M.; Vujcic, S.; Kononov, E.; Toshkov, I.; et al. OT-82, a Novel Anticancer Drug Candidate That Targets the Strong Dependence of Hematological Malignancies on NAD Biosynthesis. Leukemia 2020, 34, 1828–1839. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Bauer, P.; Baumeister, T.; Buckmelter, A.J.; Caligiuri, M.; Clodfelter, K.H.; Han, B.; Ho, Y.-C.; Kley, N.; Lin, J.; et al. Structure-Based Discovery of Novel Amide-Containing Nicotinamide Phosphoribosyltransferase (Nampt) Inhibitors. J. Med. Chem. 2013, 56, 6413–6433. [Google Scholar] [CrossRef]
- Zheng, X.; Bair, K.W.; Bauer, P.; Baumeister, T.; Bowman, K.K.; Buckmelter, A.J.; Caligiuri, M.; Clodfelter, K.H.; Feng, Y.; Han, B.; et al. Identification of Amides Derived from 1H-Pyrazolo[3,4-b]Pyridine-5-Carboxylic Acid as Potent Inhibitors of Human Nicotinamide Phosphoribosyltransferase (NAMPT). Bioorg. Med. Chem. Lett. 2013, 23, 5488–5497. [Google Scholar] [CrossRef]
- Travelli, C.; Aprile, S.; Rahimian, R.; Grolla, A.A.; Rogati, F.; Bertolotti, M.; Malagnino, F.; di Paola, R.; Impellizzeri, D.; Fusco, R.; et al. Identification of Novel Triazole-Based Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors Endowed with Antiproliferative and Antiinflammatory Activity. J. Med. Chem. 2017, 60, 1768–1792. [Google Scholar] [CrossRef]
- Wilsbacher, J.L.; Cheng, M.; Cheng, D.; Trammell, S.A.J.; Shi, Y.; Guo, J.; Koeniger, S.L.; Kovar, P.J.; He, Y.; Selvaraju, S.; et al. Discovery and Characterization of Novel Nonsubstrate and Substrate NAMPT Inhibitors. Mol. Cancer Ther. 2017, 16, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Matheny, C.J.; Wei, M.C.; Bassik, M.C.; Donnelly, A.J.; Kampmann, M.; Iwasaki, M.; Piloto, O.; Solow-Cordero, D.E.; Bouley, D.M.; Rau, R.; et al. Next-Generation NAMPT Inhibitors Identified by Sequential High-Throughput Phenotypic Chemical and Functional Genomic Screens. Chem. Biol. 2013, 20, 1352–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Green, C.F.; Hui, Y.-H.; Prieto, L.; Shepard, R.; Dong, S.; Wang, T.; Tan, B.; Gong, X.; Kays, L.; et al. Discovery of a Highly Selective NAMPT Inhibitor That Demonstrates Robust Efficacy and Improved Retinal Toxicity with Nicotinic Acid Coadministration. Mol. Cancer Ther. 2017, 16, 2677–2688. [Google Scholar] [CrossRef] [Green Version]
- Neumann, C.S.; Olivas, K.C.; Anderson, M.E.; Cochran, J.H.; Jin, S.; Li, F.; Loftus, L.V.; Meyer, D.W.; Neale, J.; Nix, J.C.; et al. Targeted Delivery of Cytotoxic NAMPT Inhibitors Using Antibody–Drug Conjugates. Mol. Cancer Ther. 2018, 17, 2633–2642. [Google Scholar] [CrossRef] [Green Version]
- Karpov, A.S.; Abrams, T.; Clark, S.; Raikar, A.; D’Alessio, J.A.; Dillon, M.P.; Gesner, T.G.; Jones, D.; Lacaud, M.; Mallet, W.; et al. Nicotinamide Phosphoribosyltransferase Inhibitor as a Novel Payload for Antibody–Drug Conjugates. ACS Med. Chem. Lett. 2018, 9, 838–842. [Google Scholar] [CrossRef]
- Abu Aboud, O.; Chen, C.-H.; Senapedis, W.; Baloglu, E.; Argueta, C.; Weiss, R.H. Dual and Specific Inhibition of NAMPT and PAK4 By KPT-9274 Decreases Kidney Cancer Growth. Mol. Cancer Ther. 2016, 15, 2119–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.-T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the Warburg Effect in Renal Cell Carcinoma by Chemical Synthetic Lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, D.J.; Ito, D.; Rees, M.G.; Seashore-Ludlow, B.; Puyang, X.; Ramos, A.H.; Cheah, J.H.; Clemons, P.A.; Warmuth, M.; Zhu, P.; et al. NAMPT Is the Cellular Target of STF-31-Like Small-Molecule Probes. ACS Chem. Biol. 2014, 9, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, A.J.; Horwitz, S.M. Targeting Histone Deacetylases in T-Cell Lymphoma. Leuk. Lymphoma 2017, 58, 1306–1319. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, L.; Huang, Y.; Chen, S.; Wu, S.; Dong, G.; Sheng, C. Nicotinamide Phosphoribosyltransferase (NAMPT) Is a New Target of Antitumor Agent Chidamide. ACS Med. Chem. Lett. 2019, 11, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Chen, W.; Wang, X.; Yang, X.; Xu, T.; Wang, P.; Zhang, W.; Rao, Y.; Miao, C.; Sheng, C. Small Molecule Inhibitors Simultaneously Targeting Cancer Metabolism and Epigenetics: Discovery of Novel Nicotinamide Phosphoribosyltransferase (NAMPT) and Histone Deacetylase (HDAC) Dual Inhibitors. J. Med. Chem. 2017, 60, 7965–7983. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dong, G.; Wu, Y.; Zhang, W.; Miao, C.; Sheng, C. Dual NAMPT/HDAC Inhibitors as a New Strategy for Multitargeting Antitumor Drug Discovery. ACS Med. Chem. Lett. 2018, 9, 34–38. [Google Scholar] [CrossRef] [PubMed]
- LeWitt, P.A. The Neurotoxicity of the Rat Poison Vacor. A Clinical Study of 12 Cases. N. Engl. J. Med. 1980, 302, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Buonvicino, D.; Mazzola, F.; Zamporlini, F.; Resta, F.; Ranieri, G.; Camaioni, E.; Muzzi, M.; Zecchi, R.; Pieraccini, G.; Dölle, C.; et al. Identification of the Nicotinamide Salvage Pathway as a New Toxification Route for Antimetabolites. Cell Chem. Biol. 2018, 25, 471–482.e7. [Google Scholar] [CrossRef] [Green Version]
- Gaut, Z.N.; Solomon, H.M. Inhibition of Nicotinate Phosphoribosyltransferase in Human Platelet Lysate by Nicotinic Acid Analogs. Biochem. Pharmacol. 1971, 20, 2903–2906. [Google Scholar] [CrossRef]
- Gaut, Z.N.; Solomon, H.M. Uptake and Metabolism of Nicotinic Acid by Human Blood Platelets. Effects of Structure Analogs and Metabolic Inhibitors. Biochim. Biophys. Acta 1970, 201, 316–322. [Google Scholar] [CrossRef]
- Gaut, Z.N.; Solomon, H.M. Inhibition of Nicotinate Phosphoribosyl Transferase by Nonsteroidal Anti-Inflammatory Drugs: A Possible Mechanism of Action. J. Pharm. Sci. 1971, 60, 1887–1888. [Google Scholar] [CrossRef]
- Galassi, L.; Di Stefano, M.; Brunetti, L.; Orsomando, G.; Amici, A.; Ruggieri, S.; Magni, G. Characterization of Human Nicotinate Phosphoribosyltransferase: Kinetic Studies, Structure Prediction and Functional Analysis by Site-Directed Mutagenesis. Biochimie 2012, 94, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Velu, S.E.; Cristofoli, W.A.; Garcia, G.J.; Brouillette, C.G.; Pierson, M.C.; Luan, C.-H.; DeLucas, L.J.; Brouillette, W.J. Tethered Dimers as NAD Synthetase Inhibitors with Antibacterial Activity. J. Med. Chem. 2003, 46, 3371–3381. [Google Scholar] [CrossRef] [PubMed]
