
Dietary Carbohydrate as Glycemic Load, Not Fat, Coupled with Genetic Permissiveness Favoring Rapid Growth and Extra Calories, Dictate Metabolic Syndrome and Diabetes Induction in Nile Rats (Arvicanthis niloticus)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model (Nile Rat, Arvicanthis niloticus)
2.2. Semipurified Diets
Fatty Acid Profiles
2.3. Experimental Design
2.4. Calculations, Measurements and Data Analysis
2.4.1. Body Weight, Growth and BMI
2.4.2. Food (Caloric) and Water Intake
2.4.3. Food Efficiency
2.4.4. Glycemic Index
2.4.5. Glycemic Load
2.4.6. Blood Glucose
Random Blood Glucose (RBG)
Diabetes Severity and Susceptibility
Fasting Blood Glucose (FBG)
OGTT
2.4.7. Organ Weights
2.4.8. Plasma Triglycerides (TG) and Total Cholesterol (TC)
2.4.9. Statistical Analyses
2.4.10. Ethics Statement
2.5. Data Analysis
- (1)
- By assigned diet groups to determine if any particular diet composition or the macronutrient CHO:fat ratio might explain the majority of the diabetes induced.
- (2)
- By quintiles of caloric intake. The data were reassessed by quintiles based on average kcal/day consumed from lowest (Q1kcal) to highest (Q5kcal) to determine if kcal/day intake, regardless of macronutrient ratio, would be more revealing than the sort by diet composition that identified rats as resistant (<75 mg/dL) or susceptible (>75 mg/dL) subgroups.
- (3)
- By quintiles of 10-week RBG. The RBG for rats in each experiment were pooled and sorted into quintiles (Q1RBG to Q5RBG) from lowest to highest to determine whether RBG would expose additional aspects of diabetes established following the first two analyses.
