
Visceral Adiposity and Cancer: Role in Pathogenesis and Prognosis



Abstract
:1. Obesity and Cancer
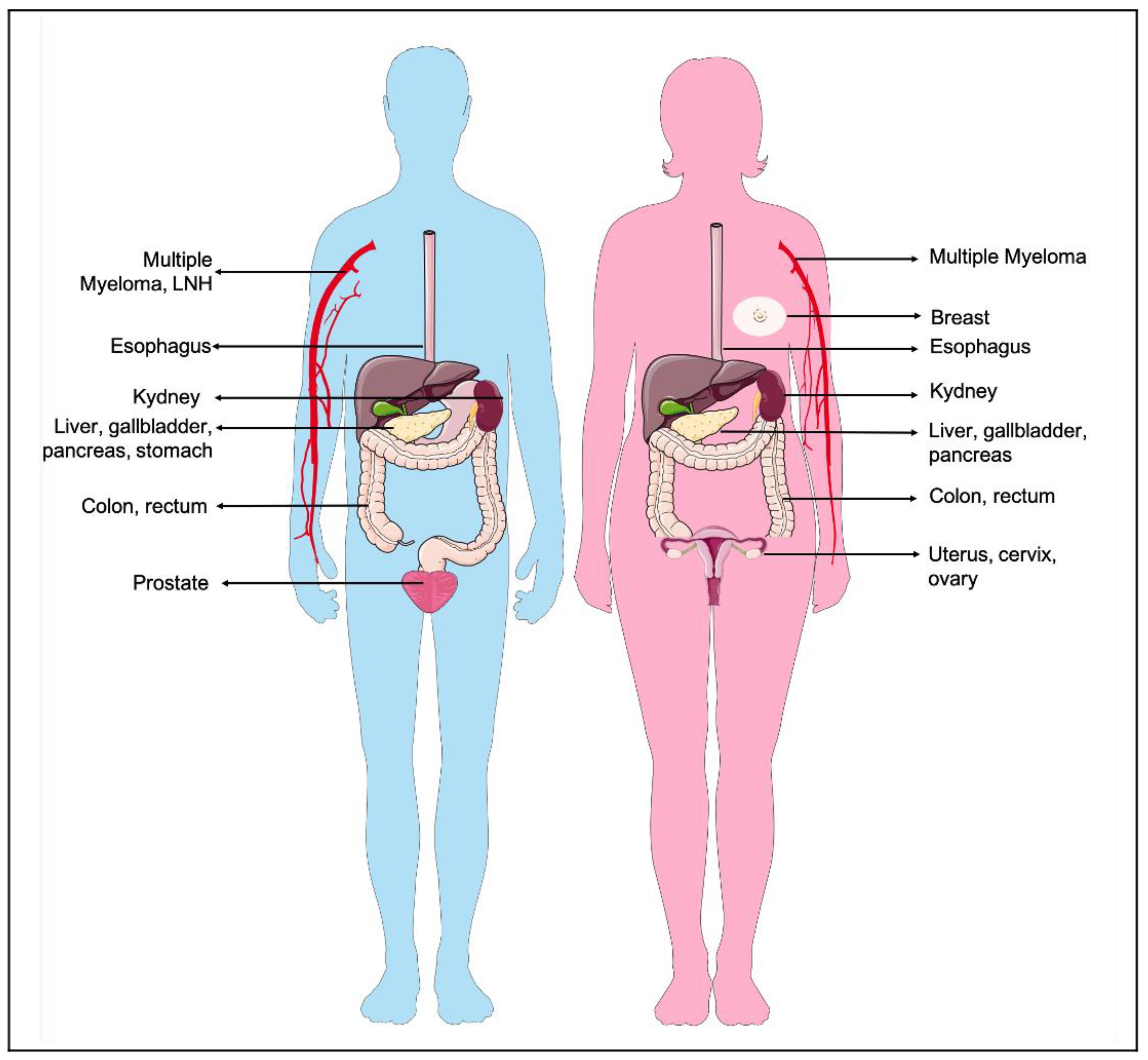
1.1. Incidence: When Obesity Promotes Cancer
1.2. Obesity and Cancer: Prognosis and Mortality
2. Obesity and Cancer: Deepening the Relationship
2.1. Adiposity
2.2. Adipokines and Chronic Low-Grade Inflammation
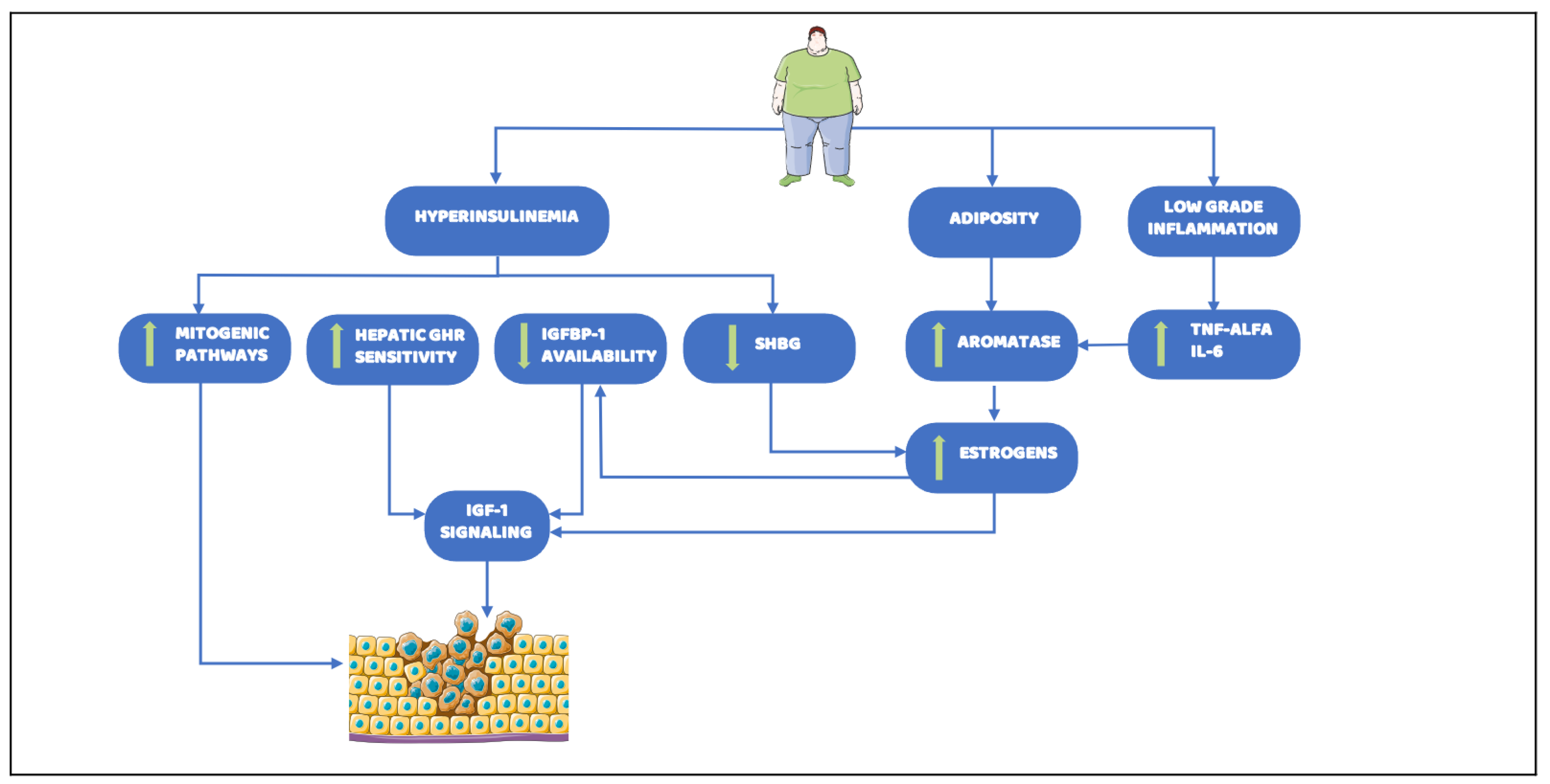
2.3. Hyperinsulinemia
3. Perspectives into Visceral Adiposity and Cancer
3.1. Anthropometric Assessment: How to Identify Visceral Obesity
3.2. Browning of Adipose Tissue: Remodeling Fat Amount
3.3. Lifestyle Interventions—Let Food Be Your Medicine
4. Conclusions and Recommendations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Obesity: Preventing and Managing the Global Epidemic; WHO Technical Report Series 894; World Health Organisation: Geneva, Switzerland, 2000; 253p. [Google Scholar]
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 31 December 2020).