- Velu, S.E.; Luan, C.-H.; Delucas, L.J.; Brouillette, C.G.; Brouillette, W.J. Tethered Dimer Inhibitors of NAD Synthetase: Parallel Synthesis of an Aryl-Substituted SAR Library. J. Comb. Chem. 2005, 7, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Moro, W.B.; Yang, Z.; Kane, T.A.; Brouillette, C.G.; Brouillette, W.J. Virtual Screening to Identify Lead Inhibitors for Bacterial NAD Synthetase (NADs). Bioorg. Med. Chem. Lett. 2009, 19, 2001–2005. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ahn, Y.-M.; Lentscher, A.G.; Lister, J.S.; Brothers, R.C.; Kneen, M.M.; Gerratana, B.; Boshoff, H.I.; Dowd, C.S. Design, Synthesis, and Evaluation of Substituted Nicotinamide Adenine Dinucleotide (NAD+) Synthetase Inhibitors as Potential Antitubercular Agents. Bioorg. Med. Chem. Lett. 2017, 27, 4426–4430. [Google Scholar] [CrossRef]
- Gehrke, I.; Bouchard, E.D.J.; Beiggi, S.; Poeppl, A.G.; Johnston, J.B.; Gibson, S.B.; Banerji, V. On-Target Effect of FK866, a Nicotinamide Phosphoribosyl Transferase Inhibitor, by Apoptosis-Mediated Death in Chronic Lymphocytic Leukemia Cells. Clin. Cancer Res. 2014, 20, 4861–4872. [Google Scholar] [CrossRef] [Green Version]
- Cloux, A.-J.; Aubry, D.; Heulot, M.; Widmann, C.; ElMokh, O.; Piacente, F.; Cea, M.; Nencioni, A.; Bellotti, A.; Bouzourène, K.; et al. Reactive Oxygen/Nitrogen Species Contribute Substantially to the Antileukemia Effect of APO866, a NAD Lowering Agent. Oncotarget 2019, 10, 6723–6738. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, T.; Penke, M.; Petzold-Quinque, S.; Schuster, S.; Richter, S.; Kiess, W.; Garten, A. Inhibition of NAMPT Sensitizes MOLT4 Leukemia Cells for Etoposide Treatment through the SIRT2-P53 Pathway. Leuk. Res. 2018, 69, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Travelli, C.; Drago, V.; Maldi, E.; Kaludercic, N.; Galli, U.; Boldorini, R.; Lisa, F.D.; Tron, G.C.; Canonico, P.L.; Genazzani, A.A. Reciprocal Potentiation of the Antitumoral Activities of FK866, an Inhibitor of Nicotinamide Phosphoribosyltransferase, and Etoposide or Cisplatin in Neuroblastoma Cells. J. Pharmacol. Exp. Ther. 2011, 338, 829–840. [Google Scholar] [CrossRef]
- Nacarelli, T.; Fukumoto, T.; Zundell, J.A.; Fatkhutdinov, N.; Jean, S.; Cadungog, M.G.; Borowsky, M.E.; Zhang, R. NAMPT Inhibition Suppresses Cancer Stem-like Cells Associated with Therapy-Induced Senescence in Ovarian Cancer. Cancer Res. 2020, 80, 890–900. [Google Scholar] [CrossRef] [Green Version]
- Cagnetta, A.; Caffa, I.; Acharya, C.; Soncini, D.; Acharya, P.; Adamia, S.; Pierri, I.; Bergamaschi, M.; Garuti, A.; Fraternali, G.; et al. APO866 Increases Antitumor Activity of Cyclosporin-A by Inducing Mitochondrial and Endoplasmic Reticulum Stress in Leukemia Cells. Clin. Cancer Res. 2015, 21, 3934–3945. [Google Scholar] [CrossRef] [Green Version]
- Cagnetta, A.; Cea, M.; Calimeri, T.; Acharya, C.; Fulciniti, M.; Tai, Y.-T.; Hideshima, T.; Chauhan, D.; Zhong, M.Y.; Patrone, F.; et al. Intracellular NAD+ Depletion Enhances Bortezomib-Induced Anti-Myeloma Activity. Blood 2013, 122, 1243–1255. [Google Scholar] [CrossRef] [Green Version]
- Cea, M.; Cagnetta, A.; Acharya, C.; Acharya, P.; Tai, Y.-T.; Yang, G.; Lovera, D.; Soncini, D.; Miglino, M.; Fraternali-Orcioni, G.; et al. Dual NAMPT and BTK Targeting Leads to Synergistic Killing of Waldenstrom’s Macroglobulinemia Cells Regardless of MYD88 and CXCR4 Somatic Mutations Status. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 6099–6109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, M.; Gravel, M.; Bramoullé, A.; Bridon, G.; Avizonis, D.; Shore, G.C.; Roulston, A. Synergy between the NAMPT Inhibitor GMX1777(8) and Pemetrexed in Non-Small Cell Lung Cancer Cells Is Mediated by PARP Activation and Enhanced NAD Consumption. Cancer Res. 2014, 74, 5948–5954. [Google Scholar] [CrossRef] [Green Version]
- Barraud, M.; Garnier, J.; Loncle, C.; Gayet, O.; Lequeue, C.; Vasseur, S.; Bian, B.; Duconseil, P.; Gilabert, M.; Bigonnet, M.; et al. A Pancreatic Ductal Adenocarcinoma Subpopulation Is Sensitive to FK866, an Inhibitor of NAMPT. Oncotarget 2016, 7, 53783–53796. [Google Scholar] [CrossRef]
- Espindola-Netto, J.M.; Chini, C.C.S.; Tarragó, M.; Wang, E.; Dutta, S.; Pal, K.; Mukhopadhyay, D.; Sola-Penna, M.; Chini, E.N. Preclinical Efficacy of the Novel Competitive NAMPT Inhibitor STF-118804 in Pancreatic Cancer. Oncotarget 2017, 8, 85054–85067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cea, M.; Soncini, D.; Fruscione, F.; Raffaghello, L.; Garuti, A.; Emionite, L.; Moran, E.; Magnone, M.; Zoppoli, G.; Reverberi, D.; et al. Synergistic Interactions between HDAC and Sirtuin Inhibitors in Human Leukemia Cells. PLoS ONE 2011, 6, e22739. [Google Scholar] [CrossRef]
- Elf, A.-K.; Bernhardt, P.; Hofving, T.; Arvidsson, Y.; Forssell-Aronsson, E.; Wängberg, B.; Nilsson, O.; Johanson, V. NAMPT Inhibitor GMX1778 Enhances the Efficacy of 177Lu-DOTATATE Treatment of Neuroendocrine Tumors. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2017, 58, 288–292. [Google Scholar] [CrossRef] [Green Version]
- Nahimana, A.; Aubry, D.; Breton, C.S.; Majjigapu, S.R.; Sordat, B.; Vogel, P.; Duchosal, M.A. The Anti-Lymphoma Activity of APO866, an Inhibitor of Nicotinamide Adenine Dinucleotide Biosynthesis, Is Potentialized When Used in Combination with Anti-CD20 Antibody. Leuk. Lymphoma 2014, 55, 2141–2150. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Yan, P.-F.; Zhao, H.; Zhang, F.-C.; Zhao, W.-H.; Feng, M. Inhibitor of Nicotinamide Phosphoribosyltransferase Sensitizes Glioblastoma Cells to Temozolomide via Activating ROS/JNK Signaling Pathway. BioMed Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Higuchi, F.; Miller, J.J.; Koerner, M.V.A.; Lelic, N.; Shankar, G.M.; Tanaka, S.; Fisher, D.E.; Batchelor, T.T.; Iafrate, A.J.; et al. The Alkylating Chemotherapeutic Temozolomide Induces Metabolic Stress in IDH1 -Mutant Cancers and Potentiates NAD+ Depletion–Mediated Cytotoxicity. Cancer Res. 2017, 77, 4102–4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajrami, I.; Kigozi, A.; Van Weverwijk, A.; Brough, R.; Frankum, J.; Lord, C.J.; Ashworth, A. Synthetic Lethality of PARP and NAMPT Inhibition in Triple-Negative Breast Cancer Cells. EMBO Mol. Med. 2012, 4, 1087–1096. [Google Scholar] [CrossRef]
- Heske, C.M.; Davis, M.I.; Baumgart, J.T.; Wilson, K.; Gormally, M.V.; Chen, L.; Zhang, X.; Ceribelli, M.; Duveau, D.Y.; Guha, R.; et al. Matrix Screen Identifies Synergistic Combination of PARP Inhibitors and Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors in Ewing Sarcoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 7301–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, A.E.; Yeung, C.; Issaq, S.H.; Collins, V.J.; Gouzoulis, M.; Zhang, Y.; Ji, J.; Mendoza, A.; Heske, C.M. Inhibition of Nicotinamide Phosphoribosyltransferase (NAMPT) with OT-82 Induces DNA Damage, Cell Death, and Suppression of Tumor Growth in Preclinical Models of Ewing Sarcoma. Oncogenesis 2020, 9, 80. [Google Scholar] [CrossRef] [PubMed]
- Somers, K.; Evans, K.; Cheung, L.; Karsa, M.; Pritchard, T.; Kosciolek, A.; Bongers, A.; El-Ayoubi, A.; Forgham, H.; Middlemiss, S.; et al. Effective Targeting of NAMPT in Patient-Derived Xenograft Models of High-Risk Pediatric Acute Lymphoblastic Leukemia. Leukemia 2020, 34, 1524–1539. [Google Scholar] [CrossRef]
- Li, M.; Kirtane, A.R.; Kiyokawa, J.; Nagashima, H.; Lopes, A.; Tirmizi, Z.A.; Lee, C.K.; Traverso, G.; Cahill, D.P.; Wakimoto, H. Local Targeting of NAD+ Salvage Pathway Alters the Immune Tumor Microenvironment and Enhances Checkpoint Immunotherapy in Glioblastoma. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Travelli, C.; Consonni, F.M.; Sangaletti, S.; Storto, M.; Morlacchi, S.; Grolla, A.A.; Galli, U.; Tron, G.C.; Portararo, P.; Rimassa, L.; et al. Nicotinamide Phosphoribosyltransferase Acts as a Metabolic Gate for Mobilization of Myeloid-Derived Suppressor Cells. Cancer Res. 2019, 79, 1938–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, J.F.; Abu Aboud, O.; McLaughlin, B.; Anderson, K.L.; Modiano, J.F.; Kim, K.; Jen, K.-Y.; Senapedis, W.; Chang, H.; Landesman, Y.; et al. Anti-Cancer Activity of PAK4/NAMPT Inhibitor and Programmed Cell Death Protein-1 Antibody in Kidney Cancer. Kidney360 2020, 1, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Abril-Rodriguez, G.; Torrejon, D.Y.; Liu, W.; Zaretsky, J.M.; Nowicki, T.S.; Tsoi, J.; Puig-Saus, C.; Baselga-Carretero, I.; Medina, E.; Quist, M.J.; et al. PAK4 Inhibition Improves PD-1 Blockade Immunotherapy. Nat. Cancer 2020, 1, 46–58. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Lopez, M.A.; Linares, M.; Kumar, S.; Oliva, S.; Martinez-Lopez, J.; Xu, L.; Xu, Y.; Perini, T.; Senapedis, W.; et al. Dual PAK4-NAMPT Inhibition Impacts Growth and Survival, and Increases Sensitivity to DNA-Damaging Agents in Waldenström Macroglobulinemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Mpilla, G.; Aboukameel, A.; Muqbil, I.; Kim, S.; Beydoun, R.; Philip, P.A.; Mohammad, R.M.; Kamgar, M.; Shidham, V.; Senapedis, W.; et al. PAK4-NAMPT Dual Inhibition as a Novel Strategy for Therapy Resistant Pancreatic Neuroendocrine Tumors. Cancers 2019, 11, 1902. [Google Scholar] [CrossRef] [Green Version]
- Aboukameel, A.; Muqbil, I.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Shacham, S.; Kauffman, M.; Philip, P.A.; Mohammad, R.M.; Azmi, A.S. Novel P21-Activated Kinase 4 (PAK4) Allosteric Modulators Overcome Drug Resistance and Stemness in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 76–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoppoli, G.; Cea, M.; Soncini, D.; Fruscione, F.; Rudner, J.; Moran, E.; Caffa, I.; Bedognetti, D.; Motta, G.; Ghio, R.; et al. Potent Synergistic Interaction between the Nampt Inhibitor APO866 and the Apoptosis Activator TRAIL in Human Leukemia Cells. Exp. Hematol. 2010, 38, 979–988. [Google Scholar] [CrossRef]
- Yang, H.-J.; Yen, M.-C.; Lin, C.-C.; Lin, C.-M.; Chen, Y.-L.; Weng, T.-Y.; Huang, T.-T.; Wu, C.-L.; Lai, M.-D. A Combination of the Metabolic Enzyme Inhibitor APO866 and the Immune Adjuvant L-1-Methyl Tryptophan Induces Additive Antitumor Activity. Exp. Biol. Med. 2010, 235, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Moore, Z.; Chakrabarti, G.; Luo, X.; Ali, A.; Hu, Z.; Fattah, F.J.; Vemireddy, R.; DeBerardinis, R.J.; Brekken, R.A.; Boothman, D.A. NAMPT Inhibition Sensitizes Pancreatic Adenocarcinoma Cells to Tumor-Selective, PAR-Independent Metabolic Catastrophe and Cell Death Induced by β-Lapachone. Cell Death Dis. 2015, 6, e1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.-Y.; Li, Q.-R.; Cheng, X.-F.; Wang, G.-J.; Hao, H.-P. NAMPT Inhibition Synergizes with NQO1-Targeting Agents in Inducing Apoptotic Cell Death in Non-Small Cell Lung Cancer Cells. Chin. J. Nat. Med. 2016, 14, 582–589. [Google Scholar] [CrossRef]
- Breton, C.S.; Aubry, D.; Ginet, V.; Puyal, J.; Heulot, M.; Widmann, C.; Duchosal, M.A.; Nahimana, A. Combinative Effects of β-Lapachone and APO866 on Pancreatic Cancer Cell Death through Reactive Oxygen Species Production and PARP-1 Activation. Biochimie 2015, 116, 141–153. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of Lactate Dehydrogenase A Induces Oxidative Stress and Inhibits Tumor Progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pogrebniak, A.; Schemainda, I.; Azzam, K.; Pelka-Fleischer, R.; Nüssler, V.; Hasmann, M. Chemopotentiating Effects of a Novel NAD Biosynthesis Inhibitor, FK866, in Combination with Antineoplastic Agents. Eur. J. Med. Res. 2006, 11, 313–321. [Google Scholar]
- Mitchell, S.R.; Larkin, K.; Grieselhuber, N.R.; Lai, T.-H.; Cannon, M.; Orwick, S.; Sharma, P.; Asemelash, Y.; Zhang, P.; Goettl, V.M.; et al. Selective Targeting of NAMPT by KPT-9274 in Acute Myeloid Leukemia. Blood Adv. 2019, 3, 242–255. [Google Scholar] [CrossRef] [Green Version]
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.-T.; Hideshima, T.; Chauhan, D.; Roccaro, A.; Sacco, A.; Calimeri, T.; Cottini, F.; et al. Targeting NAD+ Salvage Pathway Induces Autophagy in Multiple Myeloma Cells via MTORC1 and Extracellular Signal-Regulated Kinase (ERK1/2) Inhibition. Blood 2012, 120, 3519–3529. [Google Scholar] [CrossRef] [Green Version]
- Cea, M.; Cagnetta, A.; Patrone, F.; Nencioni, A.; Gobbi, M.; Anderson, K.C. Intracellular NAD+ Depletion Induces Autophagic Death in Multiple Myeloma Cells. Autophagy 2013, 9, 410–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billington, R.A.; Genazzani, A.A.; Travelli, C.; Condorelli, F. NAD Depletion by FK866 Induces Autophagy. Autophagy 2008, 4, 385–387. [Google Scholar] [CrossRef] [Green Version]
- Nahimana, A.; Attinger, A.; Aubry, D.; Greaney, P.; Ireson, C.; Thougaard, A.V.; Tjørnelund, J.; Dawson, K.M.; Dupuis, M.; Duchosal, M.A. The NAD Biosynthesis Inhibitor APO866 Has Potent Antitumor Activity against Hematologic Malignancies. Blood 2009, 113, 3276–3286. [Google Scholar] [CrossRef]