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Tchernof, A.; Despres, J.P. Pathophysiology of human visceral obesity: An update. Physiol. Rev. 2013, 93, 359–404. [Google Scholar] [CrossRef]
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, P.A.G.; Stevens, G.A.; Ezzati, P.M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R.; et al. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36–46. [Google Scholar] [CrossRef]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Bull, C.J.; Bell, J.A.; Murphy, N.; Sanderson, E.; Davey Smith, G.; Timpson, N.J.; Banbury, B.L.; Albanes, D.; Berndt, S.I.; Bezieau, S.; et al. Adiposity, metabolites, and colorectal cancer risk: Mendelian randomization study. BMC Med. 2020, 18, 396. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K.; International Agency for Research on Cancer Handbook Working Group. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Hidayat, K.; Shi, B.M. Abdominal obesity and gastroesophageal cancer risk: Systematic review and meta-analysis of prospective studies. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Zimta, A.A.; Tigu, A.B.; Muntean, M.; Cenariu, D.; Slaby, O.; Berindan-Neagoe, I. Molecular Links between Central Obesity and Breast Cancer. Int. J. Mol. Sci. 2019, 20, 5364. [Google Scholar] [CrossRef] [Green Version]
- Renehan, A.G.; Soerjomataram, I.; Leitzmann, M.F. Interpreting the epidemiological evidence linking obesity and cancer: A framework for population-attributable risk estimations in Europe. Eur. J. Cancer 2010, 46, 2581–2592. [Google Scholar] [CrossRef]
- Evans, T.; Sany, O.; Pearmain, P.; Ganesan, R.; Blann, A.; Sundar, S. Differential trends in the rising incidence of endometrial cancer by type: Data from a UK population-based registry from 1994 to 2006. Br. J. Cancer 2011, 104, 1505–1510. [Google Scholar] [CrossRef] [Green Version]
- Leitzmann, M.F.; Brenner, A.; Moore, S.C.; Koebnick, C.; Park, Y.; Hollenbeck, A.; Schatzkin, A.; Ron, E. Prospective study of body mass index, physical activity and thyroid cancer. Int. J. Cancer 2010, 126, 2947–2956. [Google Scholar] [CrossRef] [Green Version]
- Abnet, C.C.; Freedman, N.D.; Hollenbeck, A.R.; Fraumeni, J.F., Jr.; Leitzmann, M.; Schatzkin, A. A prospective study of BMI and risk of oesophageal and gastric adenocarcinoma. Eur. J. Cancer 2008, 44, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yang, D.L.; Chen, Z.Z.; Gou, B.F. Associations of body mass index with cancer incidence among populations, genders, and menopausal status: A systematic review and meta-analysis. Cancer Epidemiol. 2016, 42, 1–8. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Lennon, H.; Sperrin, M.; Badrick, E.; Renehan, A.G. The Obesity Paradox in Cancer: A Review. Curr. Oncol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef]
- Stephenson, G.D.; Rose, D.P. Breast cancer and obesity: An update. Nutr. Cancer 2003, 45, 1–16. [Google Scholar] [CrossRef]
- Ma, J.; Pollak, M.; Giovannucci, E.; Chan, J.M.; Tao, Y.; Hennekens, C.; Stampfer, M.J. A prospective study of plasma levels of insulin-like growth factor I (IGF-I) and IGF-binding protein-3, and colorectal cancer risk among men. Growth Horm. IGF Res. 2000, 10 (Suppl. A), S28–S29. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, L.; Zhou, Q.; Imam, M.U.; Cai, J.; Wang, Y.; Qi, M.; Sun, P.; Ping, Z.; Fu, X. Body mass index had different effects on premenopausal and postmenopausal breast cancer risks: A dose-response meta-analysis with 3,318,796 subjects from 31 cohort studies. BMC Public Health 2017, 17, 936. [Google Scholar] [CrossRef]
- Gong, Z.; Neuhouser, M.L.; Goodman, P.J.; Albanes, D.; Chi, C.; Hsing, A.W.; Lippman, S.M.; Platz, E.A.; Pollak, M.N.; Thompson, I.M.; et al. Obesity, diabetes, and risk of prostate cancer: Results from the prostate cancer prevention trial. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1977–1983. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef] [Green Version]
- Ligibel, J.A.; Wollins, D. American Society of Clinical Oncology Obesity Initiative: Rationale, Progress, and Future Directions. J. Clin. Oncol. 2016, 34, 4256–4260. [Google Scholar] [CrossRef]
- Greenlee, H.; Unger, J.M.; LeBlanc, M.; Ramsey, S.; Hershman, D.L. Association between Body Mass Index and Cancer Survival in a Pooled Analysis of 22 Clinical Trials. Cancer Epidemiol. Biomark. Prev. 2017, 26, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligibel, J.A.; Alfano, C.M.; Courneya, K.S.; Demark-Wahnefried, W.; Burger, R.A.; Chlebowski, R.T.; Fabian, C.J.; Gucalp, A.; Hershman, D.L.; Hudson, M.M.; et al. American Society of Clinical Oncology position statement on obesity and cancer. J. Clin. Oncol. 2014, 32, 3568–3574. [Google Scholar] [CrossRef]
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, P.J.; Ennis, M.; Pritchard, K.I.; Trudeau, M.E.; Koo, J.; Madarnas, Y.; Hartwick, W.; Hoffman, B.; Hood, N. Fasting insulin and outcome in early-stage breast cancer: Results of a prospective cohort study. J. Clin. Oncol. 2002, 20, 42–51. [Google Scholar] [CrossRef]
- Pollak, M.N.; Chapman, J.W.; Shepherd, L.; Meng, D.; Richardson, P.; Wilson, C.; Orme, B.; Pritchard, K.I. Insulin resistance, estimated by serum C-peptide level, is associated with reduced event-free survival for postmenopausal women in NCIC CTG MA.14 adjuvant breast cancer trial. J. Clin. Oncol. 2006, 24, 524. [Google Scholar] [CrossRef]
- Alberti, K.G.; Zimmet, P.; Shaw, J. Metabolic syndrome—A new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet. Med. 2006, 23, 469–480. [Google Scholar] [CrossRef]
- Despres, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, K.S.; Kim, H.; Jeong, H.; Choi, M.J.; Yoo, H.W.; Han, T.H.; Lee, H. The relationship between metabolic syndrome and the incidence of colorectal cancer. Environ. Health Prev. Med. 2020, 25, 6. [Google Scholar] [CrossRef]
- Stocks, T.; Lukanova, A.; Bjorge, T.; Ulmer, H.; Manjer, J.; Almquist, M.; Concin, H.; Engeland, A.; Hallmans, G.; Nagel, G.; et al. Metabolic factors and the risk of colorectal cancer in 580,000 men and women in the metabolic syndrome and cancer project (Me-Can). Cancer 2011, 117, 2398–2407. [Google Scholar] [CrossRef]
- Akinyemiju, T.; Moore, J.X.; Pisu, M.; Judd, S.E.; Goodman, M.; Shikany, J.M.; Howard, V.J.; Safford, M.; Gilchrist, S.C. A Prospective Study of Obesity, Metabolic Health, and Cancer Mortality. Obesity (Silver Spring) 2018, 26, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Slawinski, C.G.V.; Barriuso, J.; Guo, H.; Renehan, A.G. Obesity and Cancer Treatment Outcomes: Interpreting the Complex Evidence. Clin. Oncol. 2020, 32, 591–608. [Google Scholar] [CrossRef]
- Griggs, J.J.; Mangu, P.B.; Anderson, H.; Balaban, E.P.; Dignam, J.J.; Hryniuk, W.M.; Morrison, V.A.; Pini, T.M.; Runowicz, C.D.; Rosner, G.L.; et al. Appropriate chemotherapy dosing for obese adult patients with cancer: American Society of Clinical Oncology clinical practice guideline. J. Clin. Oncol. 2012, 30, 1553–1561. [Google Scholar] [CrossRef]
- Jayne, D.G.; Thorpe, H.C.; Copeland, J.; Quirke, P.; Brown, J.M.; Guillou, P.J. Five-year follow-up of the Medical Research Council CLASICC trial of laparoscopically assisted versus open surgery for colorectal cancer. Br. J. Surg. 2010, 97, 1638–1645. [Google Scholar] [CrossRef]
- Wu, J.; Cohen, P.; Spiegelman, B.M. Adaptive thermogenesis in adipocytes: Is beige the new brown? Genes Dev. 2013, 27, 234–250. [Google Scholar] [CrossRef] [Green Version]
- Cypess, A.M.; White, A.P.; Vernochet, C.; Schulz, T.J.; Xue, R.; Sass, C.A.; Huang, T.L.; Roberts-Toler, C.; Weiner, L.S.; Sze, C.; et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat. Med. 2013, 19, 635–639. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Linfoot, P.; Dare, D.; Aghajanian, K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am. J. Clin. Nutr. 2003, 77, 43–50. [Google Scholar] [CrossRef]
- Minehira, K.; Bettschart, V.; Vidal, H.; Vega, N.; Di Vetta, V.; Rey, V.; Schneiter, P.; Tappy, L. Effect of carbohydrate overfeeding on whole body and adipose tissue metabolism in humans. Obes. Res. 2003, 11, 1096–1103. [Google Scholar] [CrossRef]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Warburg, O. Iron, the Oxygen-Carrier of Respiration-Ferment. Science 1925, 61, 575–582. [Google Scholar] [CrossRef]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [Green Version]