- Kozako, T.; Aikawa, A.; Ohsugi, T.; Uchida, Y.; Kato, N.; Sato, K.; Ishitsuka, K.; Yoshimitsu, M.; Honda, S. High Expression of NAMPT in Adult T-Cell Leukemia/Lymphoma and Anti-Tumor Activity of a NAMPT Inhibitor. Eur. J. Pharmacol. 2019, 865, 172738. [Google Scholar] [CrossRef]
- Del Nagro, C.; Xiao, Y.; Rangell, L.; Reichelt, M.; O’Brien, T. Depletion of the Central Metabolite NAD Leads to Oncosis-Mediated Cell Death. J. Biol. Chem. 2014, 289, 35182–35192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Sauve, A.A. NAD+ Metabolism: Bioenergetics, Signaling and Manipulation for Therapy. Biochim. Biophys. Acta 2016, 1864, 1787–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Du, W.; Wu, M. Regulation of the Pentose Phosphate Pathway in Cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Hay, N. The Pentose Phosphate Pathway and Cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Nagaya, M.; Hara, H.; Kamiya, T.; Adachi, T. Inhibition of NAMPT Markedly Enhances Plasma-Activated Medium-Induced Cell Death in Human Breast Cancer MDA-MB-231 Cells. Arch. Biochem. Biophys. 2019, 676, 108155. [Google Scholar] [CrossRef]
- Cerna, D.; Li, H.; Flaherty, S.; Takebe, N.; Coleman, C.N.; Yoo, S.S. Inhibition of Nicotinamide Phosphoribosyltransferase (NAMPT) Activity by Small Molecule GMX1778 Regulates Reactive Oxygen Species (ROS)-Mediated Cytotoxicity in a P53- and Nicotinic Acid Phosphoribosyltransferase1 (NAPRT1)-Dependent Manner. J. Biol. Chem. 2012, 287, 22408–22417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-J.; Oh, G.-S.; Shen, A.; Lee, S.-B.; Choe, S.-K.; Kwon, K.-B.; Lee, S.; Seo, K.-S.; Kwak, T.H.; Park, R.; et al. Augmentation of NAD+ by NQO1 Attenuates Cisplatin-Mediated Hearing Impairment. Cell Death Dis. 2014, 5, e1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvers, M.A.; Deja, S.; Singh, N.; Egnatchik, R.A.; Sudderth, J.; Luo, X.; Beg, M.S.; Burgess, S.C.; DeBerardinis, R.J.; Boothman, D.A.; et al. The NQO1 Bioactivatable Drug, β-Lapachone, Alters the Redox State of NQO1+ Pancreatic Cancer Cells, Causing Perturbation in Central Carbon Metabolism. J. Biol. Chem. 2017, 292, 18203–18216. [Google Scholar] [CrossRef] [Green Version]
- Li, L.S.; Bey, E.A.; Dong, Y.; Meng, J.; Patra, B.; Yan, J.; Xie, X.-J.; Brekken, R.A.; Barnett, C.C.; Bornmann, W.G.; et al. Modulating Endogenous NQO1 Levels Identifies Key Regulatory Mechanisms of Action of β-Lapachone for Pancreatic Cancer Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, T.P.; Björklund, M. NQO2 Is a Reactive Oxygen Species Generating Off-Target for Acetaminophen. Mol. Pharm. 2014, 11, 4395–4404. [Google Scholar] [CrossRef] [Green Version]
- Touat, M.; Sourisseau, T.; Dorvault, N.; Chabanon, R.M.; Garrido, M.; Morel, D.; Krastev, D.B.; Bigot, L.; Adam, J.; Frankum, J.R.; et al. DNA Repair Deficiency Sensitizes Lung Cancer Cells to NAD+ Biosynthesis Blockade. J. Clin. Investig. 2018, 128, 1671–1687. [Google Scholar] [CrossRef]
- Zerp, S.F.; Vens, C.; Floot, B.; Verheij, M.; van Triest, B. NAD+ Depletion by APO866 in Combination with Radiation in a Prostate Cancer Model, Results from an in Vitro and in Vivo Study. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2014, 110, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Ito, E.; Shi, W.; Alajez, N.M.; Yue, S.; Lee, C.; Chan, N.; Bhogal, N.; Coackley, C.L.; Vines, D.; et al. Efficacy of Combining GMX1777 with Radiation Therapy for Human Head and Neck Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 898–911. [Google Scholar] [CrossRef] [Green Version]
- Bruzzone, S.; Fruscione, F.; Morando, S.; Ferrando, T.; Poggi, A.; Garuti, A.; D’Urso, A.; Selmo, M.; Benvenuto, F.; Cea, M.; et al. Catastrophic NAD+ Depletion in Activated T Lymphocytes through Nampt Inhibition Reduces Demyelination and Disability in EAE. PLoS ONE 2009, 4, e7897. [Google Scholar] [CrossRef]
- Drevs, J.; Löser, R.; Rattel, B.; Esser, N. Antiangiogenic Potency of FK866/K22.175, a New Inhibitor of Intracellular NAD Biosynthesis, in Murine Renal Cell Carcinoma. Anticancer Res. 2003, 23, 4853–4858. [Google Scholar] [PubMed]
- O’Brien, T.; Oeh, J.; Xiao, Y.; Liang, X.; Vanderbilt, A.; Qin, A.; Yang, L.; Lee, L.B.; Ly, J.; Cosino, E.; et al. Supplementation of Nicotinic Acid with NAMPT Inhibitors Results in Loss of In Vivo Efficacy in NAPRT1-Deficient Tumor Models. Neoplasia 2013, 15, 1314-IN3. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, H.; Lee, C.K.; Tateishi, K.; Higuchi, F.; Subramanian, M.; Rafferty, S.; Melamed, L.; Miller, J.J.; Wakimoto, H.; Cahill, D.P. Poly(ADP-Ribose) Glycohydrolase Inhibition Sequesters NAD+ to Potentiate the Metabolic Lethality of Alkylating Chemotherapy in IDH-Mutant Tumor Cells. Cancer Discov. 2020, 10, 1672–1689. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Galván, S.; Lucena-Cacace, A.; Perez, M.; Otero-Albiol, D.; Gomez-Cambronero, J.; Carnero, A. Tumor Cell-Secreted PLD Increases Tumor Stemness by Senescence-Mediated Communication with Microenvironment. Oncogene 2019, 38, 1309–1323. [Google Scholar] [CrossRef]
- Takao, S.; Chien, W.; Madan, V.; Lin, D.-C.; Ding, L.-W.; Sun, Q.-Y.; Mayakonda, A.; Sudo, M.; Xu, L.; Chen, Y.; et al. Targeting the Vulnerability to NAD+ Depletion in B-Cell Acute Lymphoblastic Leukemia. Leukemia 2018, 32, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Rane, C.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Crochiere, M.; Das-Gupta, S.; Minden, A. A Novel Orally Bioavailable Compound KPT-9274 Inhibits PAK4, and Blocks Triple Negative Breast Cancer Tumor Growth. Sci. Rep. 2017, 7, 42555. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Sierra, L.; Tsang, S.V.; Kurenbekova, L.; Patel, T.; Rajapakshe, K.; Shuck, R.L.; Rainusso, N.; Landesman, Y.; Unger, T.; et al. Targeting PAK4 Inhibits Ras-Mediated Signaling and Multiple Oncogenic Pathways in High-Risk Rhabdomyosarcoma. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Qasim, S.L.; Sierra, L.; Shuck, R.; Kurenbekova, L.; Patel, T.D.; Rajapakshe, K.; Wulff, J.; Nakahata, K.; Kim, H.R.; Landesman, Y.; et al. P21-Activated Kinases as Viable Therapeutic Targets for the Treatment of High-Risk Ewing Sarcoma. Oncogene 2021, 1–15. [Google Scholar] [CrossRef]