- Alkhouri, N.; Gornicka, A.; Berk, M.P.; Thapaliya, S.; Dixon, L.J.; Kashyap, S.; Schauer, P.R.; Feldstein, A.E. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J. Biol. Chem. 2010, 285, 3428–3438. [Google Scholar] [CrossRef] [Green Version]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Gesta, S.; Tseng, Y.H.; Kahn, C.R. Developmental origin of fat: Tracking obesity to its source. Cell 2007, 131, 242–256. [Google Scholar] [CrossRef] [Green Version]
- Geer, E.B.; Shen, W. Gender differences in insulin resistance, body composition, and energy balance. Gend. Med. 2009, 6 (Suppl. S1), 60–75. [Google Scholar] [CrossRef] [Green Version]
- Kyrgiou, M.; Kalliala, I.; Markozannes, G.; Gunter, M.J.; Paraskevaidis, E.; Gabra, H.; Martin-Hirsch, P.; Tsilidis, K.K. Adiposity and cancer at major anatomical sites: Umbrella review of the literature. BMJ 2017, 356, j477. [Google Scholar] [CrossRef] [Green Version]
- Bjorntorp, P. “Portal” adipose tissue as a generator of risk factors for cardiovascular disease and diabetes. Arteriosclerosis 1990, 10, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, K.A.; Lonnroth, P.; Parkkola, R.; Peltoniemi, P.; Asola, M.; Viljanen, T.; Tolvanen, T.; Knuuti, J.; Ronnemaa, T.; Huupponen, R.; et al. Glucose uptake and perfusion in subcutaneous and visceral adipose tissue during insulin stimulation in nonobese and obese humans. J. Clin. Endocrinol. Metab. 2002, 87, 3902–3910. [Google Scholar] [CrossRef]
- Lee, M.-J.; Fried, S.K. Depot-Specific Biology of Adipose Tissues: Links to Fat Distribution and Metabolic Risk. In Adipose Tissue in Health and Disease; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Hafidi, M.E.; Buelna-Chontal, M.; Sanchez-Munoz, F.; Carbo, R. Adipogenesis: A Necessary but Harmful Strategy. Int. J. Mol. Sci. 2019, 20, 3657. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Rose, D.P.; Komninou, D.; Stephenson, G.D. Obesity, adipocytokines, and insulin resistance in breast cancer. Obes. Rev. 2004, 5, 153–165. [Google Scholar] [CrossRef]
- Harvie, M.; Hooper, L.; Howell, A.H. Central obesity and breast cancer risk: A systematic review. Obes. Rev. 2003, 4, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Smith, U.; Kahn, B.B. Adipose tissue regulates insulin sensitivity: Role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med. 2016, 280, 465–475. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, T.; Craig, C.; Liu, L.F.; Perelman, D.; Allister, C.; Spielman, D.; Cushman, S.W. Adipose Cell Size and Regional Fat Deposition as Predictors of Metabolic Response to Overfeeding in Insulin-Resistant and Insulin-Sensitive Humans. Diabetes 2016, 65, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Haase, J.; Weyer, U.; Immig, K.; Kloting, N.; Bluher, M.; Eilers, J.; Bechmann, I.; Gericke, M. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 2014, 57, 562–571. [Google Scholar] [CrossRef]
- Galic, S.; Oakhill, J.S.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Iyengar, N.M.; Hudis, C.A.; Dannenberg, A.J. Targeting obesity-related adipose tissue dysfunction to prevent cancer development and progression. Semin. Oncol. 2016, 43, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, N.; Terauchi, Y.; Yamauchi, T.; Kubota, T.; Moroi, M.; Matsui, J.; Eto, K.; Yamashita, T.; Kamon, J.; Satoh, H.; et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J. Biol. Chem. 2002, 277, 25863–25866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouchi, N.; Kihara, S.; Arita, Y.; Maeda, K.; Kuriyama, H.; Okamoto, Y.; Hotta, K.; Nishida, M.; Takahashi, M.; Nakamura, T.; et al. Novel modulator for endothelial adhesion molecules: Adipocyte-derived plasma protein adiponectin. Circulation 1999, 100, 2473–2476. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Xu, S.; Piao, C.; Qiu, S.; Li, H.; Du, J. Adiponectin induces CXCL1 secretion from cancer cells and promotes tumor angiogenesis by inducing stromal fibroblast senescence. Mol. Carcinog. 2016, 55, 1796–1806. [Google Scholar] [CrossRef]
- Ouchi, N.; Kihara, S.; Funahashi, T.; Nakamura, T.; Nishida, M.; Kumada, M.; Okamoto, Y.; Ohashi, K.; Nagaretani, H.; Kishida, K.; et al. Reciprocal association of C-reactive protein with adiponectin in blood stream and adipose tissue. Circulation 2003, 107, 671–674. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Woo, Y.C.; Wang, Y.; Yeung, C.Y.; Xu, A.; Lam, K.S. Obesity, adipokines and cancer: An update. Clin. Endocrinol. (Oxf.) 2015, 83, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Sarjeant, K.; Stephens, J.M. Adipogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008417. [Google Scholar] [CrossRef] [Green Version]
- Macis, D.; Guerrieri-Gonzaga, A.; Gandini, S. Circulating adiponectin and breast cancer risk: A systematic review and meta-analysis. Int. J. Epidemiol. 2014, 43, 1226–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, R.K.; Kim, W.J.; Lee, S.A. Association between obesity-related adipokines and colorectal cancer: A case-control study and meta-analysis. World J. Gastroenterol. 2014, 20, 7941–7949. [Google Scholar] [CrossRef]
- Lonnqvist, F.; Nordfors, L.; Jansson, M.; Thorne, A.; Schalling, M.; Arner, P. Leptin secretion from adipose tissue in women. Relationship to plasma levels and gene expression. J. Clin. Invest. 1997, 99, 2398–2404. [Google Scholar] [CrossRef] [Green Version]
- Palhinha, L.; Liechocki, S.; Hottz, E.D.; Pereira, J.; de Almeida, C.J.; Moraes-Vieira, P.M.M.; Bozza, P.T.; Maya-Monteiro, C.M. Leptin Induces Proadipogenic and Proinflammatory Signaling in Adipocytes. Front. Endocrinol. (Lausanne) 2019, 10, 841. [Google Scholar] [CrossRef] [Green Version]
- Sierra-Honigmann, M.R.; Nath, A.K.; Murakami, C.; Garcia-Cardena, G.; Papapetropoulos, A.; Sessa, W.C.; Madge, L.A.; Schechner, J.S.; Schwabb, M.B.; Polverini, P.J.; et al. Biological action of leptin as an angiogenic factor. Science 1998, 281, 1683–1686. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Huang, Y.; Wang, L.; Wang, H.; Pang, X.; Li, K.; Dang, W.; Tang, H.; Wei, L.; Su, M.; et al. Leptin promotes migration and invasion of breast cancer cells by stimulating IL-8 production in M2 macrophages. Oncotarget 2016, 7, 65441–65453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delort, L.; Rossary, A.; Farges, M.C.; Vasson, M.P.; Caldefie-Chezet, F. Leptin, adipocytes and breast cancer: Focus on inflammation and anti-tumor immunity. Life Sci. 2015, 140, 37–48. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Ennis, M.; Fantus, I.G.; Pritchard, K.I.; Trudeau, M.E.; Koo, J.; Hood, N. Is leptin a mediator of adverse prognostic effects of obesity in breast cancer? J. Clin. Oncol. 2005, 23, 6037–6042. [Google Scholar] [CrossRef]
- Lazar, M.A. How obesity causes diabetes: Not a tall tale. Science 2005, 307, 373–375. [Google Scholar] [CrossRef] [Green Version]
- Bachelot, T.; Ray-Coquard, I.; Menetrier-Caux, C.; Rastkha, M.; Duc, A.; Blay, J.Y. Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. Br. J. Cancer 2003, 88, 1721–1726. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 2007, 117, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Shivappa, N.; Hebert, J.R.; Rietzschel, E.R.; De Buyzere, M.L.; Langlois, M.; Debruyne, E.; Marcos, A.; Huybrechts, I. Associations between dietary inflammatory index and inflammatory markers in the Asklepios Study. Br. J. Nutr. 2015, 113, 665–671. [Google Scholar] [CrossRef] [Green Version]
- Yeung, C.Y.; Tso, A.W.; Xu, A.; Wang, Y.; Woo, Y.C.; Lam, T.H.; Lo, S.V.; Fong, C.H.; Wat, N.M.; Woo, J.; et al. Pro-inflammatory adipokines as predictors of incident cancers in a Chinese cohort of low obesity prevalence in Hong Kong. PLoS ONE 2013, 8, e78594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Cassol, E.; Poli, G. Macrophage polarization in health and disease. Sci. World J. 2011, 11, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Laurent, V.; Guerard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y.; et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Bulun, S.E.; Mahendroo, M.S.; Simpson, E.R. Aromatase gene expression in adipose tissue: Relationship to breast cancer. J. Steroid Biochem. Mol. Biol. 1994, 49, 319–326. [Google Scholar] [CrossRef]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Invest. 2000, 106, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Druta, M.; Shibata, D.; Coppola, D.; Boler, I.; Elahi, A.; Reich, R.R.; Siegel, E.; Extermann, M. Metabolic syndrome and colorectal cancer: Is hyperinsulinemia/insulin receptor-mediated angiogenesis a critical process? J. Geriatr. Oncol. 2014, 5, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Papa, V.; Belfiore, A. Insulin receptors in breast cancer: Biological and clinical role. J. Endocrinol. Invest. 1996, 19, 324–333. [Google Scholar] [CrossRef]