- Fulciniti, M.; Martinez-Lopez, J.; Senapedis, W.; Oliva, S.; Lakshmi Bandi, R.; Amodio, N.; Xu, Y.; Szalat, R.; Gulla, A.; Samur, M.K.; et al. Functional Role and Therapeutic Targeting of P21-Activated Kinase 4 in Multiple Myeloma. Blood 2017, 129, 2233–2245. [Google Scholar] [CrossRef]
- Cordover, E.; Wei, J.; Patel, C.; Shan, N.L.; Gionco, J.; Sargsyan, D.; Wu, R.; Cai, L.; Kong, A.-N.; Jacinto, E.; et al. KPT-9274, an Inhibitor of PAK4 and NAMPT, Leads to Downregulation of MTORC2 in Triple Negative Breast Cancer Cells. Chem. Res. Toxicol. 2020, 33, 482–491. [Google Scholar] [CrossRef]
- Audrito, V.; Serra, S.; Brusa, D.; Mazzola, F.; Arruga, F.; Vaisitti, T.; Coscia, M.; Maffei, R.; Rossi, D.; Wang, T.; et al. Extracellular Nicotinamide Phosphoribosyltransferase (NAMPT) Promotes M2 Macrophage Polarization in Chronic Lymphocytic Leukemia. Blood 2015, 125, 111–123. [Google Scholar] [CrossRef]
- Lv, H.; Lv, G.; Chen, C.; Zong, Q.; Jiang, G.; Ye, D.; Cui, X.; He, Y.; Xiang, W.; Han, Q.; et al. NAD+ Metabolism Maintains Inducible PD-L1 Expression to Drive Tumor Immune Evasion. Cell Metab. 2020. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, K.; Yao, Y.; Liu, Y.; Ni, Y.; Liao, C.; Tu, Z.; Qiu, Y.; Wang, D.; Chen, D.; et al. Dual Nicotinamide Phosphoribosyltransferase and Epidermal Growth Factor Receptor Inhibitors for the Treatment of Cancer. Eur. J. Med. Chem. 2020, 113022. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.; Roulston, A.; Bélec, L.; Billot, X.; Marcellus, R.; Bédard, D.; Bernier, C.; Branchaud, S.; Chan, H.; Dairi, K.; et al. The Small Molecule GMX1778 Is a Potent Inhibitor of NAD+ Biosynthesis: Strategy for Enhanced Therapy in Nicotinic Acid Phosphoribosyltransferase 1-Deficient Tumors. Mol. Cell. Biol. 2009, 29, 5872–5888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, U.H.; Thougaard, A.V.; Jensen, P.B.; Sehested, M. A Preclinical Study on the Rescue of Normal Tissue by Nicotinic Acid in High-Dose Treatment with APO866, a Specific Nicotinamide Phosphoribosyltransferase Inhibitor. Mol. Cancer Ther. 2010, 9, 1609–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauparlant, P.; Bédard, D.; Bernier, C.; Chan, H.; Gilbert, K.; Goulet, D.; Gratton, M.-O.; Lavoie, M.; Roulston, A.; Turcotte, E.; et al. Preclinical Development of the Nicotinamide Phosphoribosyl Transferase Inhibitor Prodrug GMX1777. Anticancer. Drugs 2009, 20, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Tarrant, J.M.; Dhawan, P.; Singh, J.; Zabka, T.S.; Clarke, E.; DosSantos, G.; Dragovich, P.S.; Sampath, D.; Lin, T.; McCray, B.; et al. Preclinical Models of Nicotinamide Phosphoribosyltransferase Inhibitor-Mediated Hematotoxicity and Mitigation by Co-Treatment with Nicotinic Acid. Toxicol. Mech. Methods 2015, 25, 201–211. [Google Scholar] [CrossRef]
- Zabka, T.S.; Singh, J.; Dhawan, P.; Liederer, B.M.; Oeh, J.; Kauss, M.A.; Xiao, Y.; Zak, M.; Lin, T.; McCray, B.; et al. Retinal Toxicity, in Vivo and in Vitro, Associated with Inhibition of Nicotinamide Phosphoribosyltransferase. Toxicol. Sci. 2015, 144, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Misner, D.L.; Kauss, M.A.; Singh, J.; Uppal, H.; Bruening-Wright, A.; Liederer, B.M.; Lin, T.; McCray, B.; La, N.; Nguyen, T.; et al. Cardiotoxicity Associated with Nicotinamide Phosphoribosyltransferase Inhibitors in Rodents and in Rat and Human-Derived Cells Lines. Cardiovasc. Toxicol. 2017, 17, 307–318. [Google Scholar] [CrossRef]
- Cassar, S.; Dunn, C.; Olson, A.; Buck, W.; Fossey, S.; Ramos, M.F.; Sancheti, P.; Stolarik, D.; Britton, H.; Cole, T.; et al. From the Cover: Inhibitors of Nicotinamide Phosphoribosyltransferase Cause Retinal Damage in Larval Zebrafish. Toxicol. Sci. 2018, 161, 300–309. [Google Scholar] [CrossRef]
- Ravaud, A.; Cerny, T.; Terret, C.; Wanders, J.; Bui, B.N.; Hess, D.; Droz, J.-P.; Fumoleau, P.; Twelves, C. Phase I Study and Pharmacokinetic of CHS-828, a Guanidino-Containing Compound, Administered Orally as a Single Dose Every 3 Weeks in Solid Tumours: An ECSG/EORTC Study. Eur. J. Cancer Oxf. Engl. 1990 2005, 41, 702–707. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Marshall, J.L.; Hwang, J.H.; Malik, S.M.; He, A.R.; Deeken, J.F.; Kelso, C.B.; Dorsch-Vogel, K.; Berger, M.S. A Phase 1 Trial of GMX1777: An Inhibitor of Nicotinamide Phosphoribosyl Transferase (NAMPRT). J. Clin. Oncol. 2008, 26, 14568. [Google Scholar] [CrossRef]
- Holen, K.; Saltz, L.B.; Hollywood, E.; Burk, K.; Hanauske, A.-R. The Pharmacokinetics, Toxicities, and Biologic Effects of FK866, a Nicotinamide Adenine Dinucleotide Biosynthesis Inhibitor. Investig. New Drugs 2008, 26, 45–51. [Google Scholar] [CrossRef]
- von Heideman, A.; Berglund, Å.; Larsson, R.; Nygren, P. Safety and Efficacy of NAD Depleting Cancer Drugs: Results of a Phase I Clinical Trial of CHS 828 and Overview of Published Data. Cancer Chemother. Pharmacol. 2010, 65, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Pishvaian, M.J.; Marshall, J.L.; Hwang, J.J.; Malik, S.; He, A.R.; Deeken, J.F.; Kelso, C.B.; Cotarla, I.; Berger, M.S. A Phase I Trial of GMX1777, an Inhibitor of Nicotinamide Phosphoribosyl Transferase (NAMPRT), given as a 24-Hour Infusion. J. Clin. Oncol. 2009, 27, 3581. [Google Scholar] [CrossRef]
- Goldinger, S.M.; Gobbi Bischof, S.; Fink-Puches, R.; Klemke, C.-D.; Dréno, B.; Bagot, M.; Dummer, R. Efficacy and Safety of APO866 in Patients With Refractory or Relapsed Cutaneous T-Cell Lymphoma: A Phase 2 Clinical Trial. JAMA Dermatol. 2016, 152, 837–839. [Google Scholar] [CrossRef] [Green Version]
- Heske, C.M. Beyond Energy Metabolism: Exploiting the Additional Roles of NAMPT for Cancer Therapy. Front. Oncol. 2019, 9, 1514. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Deik, A.; Gonzalez, C.; Gonzalez, M.E.; Fu, F.; Ferrari, M.; Churchhouse, C.L.; Florez, J.C.; Jacobs, S.B.R.; Clish, C.B.; et al. Polyunsaturated Fatty Acid Desaturation Is a Mechanism for Glycolytic NAD+ Recycling. Cell Metab. 2019, 29, 856–870.e7. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulator | Target | Mechanism | Effect | Cancer/Tissue Type |
|---|---|---|---|---|
| c-MYC and Max [37] | NAMPT | -Binding to and regulating the activity of the distal 4.6 kb putative NAMPT enhancer 65 kb downstream the NAMPT transcription start site specifically through the 1 kb “B-region” within the NAMPT enhancer. | Upregulation | Salvage-dependent cancer cells |
| c-MYC [38] | NAMPT | -Binding to the NAMPT promoter. | Upregulation | MCF-7 cells (breast cancer) |
| C/EBPβ [36] | NAMPT | -Interaction with NAMPT regulatory regions. | Upregulation | Mesenchymal GSCs |
| HMGA proteins [39] | NAMPT | -Binding to an NAMPT enhancer element during oncogene-induced senescence (OIS). | Upregulation | Oncogenic Ras-induced senescent IMR90 cells (lung fibroblasts) |
| SIRT6 [40] | NAMPT | -Regulation of NAMPT enzymatic activity through lysine deacetylation. | Upregulation | HEK293 cells (human embryonic kidney cells) |
| SIRT1 [41] | NAMPT | -Regulation of NAMPT activity through lysine deacetylation and secretion of eNAMPT. | Upregulation | Adipocytes |
| Foxo1 [42] | NAMPT | -Binding to conserved insulin response elements (IREs) in the NAMPT 5′-flanking promoter region. | Downregulation | MCF-7 cells (breast cancer) |
| NAMPT-AS “RP11-22N19.2” Lnc-RNA [43] | NAMPT | -Recruitment of the transcription factor POU2F2 to the promoter region of NAMPT to enhance NAMPT transcription. -Competitive binding to miR-548b-3p leading to increasing the NAMPT mRNA pool. | Upregulation | MDA-MB-231 and MDA-MB-468 cells (triple-negative breast cancer) |
| GACAT3 [44] Lnc-RNA | NAMPT | -Competitive binding to miR-135a, whose target gene is NAMPT. | Upregulation | U87 and U251 cells (glioma) |
| miR-381 [45] | NAMPT | -Post-transcriptional binding to the 3′- untranslated region (UTR) of NAMPT. | Downregulation | MDA-MB-231 and MCF-7 cells (breast cancer) |
| miR-206 [46] | NAMPT | -Binding to the 3′-UTR of NAMPT. | Downregulation | MDA-MB-231 and MCF-7 cells (breast cancer) |
| miR-494 [47] | NAMPT | -Binding to the 3’-UTR of NAMPT. | Downregulation | MDA-MB-231 and MCF-7 cells (breast cancer) |
| miR-154 [48] | NAMPT | -Binding to the 3’-UTR of NAMPT. | Downregulation | MDA-MB-231 and MCF-7 cells (breast cancer) |
| miR-26b [49] | NAMPT | -Binding to the 3′-UTR of NAMPT. | Downregulation | SW480 cells (colorectal cancer) |
| miR-206 [50] | NAMPT | -Regulation of NAMPT expression most probably through targeting the 3′-UTR of NAMPT. | Downregulation | MiaPaCa-2 and Panc-1 cells (pancreatic cancer) |
| miR-23b [51] | NAMPT | -Regulation of NAMPT expression. | Downregulation | melanoma cells |
| Gene Amplification [37] | NAPRT/ NADSYN | -Regulation of NAPRT or NADSYN expression. | Upregulation | PH-dependent tumors and cancer cell lines |
| Gene Silencing [52] | NAPRT | -Hypermethylation of NAPRT promoter region. | Downregulation | Several cancer cell lines |
| Mutant IDH1 [53] | NAPRT | -Hypermethylation of the CpG islands in the NAPRT promoter region and thus reprogramming NAD metabolism. -IDH1-mutant cancers are uniquely sensitive to NAMPT inhibitors via NAD depletion. | Downregulation | IDH1-mutant cancer cells (MGG119, MGG152, BT142, BT142, HT1080, 30T, SW1353) |
| Mutant PPM1D [54] | NAPRT | -Hypermethylation of the CpG islands in the genome and epigenetic silencing of NAPRT gene. -PPM1D mutant cancer cells are uniquely sensitive to NAMPT inhibitors. | Downregulation | PPM1D mutant astrocytes and diffuse intrinsic pontine glioma (DIPG) cell lines |
| SIRT3 [55] | NMNAT2 | -Regulation of NMNAT2 activity through deacetylation. | Upregulation | A549 cells (non-small cell lung cancer) |
| miR-654-3p [56] | QAPRT | -Binding to the 3′-UTR of QAPRT. | Downregulation | Igrov-1 cells (ovarian cancer) |
| DSCAM-AS1 [57] Lnc-RNA | QAPRT | -Competitive binding of miRNA-150-5p and miRNA-2467-3p. | Upregulation | T47D and MCF-7 cells (breast cancer) |
| WT1 [58] | QAPRT | -Binding to a conserved site on the QAPRT promoter. | Upregulation | K562 cells (leukemia) |
| NAMPT Inhibitor | The Combination Agent/Drug | Class | Approval as an Anti-Cancer | Cancer Type | In Vitro Efficacy | In Vivo Efficacy | Ref. |
|---|---|---|---|---|---|---|---|
| FK866 | 5-fluorouracil | Antimetabolite | Yes | Gastric cancer | Yes | n/a 1 | [71] |
| FK866 | Fludarabine | Antimetabolite | Yes | Leukemia (CLL) | Yes | n/a | [135] |
| FK866 | Etoposide | Topoisomerase II inhibitor | Yes | Leukemia | Yes | n/a | [136,137] |
| GMX1777 | Etoposide | Topoisomerase II inhibitor | Yes | Lung cancer | n/a | Yes | [108] |
| FK866 | Etoposide | Topoisomerase II inhibitor | Yes | Neuroblastoma | Yes | n/a | [138] |
| FK866 | Cisplatin | Alkylating agent | Yes | Neuroblastoma | Yes | n/a | [138] |
| FK866 GMX1778 | Cisplatin | Alkylating agent | Yes | Ovarian cancer | Yes | Yes (FK866) | [139] |
| FK866 | Cyclosporin A Verapamil | Pgp inhibitor Pgp inhibitor | No No | Leukemia Leukemia | Yes Yes | n/a n/a | [140] |
| FK866 | Bortezomib | Proteasome inhibitor | Yes | Multiple myeloma | Yes | Yes | [141] |
| FK866 | Ibrutinib | Bruton’s tyrosine kinase inhibitor | Yes | Waldenstrom macroglobulinemia | Yes | Yes | [142] |
| GMX1777 | Pemetrexed | Antimetabolite (Antifolate) | Yes | Non-small-cell lung cancer (NSCLC) | Yes | Yes | [143] |
| FK866 | Gemcitabine | Antimetabolite | Yes | Pancreatic cancer (PDAC) | Yes | n/a | [144] |
| FK866 | Gemcitabine | Antimetabolite | Yes | PDAC | Yes | Yes | [50] |
| STF-118804 | Gemcitabine | Antimetabolite Topoisomerase II inhibitor Antimicrotubular agent | Yes | PDAC | Yes | n/a | [145] |
| Etoposide | Yes | PDAC | Yes | n/a | |||
| Paclitaxel | Yes | PDAC | Yes | n/a | |||
| FK866 | Vorinostat Valproic acid | HDAC inhibitor HDAC inhibitor | Yes No | Leukemia Leukemia | Yes Yes | n/a n/a | [146] |
| GMX1778 | 177Lu-DOTATATE | Radiolabeled somatostatin analog | Yes | Neuroendocrine tumors | n/a | Yes | [147] |
| FK866 | Rituximab | Anti-CD20 | Yes | Lymphoma | Yes | Yes | [148] |
| FK866 GMX1778 | Temozolomide | Alkylating agent | Yes | Glioblastoma | Yes | n/a | [149] |
| FK866 GMX1778 | Temozolomide | Alkylating agent | Yes | IDH1-mutant cancers | Yes | Yes (FK866) | [150] |
| FK866 | Olaparib | PARP inhibitor | Yes | Triple-negative breast cancer (TNBC) | Yes | Yes | [151] |
| GNE-618 FK866 GMX1778 | Niraparib | PARP inhibitor | Yes | Ewing sarcoma | Yes | Yes (GNE-618) | [152] |
| OT-82 | Niraparib | PARP Inhibitor | Yes | Ewing sarcoma | Yes | Yes | [153] |
| OT-82 | Irinotecan & its metabolite SN-38 | topoisomerase I inhibitors | Yes (Irinotecan) | Ewing sarcoma | Yes (SN-38) | Yes (Irinotecan) | [153] |
| OT-82 | Cytarabine | Antimetabolite | Yes | Acute lymphoblastic leukemia (ALL) | Yes | Yes | [154] |
| OT-82 | Dasatinib | Tyrosine kinase inhibitor | Yes | ALL | n/a | Yes | [154] |