- Bianchini, F.; Kaaks, R.; Vainio, H. Weight control and physical activity in cancer prevention. Obes. Rev. 2002, 3, 5–8. [Google Scholar] [CrossRef]
- Yu, H.; Rohan, T. Role of the insulin-like growth factor family in cancer development and progression. J. Natl. Cancer Inst. 2000, 92, 1472–1489. [Google Scholar] [CrossRef]
- Khandwala, H.M.; McCutcheon, I.E.; Flyvbjerg, A.; Friend, K.E. The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocr. Rev. 2000, 21, 215–244. [Google Scholar] [CrossRef]
- Kawai, M.; Namba, N.; Mushiake, S.; Etani, Y.; Nishimura, R.; Makishima, M.; Ozono, K. Growth hormone stimulates adipogenesis of 3T3-L1 cells through activation of the Stat5A/5B-PPARgamma pathway. J. Mol. Endocrinol. 2007, 38, 19–34. [Google Scholar] [CrossRef] [Green Version]
- Leung, K.C.; Doyle, N.; Ballesteros, M.; Waters, M.J.; Ho, K.K. Insulin regulation of human hepatic growth hormone receptors: Divergent effects on biosynthesis and surface translocation. J. Clin. Endocrinol. Metab. 2000, 85, 4712–4720. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Renehan, A.G.; Frystyk, J.; Flyvbjerg, A. Obesity and cancer risk: The role of the insulin-IGF axis. Trends Endocrinol. Metab. 2006, 17, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Carreras-Torres, R.; Song, M.; Chan, A.T.; Martin, R.M.; Papadimitriou, N.; Dimou, N.; Tsilidis, K.K.; Banbury, B.; Bradbury, K.E.; et al. Circulating Levels of Insulin-like Growth Factor 1 and Insulin-like Growth Factor Binding Protein 3 Associate With Risk of Colorectal Cancer Based on Serologic and Mendelian Randomization Analyses. Gastroenterology 2020, 158, 1300–1312.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, N.E.; Key, T.J.; Appleby, P.N.; Travis, R.C.; Roddam, A.W.; Rinaldi, S.; Egevad, L.; Rohrmann, S.; Linseisen, J.; Pischon, T.; et al. Serum insulin-like growth factor (IGF)-I and IGF-binding protein-3 concentrations and prostate cancer risk: Results from the European Prospective Investigation into Cancer and Nutrition. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1121–1127. [Google Scholar] [CrossRef] [Green Version]
- Renehan, A.G.; Zwahlen, M.; Minder, C.; O’Dwyer, S.T.; Shalet, S.M.; Egger, M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 2004, 363, 1346–1353. [Google Scholar] [CrossRef]
- Purohit, A.; Reed, M.J. Regulation of estrogen synthesis in postmenopausal women. Steroids 2002, 67, 979–983. [Google Scholar] [CrossRef]
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sestak, I.; Distler, W.; Forbes, J.F.; Dowsett, M.; Howell, A.; Cuzick, J. Effect of body mass index on recurrences in tamoxifen and anastrozole treated women: An exploratory analysis from the ATAC trial. J. Clin. Oncol. 2010, 28, 3411–3415. [Google Scholar] [CrossRef]
- Shaw, E.; Farris, M.; McNeil, J.; Friedenreich, C. Obesity and Endometrial Cancer. In Recent Results in Cancer Research; Springer: Cham, Switzerland, 2016; Volume 208, pp. 107–136. [Google Scholar] [CrossRef]
- Furberg, A.S.; Veierod, M.B.; Wilsgaard, T.; Bernstein, L.; Thune, I. Serum high-density lipoprotein cholesterol, metabolic profile, and breast cancer risk. J. Natl. Cancer Inst. 2004, 96, 1152–1160. [Google Scholar] [CrossRef]
- Kliemann, N.; Murphy, N.; Viallon, V.; Freisling, H.; Tsilidis, K.K.; Rinaldi, S.; Mancini, F.R.; Fagherazzi, G.; Boutron-Ruault, M.C.; Boeing, H.; et al. Predicted basal metabolic rate and cancer risk in the European Prospective Investigation into Cancer and Nutrition. Int. J. Cancer 2020, 147, 648–661. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Luo, J.; Margolis, K.L.; Adami, H.O.; LaCroix, A.; Ye, W.; Women’s Health Initiative Investigators. Obesity and risk of pancreatic cancer among postmenopausal women: The Women’s Health Initiative (United States). Br. J. Cancer 2008, 99, 527–531. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Punyanitya, M.; Wang, Z.; Gallagher, D.; St-Onge, M.P.; Albu, J.; Heymsfield, S.B.; Heshka, S. Total body skeletal muscle and adipose tissue volumes: Estimation from a single abdominal cross-sectional image. J. Appl. Physiol. (1985) 2004, 97, 2333–2338. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Shin, Y.; Park, J.; Kim, H.; Lee, I.S.; Seo, D.W.; Huh, J.; Lee, T.Y.; Park, T.; Lee, J.; et al. Development and Validation of a Deep Learning System for Segmentation of Abdominal Muscle and Fat on Computed Tomography. Korean J. Radiol. 2020, 21, 88–100. [Google Scholar] [CrossRef]
- Paris, M.T. Body Composition Analysis of Computed Tomography Scans in Clinical Populations: The Role of Deep Learning. Lifestyle Genom. 2020, 13, 28–31. [Google Scholar] [CrossRef]
- Wu, S.; Liu, J.; Wang, X.; Li, M.; Gan, Y.; Tang, Y. Association of obesity and overweight with overall survival in colorectal cancer patients: A meta-analysis of 29 studies. Cancer Causes Control. 2014, 25, 1489–1502. [Google Scholar] [CrossRef]
- Campbell, P.T.; Newton, C.C.; Dehal, A.N.; Jacobs, E.J.; Patel, A.V.; Gapstur, S.M. Impact of body mass index on survival after colorectal cancer diagnosis: The Cancer Prevention Study-II Nutrition Cohort. J. Clin. Oncol. 2012, 30, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Charvat, H.; Freisling, H.; Noh, H.; Adami, H.O.; Soerjomataram, I.; Weiderpass, E. Adult Overweight and Survival from Breast and Colorectal Cancer in Swedish Women. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1518–1524. [Google Scholar] [CrossRef]
- Yumuk, V.; Tsigos, C.; Fried, M.; Schindler, K.; Busetto, L.; Micic, D.; Toplak, H. European Guidelines for Obesity Management in Adults. Obes. Facts 2015, 8, 402–424. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jacobs, E.J.; Patel, A.V.; Rodriguez, C.; McCullough, M.L.; Thun, M.J.; Calle, E.E. A prospective study of waist circumference and body mass index in relation to colorectal cancer incidence. Cancer Causes Control 2008, 19, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Pischon, T.; Lahmann, P.H.; Boeing, H.; Friedenreich, C.; Norat, T.; Tjonneland, A.; Halkjaer, J.; Overvad, K.; Clavel-Chapelon, F.; Boutron-Ruault, M.C.; et al. Body size and risk of colon and rectal cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). J. Natl. Cancer Inst. 2006, 98, 920–931. [Google Scholar] [CrossRef]
- Pischon, T.; Boeing, H.; Hoffmann, K.; Bergmann, M.; Schulze, M.B.; Overvad, K.; van der Schouw, Y.T.; Spencer, E.; Moons, K.G.; Tjonneland, A.; et al. General and abdominal adiposity and risk of death in Europe. N. Engl. J. Med. 2008, 359, 2105–2120. [Google Scholar] [CrossRef] [Green Version]
- Arslan, A.A.; Helzlsouer, K.J.; Kooperberg, C.; Shu, X.O.; Steplowski, E.; Bueno-de-Mesquita, H.B.; Fuchs, C.S.; Gross, M.D.; Jacobs, E.J.; Lacroix, A.Z.; et al. Anthropometric measures, body mass index, and pancreatic cancer: A pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan). Arch. Intern. Med. 2010, 170, 791–802. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Han, C.T.; Zhang, G.M.; Zhang, C.Z.; Yang, W.Y.; Shen, Y.; Vidal, A.C.; Freedland, S.J.; Zhu, Y.; Ye, D.W. Waist-hip Ratio (WHR), a Better Predictor for Prostate Cancer than Body Mass Index (BMI): Results from a Chinese Hospital-based Biopsy Cohort. Sci. Rep. 2017, 7, 43551. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Liu, L.; Cui, S.; Tian, F.; Fan, Z.; Geng, C.; Cao, X.; Yang, Z.; Wang, X.; Liang, H.; et al. Distinct Effects of Body Mass Index and Waist/Hip Ratio on Risk of Breast Cancer by Joint Estrogen and Progestogen Receptor Status: Results from a Case-Control Study in Northern and Eastern China and Implications for Chemoprevention. Oncologist 2017, 22, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Berrington de Gonzalez, A.; Spencer, E.A.; Bueno-de-Mesquita, H.B.; Roddam, A.; Stolzenberg-Solomon, R.; Halkjaer, J.; Tjonneland, A.; Overvad, K.; Clavel-Chapelon, F.; Boutron-Ruault, M.C.; et al. Anthropometry, physical activity, and the risk of pancreatic cancer in the European prospective investigation into cancer and nutrition. Cancer Epidemiol. Biomark. Prev. 2006, 15, 879–885. [Google Scholar] [CrossRef] [Green Version]