| OT-82 | Etoposide | Topoisomerase II inhibitor | Yes | ALL | Yes | n/a | [154] |
| GMX1778 | Anti-mouse PD-1 antibody | Immune checkpoint inhibitor | Human anti-PD1: Yes | Glioblastoma | n/a | Yes | [155] |
| MV87 | Anti-mouse PD-1 antibody | Immune checkpoint inhibitor | Human anti-PD1: Yes | Fibrosarcoma | n/a | Yes | [156] |
| KPT-9274 | Anti-mouse PD-1 antibody | Immune checkpoint inhibitor | Human anti-PD1: Yes | Renal cell carcinoma | n/a | Yes | [157] |
| KPT-9274 | Anti-mouse PD-1 antibody | Immune checkpoint inhibitor | Human anti-PD1: Yes | Melanoma Colon adenocarcinoma | n/a | Yes (PAK4) | [158] |
| KPT-9274 | Bendamustine Melphalan | Alkylating agent Alkylating agent | Yes Yes | Waldenstrom macroglobulinemia | Yes Yes | Yes n/a | [159] |
| KPT-9274 | Everolimus | mTOR inhibitor | Yes | Pancreatic neuroendocrine tumor | Yes | n/a | [160] |
| KPT-9274 | Gemcitabine Oxaliplatin | Antimetabolite Alkylating agent | Yes Yes | PDAC PDAC | Yes Yes | Yes (PAK4) n/a | [161] |
| FK866 | TRAIL | Apoptosis activator | Not approved as a drug | Leukemia | Yes | n/a | [162] |
| FK866 | 2-HNA | NAPRT inhibitor | Not approved as a drug | Ovarian cancer Pancreatic cancer | Yes | Yes (sodium salt of 2-HNA) | [62] |
| FK866 | L-1-methyl-tryptophan | Indoleamine 2,3-dioxygenase (IDO) inhibitor | Not approved as a drug | Gastric cancer Bladder cancer | n/a | Yes (only in immuno-competent mice) | [163] |
| FK866 | β-Lapachone | ROS generator & NQO1 substrate | Not approved as a drug | PDAC NSCLC | Yes | n/a | [164,165,166] |
| FK866 | FX11 | Lactate dehydrogenase A (LDHA) inhibitor | Not approved as a drug | Lymphoma | Yes | Yes | [167] |
| FK866 | 1-methyl-3-nitro-1-nitrosoguanidinium (MNNG) | Alkylating agent | Not approved as a drug | Leukemia | Yes | n/a | [168] |
| Compound | Cancer Type | Cancer Cell Lines | In Vitro Effects | Mouse Model | In Vivo Model | In Vivo Effects | Reported Mode of Action |
|---|---|---|---|---|---|---|---|
| KPT-9274 | Renal cell carcinoma (RCC) [118] | RCC cell lines: 786-O ACHN Caki-1 | -Attenuation of viability, invasion, and migration in several RCC cell lines. -Limited toxicity in normal human renal epithelial cells. -Induction of apoptosis. -Decrease in G2-M transition. -Reduced NAD and SIRT1 levels. -NA rescued NAD levels in normal renal epithelial cells but not in 786-O and Caki-1 “NAPRT deficient” cells. -Reduction in nuclear β-catenin and of the Wnt/β-catenin targets c-MYC and cyclin D1 as a result of PAK4 inhibition. | Male nude mice | RCC xenograft model: 786-O cells | -Reduced tumor growth. -No significant animal weight loss. | PAK4 and NAMPT inhibition |
| KPT-9274 | Renal cell carcinoma (RCC) [157] | Ex vivo: -Reduced tumor expression of PAK4 and phospho-β-catenin. -NAD + NADH levels in tumors decreased by KPT-9274 and increased by anti-PD1 antibody. | Male BALB/cJ mice | RCC allograft model: Mouse RENCA-luciferase (RENCA-Luc) cells | -Significant reduction in tumor growth with KPT-9274 and anti-PD1 combination compared to each agent alone. -No significant animal weight loss. | PAK4 and NAMPT inhibition | |
| KPT-9274 | Pancreatic ductal adenocarcinoma (PDAC) [161] | PDAC cell lines: MiaPaCa-2 HPAC Panc1 Colo-357 L3.6pl MiaPaCa-2 cancer stem cells | -Inhibition of proliferation of PDAC cells. -Limited toxicity in normal pancreatic human epithelial cells. -Cancer-selective induction of apoptosis and cell-cycle arrest. -Suppression of cancer migration. -Overcoming stemness (PDAC cancer stem cells) and downregulation of EMT markers. -Synergistic effect with gemcitabine and oxaliplatin. | Female SCID mice | PDAC xenograft model: L3.6pl cells AsPc-1 cells PDAC cancer stem cell xenograft model: CD44+/CD133+/EpCAM+ MiaPaCa-2 cells | -Remarkable antitumor activity as a single agent. -Marginal antitumor activity in combination with gemcitabine. -No signs of toxicity. -Suppression of growth of highly resistant PDAC cancer stem cell-derived tumors. | PAK4 inhibition |
| KPT-9274 | Acute myeloid leukemia (AML) [169] | AML cell lines: HL-60 THP-1 Kasumi-1 MV4-11 OCI-AML3 MOLM13 Primary AML cells | -Inhibition of proliferation of AML cells. -Cell cycle arrest. -Induction of apoptosis. -Reduction in NAD levels, disruption of mitochondrial activity, and cellular respiration. -Limited toxicity on normal hematopoietic cells. | NSG mice | AML xenograft model: luciferase-positive MV4-11 cells Patient-derived xenograft (PDX) model of AML | -Improved overall survival. -Reduced disease progression and tumor burden. | NAMPT inhibition |
| KPT-9274 | B-cell acute lymphoblastic leukemia (B-ALL) [194] | B-ALL cell lines: KOPN-8 RS4;11 REH 697 OP-1 Nalm6 Sup-B15 SEM PDX B-ALL cells | -Strong inhibition of cell growth. -Induction of apoptosis. -Intracellular NAD depletion and modulation of NAD-dependent pathways. -NA supplementation reversed KPT-9274-mediated growth inhibition in three sensitive B-ALL cell lines. | NSG mice | PDX model of B-ALL: luciferase-transduced LAX2 cells | -Effective suppression of leukemia progression. -Significantly improved survival. -Acceptable adverse effect profile (normal mice activity, no significant difference in body weight between groups). | NAMPT inhibition |
| KPT-9274 | Triple-negative breast cancer (TNBC) [195] | BC cell lines: MDA-MB-231 MDA-MB-468 SUM159 MCF7 SkBr-3 BT-474 | -Inhibition of cell proliferation in several BC cell lines. -Reduction in viability was more pronounced in TNBC cell lines (almost complete inhibition). -Stimulation of apoptosis. | Female nude mice | TNBC xenograft models: MDA-MB-231 cells MDA-MB-468 cells SUM159 cells | -Significant reduction in tumor weights and volumes. -No significant effect on the body weights of the mice. -Reduced PAK4 protein levels in tumors. | PAK4 inhibition |
| KPT-9274 | Melanoma [158] | Melanoma cell lines: Murine B16 cells | C57BL/6 mice | Melanoma model: B16 cells | -Significant decrease in tumor growth with KPT-9274 and anti-PD1 combination compared to each agent alone. | PAK4 inhibition | |
| KPT-9274 | Colon cancer [158] | Colon cancer cell lines: Murine MC38 cells | C57BL/6 mice | Colon adenocarcinoma model: MC38 cells | -Significant decrease in tumor growth with KPT-9274 alone or combined with anti-PD1 compared to anti-PD1 alone. | PAK4 inhibition | |