- Ardesch, F.H.; Ruiter, R.; Mulder, M.; Lahousse, L.; Stricker, B.H.C.; Kiefte-de Jong, J.C. The Obesity Paradox in Lung Cancer: Associations With Body Size Versus Body Shape. Front. Oncol. 2020, 10, 591110. [Google Scholar] [CrossRef]
- Keum, N.; Greenwood, D.C.; Lee, D.H.; Kim, R.; Aune, D.; Ju, W.; Hu, F.B.; Giovannucci, E.L. Adult weight gain and adiposity-related cancers: A dose-response meta-analysis of prospective observational studies. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Ouellet, V.; Labbe, S.M.; Blondin, D.P.; Phoenix, S.; Guerin, B.; Haman, F.; Turcotte, E.E.; Richard, D.; Carpentier, A.C. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J. Clin. Invest. 2012, 122, 545–552. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef] [Green Version]
- Van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Vijgen, G.H.; Bouvy, N.D.; Teule, G.J.; Brans, B.; Hoeks, J.; Schrauwen, P.; van Marken Lichtenbelt, W.D. Increase in brown adipose tissue activity after weight loss in morbidly obese subjects. J. Clin. Endocrinol. Metab. 2012, 97, E1229–E1233. [Google Scholar] [CrossRef] [Green Version]
- Contreras, C.; Gonzalez, F.; Ferno, J.; Dieguez, C.; Rahmouni, K.; Nogueiras, R.; Lopez, M. The brain and brown fat. Ann. Med. 2015, 47, 150–168. [Google Scholar] [CrossRef] [Green Version]
- Nedergaard, J.; Cannon, B. The browning of white adipose tissue: Some burning issues. Cell Metab. 2014, 20, 396–407. [Google Scholar] [CrossRef] [Green Version]
- Shabalina, I.G.; Petrovic, N.; de Jong, J.M.; Kalinovich, A.V.; Cannon, B.; Nedergaard, J. UCP1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep. 2013, 5, 1196–1203. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Maretich, P.; Kajimura, S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends Endocrinol. Metab. 2018, 29, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Seale, P. Control of brown and beige fat development. Nat. Rev. Mol. Cell Biol. 2016, 17, 691–702. [Google Scholar] [CrossRef]
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartelt, A.; Bruns, O.T.; Reimer, R.; Hohenberg, H.; Ittrich, H.; Peldschus, K.; Kaul, M.G.; Tromsdorf, U.I.; Weller, H.; Waurisch, C.; et al. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 2011, 17, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanford, K.I.; Middelbeek, R.J.; Townsend, K.L.; An, D.; Nygaard, E.B.; Hitchcox, K.M.; Markan, K.R.; Nakano, K.; Hirshman, M.F.; Tseng, Y.H.; et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J. Clin. Invest. 2013, 123, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Commins, S.P.; Watson, P.M.; Padgett, M.A.; Dudley, A.; Argyropoulos, G.; Gettys, T.W. Induction of uncoupling protein expression in brown and white adipose tissue by leptin. Endocrinology 1999, 140, 292–300. [Google Scholar] [CrossRef]
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Munzberg, H.; Zhang, Z.Y.; Kahn, B.B.; et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 2015, 160, 88–104. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Yoo, H.; Bosco, C.; Lee, B.; Feng, G.S.; Schaack, J.; Chi, N.W.; Shao, J. Adiponectin reduces thermogenesis by inhibiting brown adipose tissue activation in mice. Diabetologia 2014, 57, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Nisoli, E.; Briscini, L.; Giordano, A.; Tonello, C.; Wiesbrock, S.M.; Uysal, K.T.; Cinti, S.; Carruba, M.O.; Hotamisligil, G.S. Tumor necrosis factor alpha mediates apoptosis of brown adipocytes and defective brown adipocyte function in obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 8033–8038. [Google Scholar] [CrossRef] [Green Version]
- Seale, P.; Conroe, H.M.; Estall, J.; Kajimura, S.; Frontini, A.; Ishibashi, J.; Cohen, P.; Cinti, S.; Spiegelman, B.M. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Invest. 2011, 121, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Montanari, T.; Poscic, N.; Colitti, M. Factors involved in white-to-brown adipose tissue conversion and in thermogenesis: A review. Obes. Rev. 2017, 18, 495–513. [Google Scholar] [CrossRef]
- Rothwell, N.J.; Stock, M.J. A role for brown adipose tissue in diet-induced thermogenesis. Obes. Res. 1997, 5, 650–656. [Google Scholar] [CrossRef]
- Lowell, B.B.; Spiegelman, B.M. Towards a molecular understanding of adaptive thermogenesis. Nature 2000, 404, 652–660. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, M.; Ning, G.; Gu, W.; Su, T.; Xu, M.; Li, B.; Wang, W. Brown adipose tissue in humans is activated by elevated plasma catecholamines levels and is inversely related to central obesity. PLoS ONE 2011, 6, e21006. [Google Scholar] [CrossRef] [Green Version]
- Cypess, A.M.; Weiner, L.S.; Roberts-Toler, C.; Franquet Elia, E.; Kessler, S.H.; Kahn, P.A.; English, J.; Chatman, K.; Trauger, S.A.; Doria, A.; et al. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab. 2015, 21, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Loprinzi, P.D.; Yang, L.; Liu, J.; Liu, S.; Zou, L. The Beneficial Effects of Traditional Chinese Exercises for Adults with Low Back Pain: A Meta-Analysis of Randomized Controlled Trials. Medicina (Kaunas) 2019, 55, 118. [Google Scholar] [CrossRef] [Green Version]
- Rachid, T.L.; Penna-de-Carvalho, A.; Bringhenti, I.; Aguila, M.B.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Fenofibrate (PPARalpha agonist) induces beige cell formation in subcutaneous white adipose tissue from diet-induced male obese mice. Mol. Cell. Endocrinol. 2015, 402, 86–94. [Google Scholar] [CrossRef]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar] [CrossRef] [Green Version]
- Beiroa, D.; Imbernon, M.; Gallego, R.; Senra, A.; Herranz, D.; Villarroya, F.; Serrano, M.; Ferno, J.; Salvador, J.; Escalada, J.; et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes 2014, 63, 3346–3358. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Goto, T.; Yu, R.; Uchida, K.; Tominaga, M.; Kano, Y.; Takahashi, N.; Kawada, T. Fish oil intake induces UCP1 upregulation in brown and white adipose tissue via the sympathetic nervous system. Sci. Rep. 2015, 5, 18013. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Liang, X.; Yang, Q.; Fu, X.; Rogers, C.J.; Zhu, M.; Rodgers, B.D.; Jiang, Q.; Dodson, M.V.; Du, M. Resveratrol induces brown-like adipocyte formation in white fat through activation of AMP-activated protein kinase (AMPK) alpha1. Int. J. Obes. (Lond.) 2015, 39, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Zamorano, N.; Fabbiano, S.; Chevalier, C.; Stojanovic, O.; Colin, D.J.; Stevanovic, A.; Veyrat-Durebex, C.; Tarallo, V.; Rigo, D.; Germain, S.; et al. Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat. Med. 2015, 21, 1497–1501. [Google Scholar] [CrossRef]
- Li, G.; Xie, C.; Lu, S.; Nichols, R.G.; Tian, Y.; Li, L.; Patel, D.; Ma, Y.; Brocker, C.N.; Yan, T.; et al. Intermittent Fasting Promotes White Adipose Browning and Decreases Obesity by Shaping the Gut Microbiota. Cell Metab. 2017, 26, 672–685.e4. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, F.; Biessy, C.; Ferrari, P.; Freisling, H.; Rinaldi, S.; Chajes, V.; Dahm, C.C.; Overvad, K.; Dossus, L.; Lagiou, P.; et al. Healthy Lifestyle and Risk of Cancer in the European Prospective Investigation Into Cancer and Nutrition Cohort Study. Medicine (Baltimore) 2016, 95, e2850. [Google Scholar] [CrossRef] [Green Version]
- Naudin, S.; Viallon, V.; Hashim, D.; Freisling, H.; Jenab, M.; Weiderpass, E.; Perrier, F.; McKenzie, F.; Bueno-de-Mesquita, H.B.; Olsen, A.; et al. Healthy lifestyle and the risk of pancreatic cancer in the EPIC study. Eur. J. Epidemiol. 2020, 35, 975–986. [Google Scholar] [CrossRef]
- Look, A.R.G.; Wing, R.R. Long-term effects of a lifestyle intervention on weight and cardiovascular risk factors in individuals with type 2 diabetes mellitus: Four-year results of the Look AHEAD trial. Arch. Intern. Med. 2010, 170, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Hendryx, M.; Manson, J.E.; Figueiredo, J.C.; LeBlanc, E.S.; Barrington, W.; Rohan, T.E.; Howard, B.V.; Reding, K.; Ho, G.Y.; et al. Intentional Weight Loss and Obesity-Related Cancer Risk. JNCI Cancer Spectr. 2019, 3, pkz054. [Google Scholar] [CrossRef]
- World Cancer Research Fund; American Institute for Cancer Research. Diet, Nutrition, Physical Activity and Cancer: A Global Perspective. Available online: https://www.wcrf.org/diet-and-cancer/ (accessed on 10 June 2021).
- Thomas, E.L.; Brynes, A.E.; McCarthy, J.; Goldstone, A.P.; Hajnal, J.V.; Saeed, N.; Frost, G.; Bell, J.D. Preferential loss of visceral fat following aerobic exercise, measured by magnetic resonance imaging. Lipids 2000, 35, 769–776. [Google Scholar] [CrossRef]
- Irving, B.A.; Davis, C.K.; Brock, D.W.; Weltman, J.Y.; Swift, D.; Barrett, E.J.; Gaesser, G.A.; Weltman, A. Effect of exercise training intensity on abdominal visceral fat and body composition. Med. Sci. Sports Exerc. 2008, 40, 1863–1872. [Google Scholar] [CrossRef] [Green Version]
- Slentz, C.A.; Duscha, B.D.; Johnson, J.L.; Ketchum, K.; Aiken, L.B.; Samsa, G.P.; Houmard, J.A.; Bales, C.W.; Kraus, W.E. Effects of the amount of exercise on body weight, body composition, and measures of central obesity: STRRIDE—A randomized controlled study. Arch. Intern. Med. 2004, 164, 31–39. [Google Scholar] [CrossRef]
- Vissers, D.; Hens, W.; Taeymans, J.; Baeyens, J.P.; Poortmans, J.; Van Gaal, L. The effect of exercise on visceral adipose tissue in overweight adults: A systematic review and meta-analysis. PLoS ONE 2013, 8, e56415. [Google Scholar] [CrossRef] [Green Version]
- Gollisch, K.S.; Brandauer, J.; Jessen, N.; Toyoda, T.; Nayer, A.; Hirshman, M.F.; Goodyear, L.J. Effects of exercise training on subcutaneous and visceral adipose tissue in normal- and high-fat diet-fed rats. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E495–E504. [Google Scholar] [CrossRef]
- WHO. Weight Control and Physical Activity, 1st ed.; IARC Publications: Lyon, France, 2002; Volume 6. [Google Scholar]
- Hojman, P.; Gehl, J.; Christensen, J.F.; Pedersen, B.K. Molecular Mechanisms Linking Exercise to Cancer Prevention and Treatment. Cell Metab. 2018, 27, 10–21. [Google Scholar] [CrossRef] [Green Version]
- Munt, A.E.; Partridge, S.R.; Allman-Farinelli, M. The barriers and enablers of healthy eating among young adults: A missing piece of the obesity puzzle: A scoping review. Obes. Rev. 2017, 18, 1–17. [Google Scholar] [CrossRef]
- Donaldson, M.S. Nutrition and cancer: A review of the evidence for an anti-cancer diet. Nutr. J. 2004, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Sofi, F.; Cesari, F.; Abbate, R.; Gensini, G.F.; Casini, A. Adherence to Mediterranean diet and health status: Meta-analysis. BMJ 2008, 337, a1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, L.; Chen, L.; White, D.L.; Tinker, L.; Chlebowski, R.T.; Van Horn, L.V.; Richardson, P.; Lane, D.; Sangi-Haghpeykar, H.; El-Serag, H.B. Low-fat Dietary Pattern and Pancreatic Cancer Risk in the Women’s Health Initiative Dietary Modification Randomized Controlled Trial. J. Natl. Cancer Inst. 2018, 110. [Google Scholar] [CrossRef] [PubMed]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Trichopoulou, A.; Martinez-Gonzalez, M.A.; Tong, T.Y.; Forouhi, N.G.; Khandelwal, S.; Prabhakaran, D.; Mozaffarian, D.; de Lorgeril, M. Definitions and potential health benefits of the Mediterranean diet: Views from experts around the world. BMC Med. 2014, 12, 112. [Google Scholar] [CrossRef] [Green Version]
- De Santis, S.; Cariello, M.; Piccinin, E.; Sabba, C.; Moschetta, A. Extra Virgin Olive Oil: Lesson from Nutrigenomics. Nutrients 2019, 11, 2085. [Google Scholar] [CrossRef] [Green Version]
- Gadgil, M.D.; Appel, L.J.; Yeung, E.; Anderson, C.A.; Sacks, F.M.; Miller, E.R., 3rd. The effects of carbohydrate, unsaturated fat, and protein intake on measures of insulin sensitivity: Results from the OmniHeart trial. Diabetes Care 2013, 36, 1132–1137. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cheng, M.; Zhao, M.; Ge, A.; Guo, F.; Zhang, M.; Yang, Y.; Liu, L.; Yang, N. Differential effects of high-fat-diet rich in lard oil or soybean oil on osteopontin expression and inflammation of adipose tissue in diet-induced obese rats. Eur. J. Nutr. 2013, 52, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Bendall, C.L.; Mayr, H.L.; Opie, R.S.; Bes-Rastrollo, M.; Itsiopoulos, C.; Thomas, C.J. Central obesity and the Mediterranean diet: A systematic review of intervention trials. Crit. Rev. Food Sci. Nutr. 2018, 58, 3070–3084. [Google Scholar] [CrossRef] [PubMed]
- Rumawas, M.E.; Meigs, J.B.; Dwyer, J.T.; McKeown, N.M.; Jacques, P.F. Mediterranean-style dietary pattern, reduced risk of metabolic syndrome traits, and incidence in the Framingham Offspring Cohort. Am. J. Clin. Nutr. 2009, 90, 1608–1614. [Google Scholar] [CrossRef] [Green Version]
- Bertoli, S.; Leone, A.; Vignati, L.; Bedogni, G.; Martinez-Gonzalez, M.A.; Bes-Rastrollo, M.; Spadafranca, A.; Vanzulli, A.; Battezzati, A. Adherence to the Mediterranean diet is inversely associated with visceral abdominal tissue in Caucasian subjects. Clin. Nutr. 2015, 34, 1266–1272. [Google Scholar] [CrossRef]
- Sofi, F.; Dinu, M.; Pagliai, G.; Cesari, F.; Gori, A.M.; Sereni, A.; Becatti, M.; Fiorillo, C.; Marcucci, R.; Casini, A. Low-Calorie Vegetarian Versus Mediterranean Diets for Reducing Body Weight and Improving Cardiovascular Risk Profile: CARDIVEG Study (Cardiovascular Prevention With Vegetarian Diet). Circulation 2018, 137, 1103–1113. [Google Scholar] [CrossRef]
- Brehm, B.J.; Lattin, B.L.; Summer, S.S.; Boback, J.A.; Gilchrist, G.M.; Jandacek, R.J.; D’Alessio, D.A. One-year comparison of a high-monounsaturated fat diet with a high-carbohydrate diet in type 2 diabetes. Diabetes Care 2009, 32, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Shai, I.; Schwarzfuchs, D.; Henkin, Y.; Shahar, D.R.; Witkow, S.; Greenberg, I.; Golan, R.; Fraser, D.; Bolotin, A.; Vardi, H.; et al. Weight loss with a low-carbohydrate, Mediterranean, or low-fat diet. N. Engl. J. Med. 2008, 359, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Estruch, R.; Martinez-Gonzalez, M.A.; Corella, D.; Salas-Salvado, J.; Fito, M.; Chiva-Blanch, G.; Fiol, M.; Gomez-Gracia, E.; Aros, F.; Lapetra, J.; et al. Effect of a high-fat Mediterranean diet on bodyweight and waist circumference: A prespecified secondary outcomes analysis of the PREDIMED randomised controlled trial. Lancet Diabetes Endocrinol. 2019, 7, e6–e17. [Google Scholar] [CrossRef]
- Smith, S.R.; Zachwieja, J.J. Visceral adipose tissue: A critical review of intervention strategies. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couto, E.; Boffetta, P.; Lagiou, P.; Ferrari, P.; Buckland, G.; Overvad, K.; Dahm, C.C.; Tjonneland, A.; Olsen, A.; Clavel-Chapelon, F.; et al. Mediterranean dietary pattern and cancer risk in the EPIC cohort. Br. J. Cancer 2011, 104, 1493–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamia, C.; Lagiou, P.; Buckland, G.; Grioni, S.; Agnoli, C.; Taylor, A.J.; Dahm, C.C.; Overvad, K.; Olsen, A.; Tjonneland, A.; et al. Mediterranean diet and colorectal cancer risk: Results from a European cohort. Eur. J. Epidemiol. 2013, 28, 317–328. [Google Scholar] [CrossRef]
- Buckland, G.; Agudo, A.; Lujan, L.; Jakszyn, P.; Bueno-de-Mesquita, H.B.; Palli, D.; Boeing, H.; Carneiro, F.; Krogh, V.; Sacerdote, C.; et al. Adherence to a Mediterranean diet and risk of gastric adenocarcinoma within the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort study. Am. J. Clin. Nutr. 2010, 91, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Schneider, L.; Su, L.J.; Arab, L.; Bensen, J.T.; Farnan, L.; Fontham, E.T.H.; Song, L.; Hussey, J.; Merchant, A.T.; Mohler, J.L.; et al. Dietary patterns based on the Mediterranean diet and DASH diet are inversely associated with high aggressive prostate cancer in PCaP. Ann. Epidemiol. 2019, 29, 16–22.e11. [Google Scholar] [CrossRef]
- Turati, F.; Carioli, G.; Bravi, F.; Ferraroni, M.; Serraino, D.; Montella, M.; Giacosa, A.; Toffolutti, F.; Negri, E.; Levi, F.; et al. Mediterranean Diet and Breast Cancer Risk. Nutrients 2018, 10, 326. [Google Scholar] [CrossRef] [Green Version]