| KPT-9274 | Pancreatic neuro-endocrine tumors (PNET) [160] | PNET cell lines: BON-1 QGP-1 | -Reduction in growth and survival of PNET cells. -Reduced NAD and ATP levels and ATP collapse was reversed by NA. -Synergistic effect with everolimus. | Female SCID mice | PNET xenograft model: BON-1 cells | -Significant reduction in tumor growth as monotherapy. | PAK4 and NAMPT inhibition |
| KPT-9274 | Waldenstrom macroglobulinemia (WM) [159] | WM cell lines: BCWM-1 MWCL-1 RPCIWM-1 Primary WM cells | -Reduction in cell viability. -NA rescued BCWM-1 cells from KPT-9274-mediated growth inhibition. -Impairment of DNA repair and induction of DNA damage. -Induction of apoptosis. -Synergistic effect in combination with DNA-damaging agents (bendamustine & melphalan). | SCID mice | WM xenograft model: BCMW-1 cells | -Significant inhibition of tumor growth as a single agent. -Significant reduction in tumor volume with KPT-9274 and bendamustine combination compared with either agent alone. | PAK4 and NAMPT inhibition |
| KPT-9274 | Multiple myeloma (MM) [198] | Many human myeloma cell lines Primary MM cells | -Reduction in cell growth and survival in a large panel of MM cell lines and primary MM cells. -Suppression of the promoting effects of the bone marrow microenvironment. -No significant effect on bone marrow mononuclear cells or PBMCS. -Induction of apoptosis and deregulation of the MEK/ERK pathway. | Nude mice | MM xenograft models: MM1S cells OPM2 cells | -Significant single-agent antitumor activity in both MM xenograft models. -Higher sensitivity to KPT-9274 was seen with t(4:14) FGFR3-mutated OPM2 tumors compared to MM1S xenografts. | PAK4 inhibition |
| KPT-9274 | Ewing sarcoma (EWS) [197] | EWS cell lines: CHLA-10 A673 TC32 | -Suppression in cell proliferation. -Reduction in invasive and migratory characteristics. -Potential synergistic effect with doxorubicin and vincristine. | NSG mice | EWS xenograft models: A673 cells TC71 cells PDX and metastatic models of EWS | -Significant reduction in tumor growth as a single agent. -Reduced metastatic burden in the EWS metastatic model. | PAK4 inhibition |
| KPT-9274 | Rhabdomyosarcoma (RMS) [196] | RMS cell lines: RH30 RD RH4 | -Reduction in cell proliferation in multiple RMS cell lines (IC50 ranges from 40 to 80 nM). -Limited toxicity in normal skeletal muscle myoblast cells (IC50 values were 8–10 times higher). -Induction of apoptosis and G1-S arrest. -Inhibition of cell motility and invasive properties. -Reduced PAK4 activity and β-catenin activation. | NSG mice | RMS orthotopic xenograft models: RH30 cells RD cells PDX model of relapsed RMS Metastatic model of RMS: RD cells | -Significant reduction in tumor growth in the orthotopic and PDX models with KPT-9274 alone compared to vehicle. -No significant changes in body weight. -Reduced metastatic burden in the liver with KPT-9274 in the metastatic model. | PAK4 inhibition |
| OT-82 | Pediatric acute lymphoblastic leukemia (ALL) [154] | ALL cell lines: PER-826A REH PER-703A CEM PER-485 RS4;11 KOPN-8 Loucy Pediatric PDX ALL cells | -Potent dose-dependent reduction in cell viability. -Reduction in NAD levels, ATP levels, and PARP activity. -Higher DNA damage. -Induction of apoptosis. -Synergistic effect with cytarabine and etoposide in PER-485 cell line. | NOD/SCID mice NSG mice | 21 PDX models of high-risk pediatric ALL | -Significant extension of the survival in 20/21 (95%) PDXs and objective response in 18/21 (86%) PDXs→ significant leukemia regression. -Therapeutic enhancement when combined with cytarabine in 2 aggressive MLLr-ALL PDX models and with dasatinib in one Ph+ ALL PDX model. | |
| OT-82 | Ewing sarcoma (EWS) [153] | EWS cell lines: TC71 TC32 RDES SK-N-MC EW8 5838 CHLA-258 | -Potent inhibition in cell growth and proliferation. -Reduction in NAD levels and PARP activity. -Higher DNA damage. -G2 arrest and induction of apoptosis. -Enhanced antiproliferative effects when combined with niraparib or SN-38. | SCID beige mice NOG mice | Orthotopic xenograft models of EWS: TC71 cells TC32 cells PDX model of EWS | -Significant reduction in tumor growth and prolongation of survival with doses of 25 mg/kg and 50 mg/kg. -Cessation of treatment resulted in tumor regrowth. -Improved OT-82 efficacy when combined with irinotecan or niraparib (slower tumor growth and prolongation of median survival). -Several unexpected deaths occurred with OT-82/niraparib combination. | |
| OT-82 | Acute myeloid leukemia (AML) [109] | AML cell lines: MV4-11 THP1 | -Potent reduction in cell viability. -Depletion of NAD and ATP levels. -Induction of apoptosis. -NA addition rescued MV4-11 cells from OT-82-mediated cytotoxicity. | SCID mice | Subcutaneous (SC) and systemic xenograft models of AML: MV4-11 cells | -Significant dose-dependent reduction in tumor volume→ SC model. -Significant prolongation of mice survival at 25 and 40 mg/kg → systemic model. -Improved survival with the optimized OT-82 regimen→ systemic model. | |
| OT-82 | Erythroleukemia [109] | Erythroleukemia cell line: HEL92.1.7 | -Potent reduction in cell viability. | SCID mice | SC and systemic xenograft models of erythroleukemia: HEL92.1.7 cells | -Significant reduction in tumor volume at 50 mg/kg dose→ SC model. -Significant prolongation of mice survival at 40 mg/kg→ systemic model. | |
| OT-82 | Burkitt lymphoma (BL) [109] | BL cell lines: Raji Ramos | -Potent reduction in cell viability. | SCID mice | SC xenograft model of BL: Ramos cells | -Significant reduction in tumor volume with the optimized OT-82 regimen. | |
| OT-82 | Multiple myeloma (MM) [109] | SCID mice | SC xenograft model of MM: RPMI 8226 cells | -Significant reduction in tumor volume with the optimized OT-82 regimen. | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghanem, M.S.; Monacelli, F.; Nencioni, A. Advances in NAD-Lowering Agents for Cancer Treatment. Nutrients 2021, 13, 1665. https://doi.org/10.3390/nu13051665
Ghanem MS, Monacelli F, Nencioni A. Advances in NAD-Lowering Agents for Cancer Treatment. Nutrients. 2021; 13(5):1665. https://doi.org/10.3390/nu13051665
Chicago/Turabian StyleGhanem, Moustafa S., Fiammetta Monacelli, and Alessio Nencioni. 2021. "Advances in NAD-Lowering Agents for Cancer Treatment" Nutrients 13, no. 5: 1665. https://doi.org/10.3390/nu13051665
APA StyleGhanem, M. S., Monacelli, F., & Nencioni, A. (2021). Advances in NAD-Lowering Agents for Cancer Treatment. Nutrients, 13(5), 1665. https://doi.org/10.3390/nu13051665