- Van den Brandt, P.A.; Schulpen, M. Mediterranean diet adherence and risk of postmenopausal breast cancer: Results of a cohort study and meta-analysis. Int. J. Cancer 2017, 140, 2220–2231. [Google Scholar] [CrossRef]
- Mitrou, P.N.; Kipnis, V.; Thiebaut, A.C.; Reedy, J.; Subar, A.F.; Wirfalt, E.; Flood, A.; Mouw, T.; Hollenbeck, A.R.; Leitzmann, M.F.; et al. Mediterranean dietary pattern and prediction of all-cause mortality in a US population: Results from the NIH-AARP Diet and Health Study. Arch. Intern. Med. 2007, 167, 2461–2468. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Schwedhelm, C.; Galbete, C.; Hoffmann, G. Adherence to Mediterranean Diet and Risk of Cancer: An Updated Systematic Review and Meta-Analysis. Nutrients 2017, 9, 1063. [Google Scholar] [CrossRef]
- Farinetti, A.; Zurlo, V.; Manenti, A.; Coppi, F.; Mattioli, A.V. Mediterranean diet and colorectal cancer: A systematic review. Nutrition 2017, 43–44, 83–88. [Google Scholar] [CrossRef]
- Bingham, S.A.; Day, N.E.; Luben, R.; Ferrari, P.; Slimani, N.; Norat, T.; Clavel-Chapelon, F.; Kesse, E.; Nieters, A.; Boeing, H.; et al. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): An observational study. Lancet 2003, 361, 1496–1501. [Google Scholar] [CrossRef]
- Bradbury, K.E.; Appleby, P.N.; Key, T.J. Fruit, vegetable, and fiber intake in relation to cancer risk: Findings from the European Prospective Investigation into Cancer and Nutrition (EPIC). Am. J. Clin. Nutr. 2014, 100 (Suppl. S1), 394S–398S. [Google Scholar] [CrossRef] [Green Version]
- Pitsavos, C.; Panagiotakos, D.B.; Tzima, N.; Chrysohoou, C.; Economou, M.; Zampelas, A.; Stefanadis, C. Adherence to the Mediterranean diet is associated with total antioxidant capacity in healthy adults: The ATTICA study. Am. J. Clin. Nutr. 2005, 82, 694–699. [Google Scholar] [CrossRef]
- Owen, R.W.; Giacosa, A.; Hull, W.E.; Haubner, R.; Spiegelhalder, B.; Bartsch, H. The antioxidant/anticancer potential of phenolic compounds isolated from olive oil. Eur. J. Cancer 2000, 36, 1235–1247. [Google Scholar] [CrossRef]
- Chrysohoou, C.; Panagiotakos, D.B.; Pitsavos, C.; Das, U.N.; Stefanadis, C. Adherence to the Mediterranean diet attenuates inflammation and coagulation process in healthy adults: The ATTICA Study. J. Am. Coll. Cardiol. 2004, 44, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Scalbert, A.; Brennan, L.; Fiehn, O.; Hankemeier, T.; Kristal, B.S.; van Ommen, B.; Pujos-Guillot, E.; Verheij, E.; Wishart, D.; Wopereis, S. Mass-spectrometry-based metabolomics: Limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics 2009, 5, 435–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, J.A.; Fensom, G.K.; Rinaldi, S.; Scalbert, A.; Appleby, P.N.; Achaintre, D.; Gicquiau, A.; Gunter, M.J.; Ferrari, P.; Kaaks, R.; et al. Patterns in metabolite profile are associated with risk of more aggressive prostate cancer: A prospective study of 3057 matched case-control sets from EPIC. Int. J. Cancer 2020, 146, 720–730. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Turati, F.; Lagiou, P.; Trichopoulos, D.; Augustin, L.S.; La Vecchia, C.; Trichopoulou, A. Mediterranean diet and glycaemic load in relation to incidence of type 2 diabetes: Results from the Greek cohort of the population-based European Prospective Investigation into Cancer and Nutrition (EPIC). Diabetologia 2013, 56, 2405–2413. [Google Scholar] [CrossRef] [Green Version]
- Kastorini, C.M.; Milionis, H.J.; Esposito, K.; Giugliano, D.; Goudevenos, J.A.; Panagiotakos, D.B. The effect of Mediterranean diet on metabolic syndrome and its components: A meta-analysis of 50 studies and 534,906 individuals. J. Am. Coll. Cardiol. 2011, 57, 1299–1313. [Google Scholar] [CrossRef] [Green Version]
- Klement, R.J.; Pazienza, V. Impact of Different Types of Diet on Gut Microbiota Profiles and Cancer Prevention and Treatment. Medicina (Kaunas) 2019, 55, 84. [Google Scholar] [CrossRef] [Green Version]
- Vitaglione, P.; Mennella, I.; Ferracane, R.; Rivellese, A.A.; Giacco, R.; Ercolini, D.; Gibbons, S.M.; La Storia, A.; Gilbert, J.A.; Jonnalagadda, S.; et al. Whole-grain wheat consumption reduces inflammation in a randomized controlled trial on overweight and obese subjects with unhealthy dietary and lifestyle behaviors: Role of polyphenols bound to cereal dietary fiber. Am. J. Clin. Nutr. 2015, 101, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Caesar, R.; Tremaroli, V.; Kovatcheva-Datchary, P.; Cani, P.D.; Backhed, F. Crosstalk between Gut Microbiota and Dietary Lipids Aggravates WAT Inflammation through TLR Signaling. Cell Metab. 2015, 22, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [Green Version]
- Frazier, T.H.; DiBaise, J.K.; McClain, C.J. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN J. Parenter. Enteral. Nutr. 2011, 35, 14S–20S. [Google Scholar] [CrossRef]
- Fabbiano, S.; Suarez-Zamorano, N.; Rigo, D.; Veyrat-Durebex, C.; Stevanovic Dokic, A.; Colin, D.J.; Trajkovski, M. Caloric Restriction Leads to Browning of White Adipose Tissue through Type 2 Immune Signaling. Cell Metab. 2016, 24, 434–446. [Google Scholar] [CrossRef]
- Rodrigues, M.O.M.; Evangelista-Silva, P.H.; Neves, N.N.; Moreno, L.G.; Santos, C.S.; Rocha, K.L.S.; Ottone, V.O.; Batista-da-Silva, B.; Dias-Peixoto, M.F.; Magalhaes, F.C.; et al. Caloric restriction-induced weight loss with a high-fat diet does not fully recover visceral adipose tissue inflammation in previously obese C57BL/6 mice. Appl. Physiol. Nutr. Metab. 2020, 45, 1353–1359. [Google Scholar] [CrossRef]
- Barquissau, V.; Leger, B.; Beuzelin, D.; Martins, F.; Amri, E.Z.; Pisani, D.F.; Saris, W.H.M.; Astrup, A.; Maoret, J.J.; Iacovoni, J.; et al. Caloric Restriction and Diet-Induced Weight Loss Do Not Induce Browning of Human Subcutaneous White Adipose Tissue in Women and Men with Obesity. Cell Rep. 2018, 22, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Pinho, C.P.S.; Diniz, A.D.S.; Arruda, I.K.G.; Leite, A.; Rodrigues, I.G. Effects of weight loss on adipose visceral and subcutaneous tissue in overweight adults. Clin. Nutr. 2018, 37, 1252–1258. [Google Scholar] [CrossRef]
- Gasteyger, C.; Larsen, T.M.; Vercruysse, F.; Pedersen, D.; Toubro, S.; Astrup, A. Visceral fat loss induced by a low-calorie diet: A direct comparison between women and men. Diabetes Obes. Metab. 2009, 11, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, E.; Peirce, G.; Brown, S.; Freemyer, J.; Rice, W.; Lee, L.; Coviello, L.; Davis, K.G. A short-duration restrictive diet reduces visceral adiposity in the morbidly obese surgical patient. Am. J. Surg. 2016, 212, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bujo, H.; Takahashi, K.; Shibasaki, M.; Zhu, Y.; Yoshida, Y.; Otsuka, Y.; Hashimoto, N.; Saito, Y. Visceral fat: Higher responsiveness of fat mass and gene expression to calorie restriction than subcutaneous fat. Exp. Biol. Med. (Maywood) 2003, 228, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, E.; Kumahara, H.; Tobina, T.; Matsuda, T.; Ayabe, M.; Kiyonaga, A.; Anzai, K.; Higaki, Y.; Tanaka, H. Lifestyle intervention involving calorie restriction with or without aerobic exercise training improves liver fat in adults with visceral adiposity. J. Obes. 2014, 2014, 197216. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, T.; Paulsen, S.K.; Bruun, J.M.; Overgaard, K.; Ringgaard, S.; Pedersen, S.B.; Positano, V.; Richelsen, B. Comparable reduction of the visceral adipose tissue depot after a diet-induced weight loss with or without aerobic exercise in obese subjects: A 12-week randomized intervention study. Eur. J. Endocrinol. 2009, 160, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, M.M.; Sabatini, D.M.; Yilmaz, O.H. Dietary and metabolic control of stem cell function in physiology and cancer. Cell Stem Cell 2014, 14, 292–305. [Google Scholar] [CrossRef] [Green Version]
- Redman, L.M.; Smith, S.R.; Burton, J.H.; Martin, C.K.; Il’yasova, D.; Ravussin, E. Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab. 2018, 27, 805–815.e4. [Google Scholar] [CrossRef] [Green Version]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Longo, V.D. Fasting vs dietary restriction in cellular protection and cancer treatment: From model organisms to patients. Oncogene 2011, 30, 3305–3316. [Google Scholar] [CrossRef] [Green Version]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms arend clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Brandhorst, S.; Longo, V.D. Fasting and Caloric Restriction in Cancer Prevention and Treatment. In Recent Results in Cancer Research; Springer: Cham, Switzerland, 2016; Volume 207, pp. 241–266. [Google Scholar] [CrossRef]
- De Cabo, R.; Mattson, M.P. Effects of Intermittent Fasting on Health, Aging, and Disease. N. Engl. J. Med. 2019, 381, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Varady, K.A.; Hellerstein, M.K. Alternate-day fasting and chronic disease prevention: A review of human and animal trials. Am. J. Clin. Nutr. 2007, 86, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, R.E.; Laughlin, G.A.; LaCroix, A.Z.; Hartman, S.J.; Natarajan, L.; Senger, C.M.; Martinez, M.E.; Villasenor, A.; Sears, D.D.; Marinac, C.R.; et al. Intermittent Fasting and Human Metabolic Health. J. Acad. Nutr. Diet. 2015, 115, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, L.; Villareal, D.T.; Das, S.K.; Smith, S.R.; Meydani, S.N.; Pittas, A.G.; Klein, S.; Bhapkar, M.; Rochon, J.; Ravussin, E.; et al. Effects of 2-year calorie restriction on circulating levels of IGF-1, IGF-binding proteins and cortisol in nonobese men and women: A randomized clinical trial. Aging Cell 2016, 15, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Panda, S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016, 23, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Zhu, X.; Wang, H.; Wang, F.; Guan, W. Roles of caloric restriction, ketogenic diet and intermittent fasting during initiation, progression and metastasis of cancer in animal models: A systematic review and meta-analysis. PLoS ONE 2014, 9, e115147. [Google Scholar] [CrossRef]
- Zou, Y.; Fineberg, S.; Pearlman, A.; Feinman, R.D.; Fine, E.J. The effect of a ketogenic diet and synergy with rapamycin in a mouse model of breast cancer. PLoS ONE 2020, 15, e0233662. [Google Scholar] [CrossRef]
- Leidy, H.J.; Clifton, P.M.; Astrup, A.; Wycherley, T.P.; Westerterp-Plantenga, M.S.; Luscombe-Marsh, N.D.; Woods, S.C.; Mattes, R.D. The role of protein in weight loss and maintenance. Am. J. Clin. Nutr. 2015, 101, 1320S–1329S. [Google Scholar] [CrossRef] [Green Version]
- Wycherley, T.P.; Buckley, J.D.; Noakes, M.; Clifton, P.M.; Brinkworth, G.D. Comparison of the effects of weight loss from a high-protein versus standard-protein energy-restricted diet on strength and aerobic capacity in overweight and obese men. Eur. J. Nutr. 2013, 52, 317–325. [Google Scholar] [CrossRef]
- Valenzano, A.; Polito, R.; Trimigno, V.; Di Palma, A.; Moscatelli, F.; Corso, G.; Sessa, F.; Salerno, M.; Montana, A.; Di Nunno, N.; et al. Effects of Very Low Calorie Ketogenic Diet on the Orexinergic System, Visceral Adipose Tissue, and ROS Production. Antioxidants (Basel) 2019, 8, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, A.I.; Gomez-Arbelaez, D.; Crujeiras, A.B.; Granero, R.; Aguera, Z.; Jimenez-Murcia, S.; Sajoux, I.; Lopez-Jaramillo, P.; Fernandez-Aranda, F.; Casanueva, F.F. Effect of A Very Low-Calorie Ketogenic Diet on Food and Alcohol Cravings, Physical and Sexual Activity, Sleep Disturbances, and Quality of Life in Obese Patients. Nutrients 2018, 10, 1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilone, V.; Tramontano, S.; Renzulli, M.; Romano, M.; Cobellis, L.; Berselli, T.; Schiavo, L. Metabolic effects, safety, and acceptability of very low-calorie ketogenic dietetic scheme on candidates for bariatric surgery. Surg. Obes. Relat. Dis. 2018, 14, 1013–1019. [Google Scholar] [CrossRef]
- Moreno, B.; Crujeiras, A.B.; Bellido, D.; Sajoux, I.; Casanueva, F.F. Obesity treatment by very low-calorie-ketogenic diet at two years: Reduction in visceral fat and on the burden of disease. Endocrine 2016, 54, 681–690. [Google Scholar] [CrossRef]
- Cohen, C.W.; Fontaine, K.R.; Arend, R.C.; Alvarez, R.D.; Leath, C.A., III.; Huh, W.K.; Bevis, K.S.; Kim, K.H.; Straughn, J.M., Jr.; Gower, B.A. A Ketogenic Diet Reduces Central Obesity and Serum Insulin in Women with Ovarian or Endometrial Cancer. J. Nutr. 2018, 148, 1253–1260. [Google Scholar] [CrossRef]
- Goldberg, E.L.; Shchukina, I.; Asher, J.L.; Sidorov, S.; Artyomov, M.N.; Dixit, V.D. Ketogenesis activates metabolically protective gammadelta T cells in visceral adipose tissue. Nat. Metab. 2020, 2, 50–61. [Google Scholar] [CrossRef]
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015, 22, 86–99. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Cancer Site or Type | Renehan et al. 1 (2008) | IARC Working Group 2 (2006) | Wang et al. 3 (2016) |
|---|---|---|---|
| Thyroid | Men (p = 0.02) | Yes | Men (p < 0.0001) |
| Women (p = 0.001) | Women (p = 0.728) | ||
| Kidney | Men (p < 0.0001) | Yes | Men (p < 0.0001) |
| Women (p < 0.0001) | Women (p < 0.0001) | ||
| Colon | Men (p < 0.0001) | Yes | Men (p < 0.0001) |
| Women (p < 0.0001) | (colorectal) | Women (p = 0.005) | |
| Rectum | Men (p < 0.0001) | (colorectal) | |
| Esophagus | Men (p < 0.0001) | Yes | Men (p < 0.0001) |
| Women (p < 0.0001) | (adenocarcinoma) | Women (p = 0.041) | |
| (adenocarcinoma) | (esophagus and stomach) | ||
| Stomach | - | Yes | |
| Multiple Myeloma | Men (p < 0.001) | Yes | - |
| Women (p < 0.0001) | |||
| Leukemia | Men (p < 0.0001) | - | - |
| Women (p = 0.01) | |||
| Non Hodgkin Lymphoma | Men (p < 0.0001) | - | - |
| Women (p = 0.01) | |||
| Melanoma | Men (p = 0.04) | - | - |
| Gallbladder | Women (p = 0.04) | Yes | - |
| Pancreas | Women (p = 0.01) | Yes | Men (p < 0.0001) |
| Women (p = 0.014) | |||
| Liver | - | Yes | Men (p < 0.0001) |
| Women (p = 0.9) | |||
| Meningioma | - | Yes | - |
| Ovary | - | Yes | Women (p = 0.009) |
| Prostate | - | - | Men (p < 0.0001) |
| Endometrium | Women (p < 0.001) | Yes | - |
| (corpus uteri) | |||
| Postmenopausal Breast cancer | Women (p < 0.0001) | Yes | Women (p < 0.0001) |
| Method | Description |
|---|---|
| Every-Other-Day Fasting | Food is withdrawn for 24 h on alternate days, with water provided ad libitum. Overall calorie intake need not be limited. |
| Time-Restricted Feeding | It restricts the timing of meals, without regard to their caloric content, to a time window of few hours in a day. |
| Periodic Fasting | It lasts 2 or more days and is separated from the next cycle by at least 1 week of normal feeding. |
| Brandhorst | It lasts 4 days and provides 10–50% of the normal caloric intake |
| Fasting Mimicking Diet | Periodic cycle of diets that provides a relatively high caloric content but mimics effects of fasting. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crudele, L.; Piccinin, E.; Moschetta, A. Visceral Adiposity and Cancer: Role in Pathogenesis and Prognosis. Nutrients 2021, 13, 2101. https://doi.org/10.3390/nu13062101
Crudele L, Piccinin E, Moschetta A. Visceral Adiposity and Cancer: Role in Pathogenesis and Prognosis. Nutrients. 2021; 13(6):2101. https://doi.org/10.3390/nu13062101
Chicago/Turabian StyleCrudele, Lucilla, Elena Piccinin, and Antonio Moschetta. 2021. "Visceral Adiposity and Cancer: Role in Pathogenesis and Prognosis" Nutrients 13, no. 6: 2101. https://doi.org/10.3390/nu13062101
APA StyleCrudele, L., Piccinin, E., & Moschetta, A. (2021). Visceral Adiposity and Cancer: Role in Pathogenesis and Prognosis. Nutrients, 13(6), 2101. https://doi.org/10.3390/nu13062101