3. Results
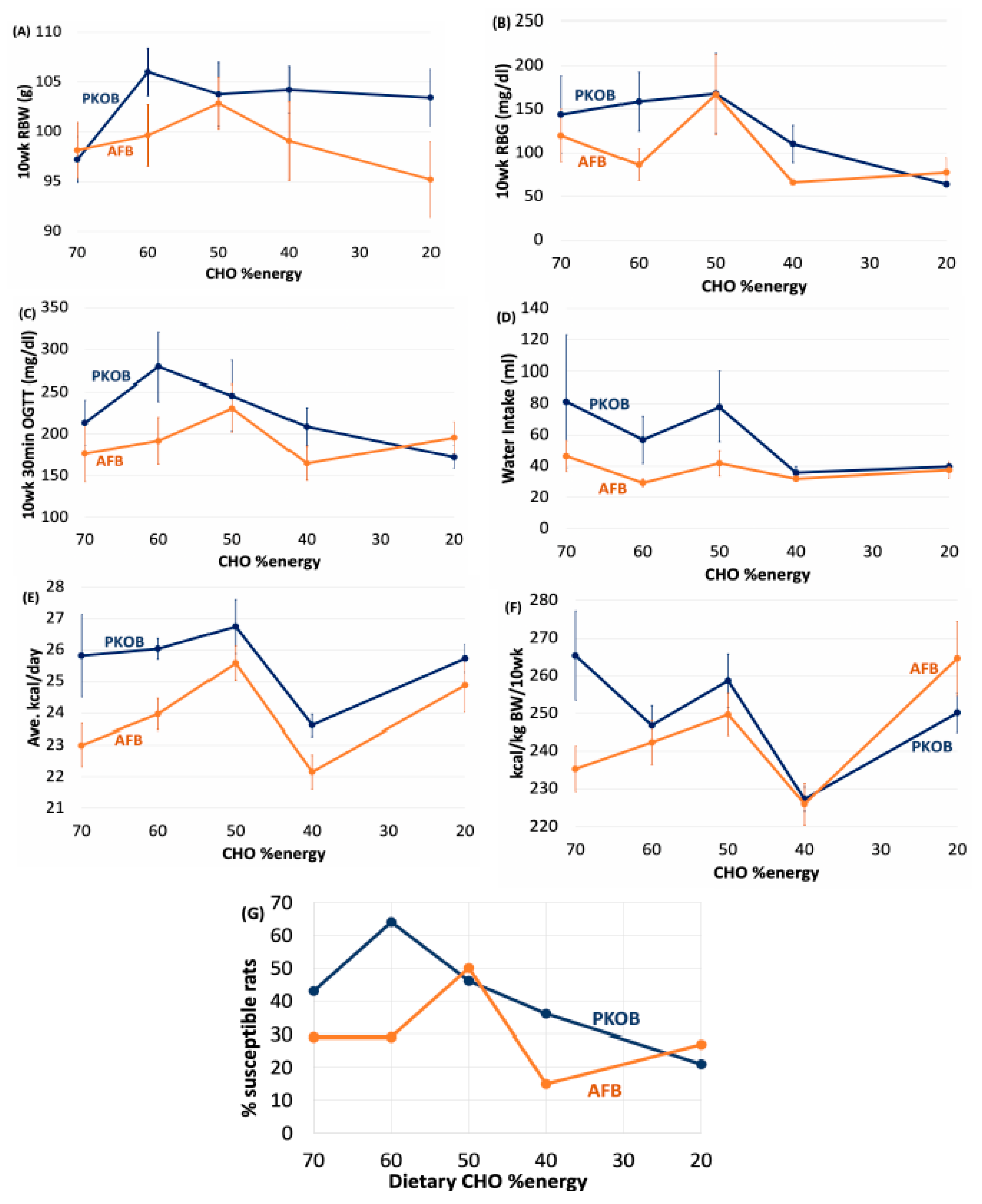
3.1. Analysis by Diet Composition
3.1.1. Diet Composition Affects T2DM Induction and Severity
3.1.2. Diet Effect on BW and Caloric Intake
3.1.3. Diet Effect on Food Efficiency
3.1.4. Diet Effect on Water Intake
3.1.5. Diet Effect on OGTT
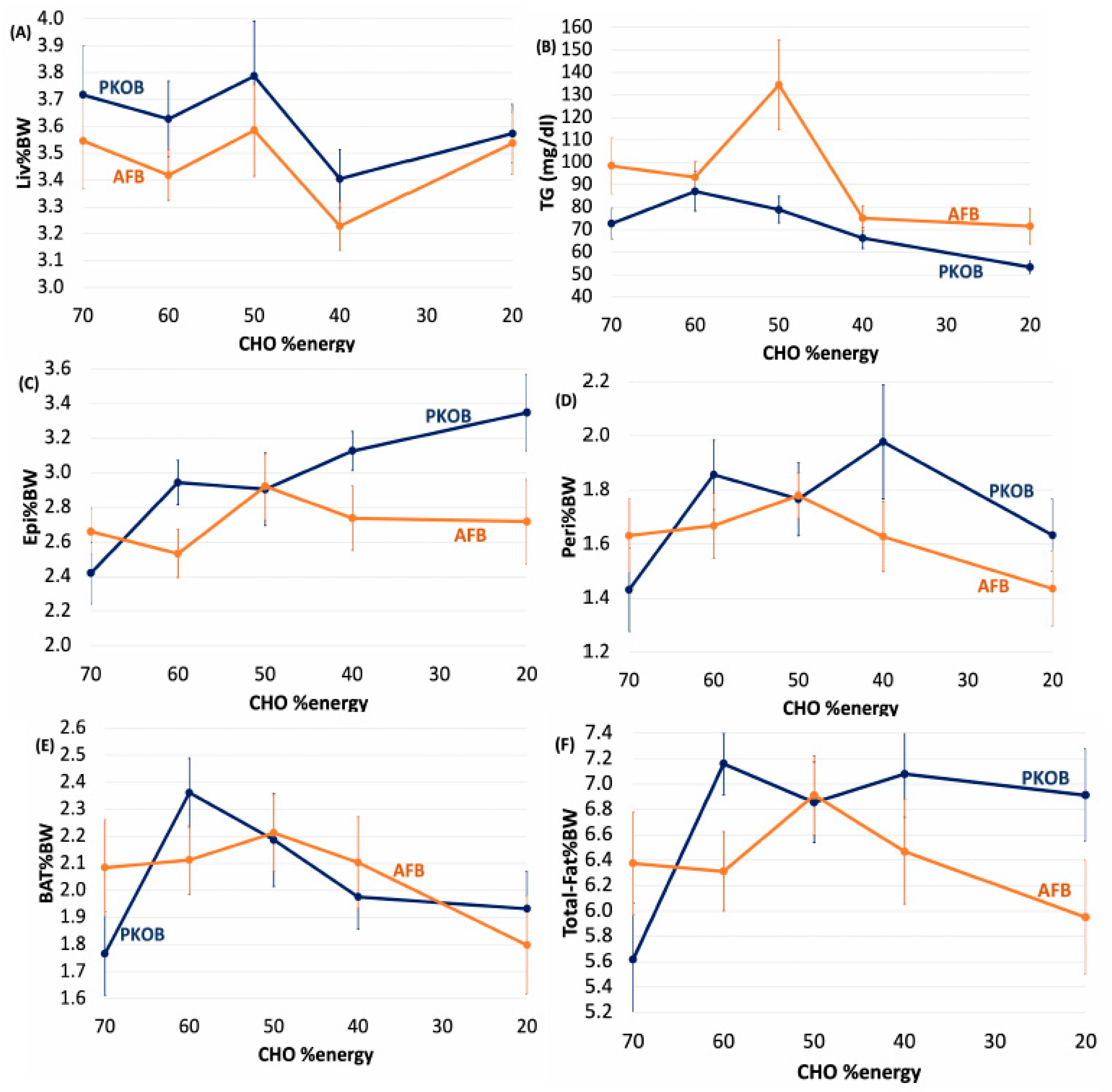
3.1.6. Diet Effect on Organ Weights
3.1.7. Diet Effect on Plasma Lipids
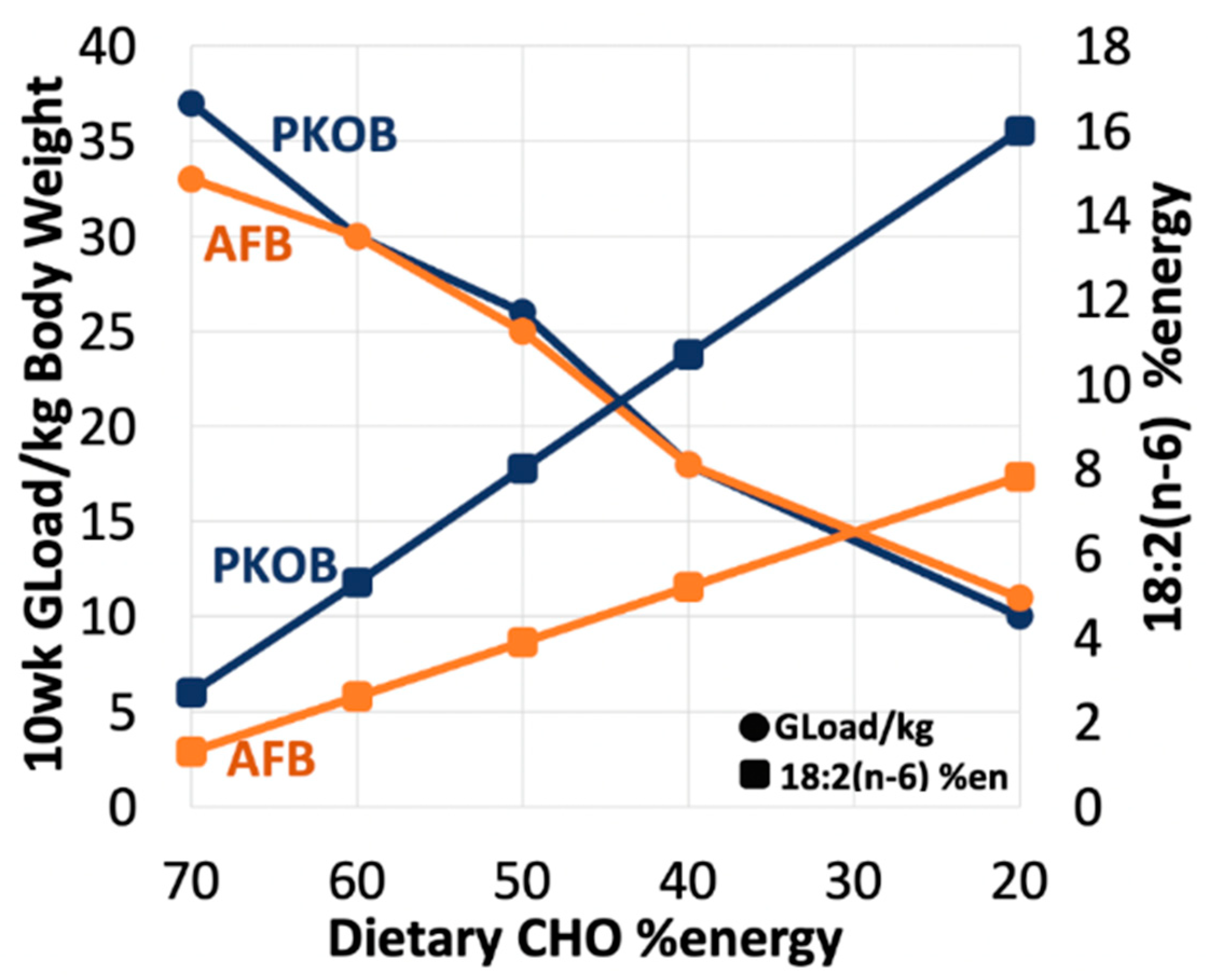
3.1.8. Diet Effect on 18:2(n-6) %Energy and Gload
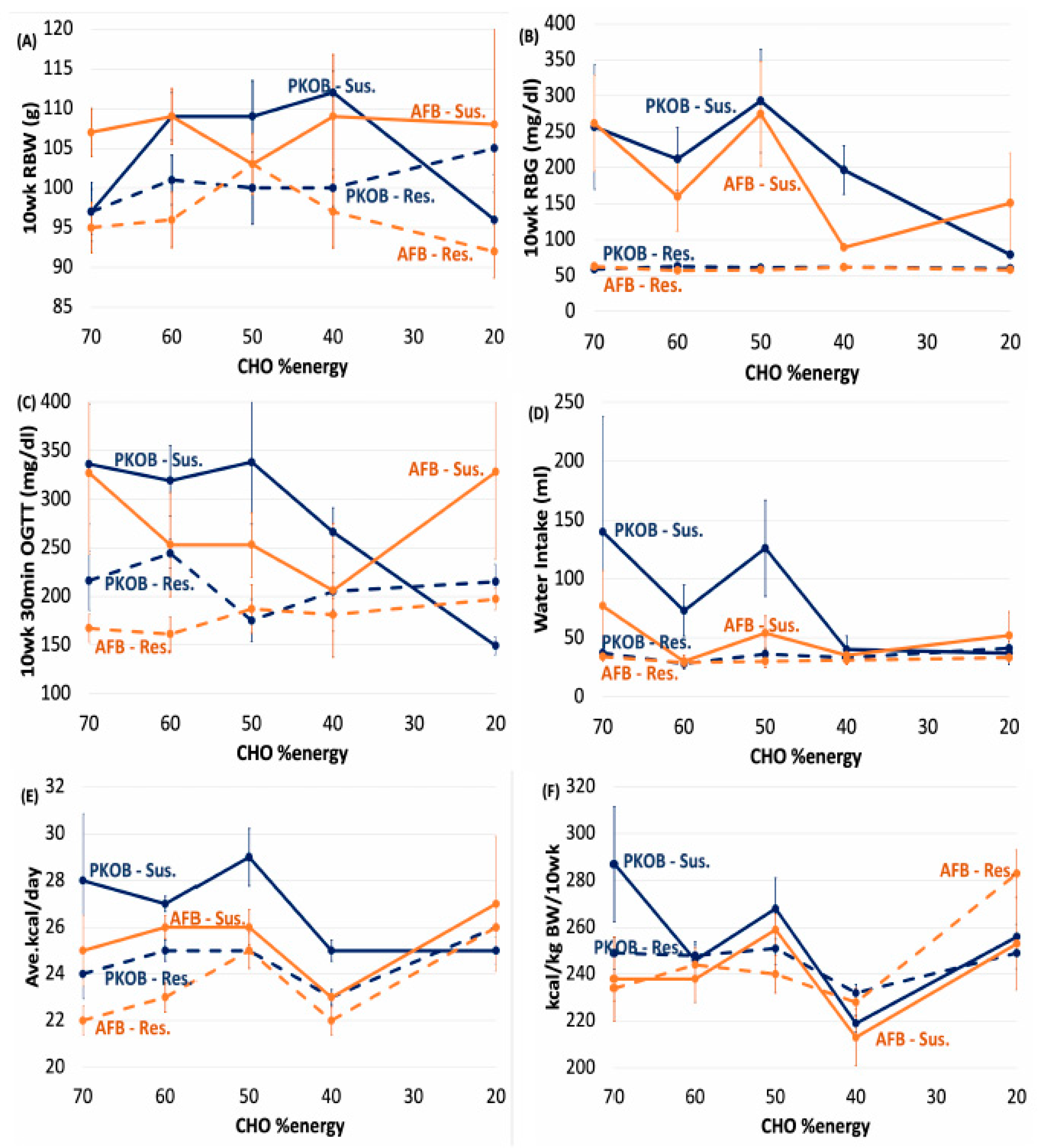
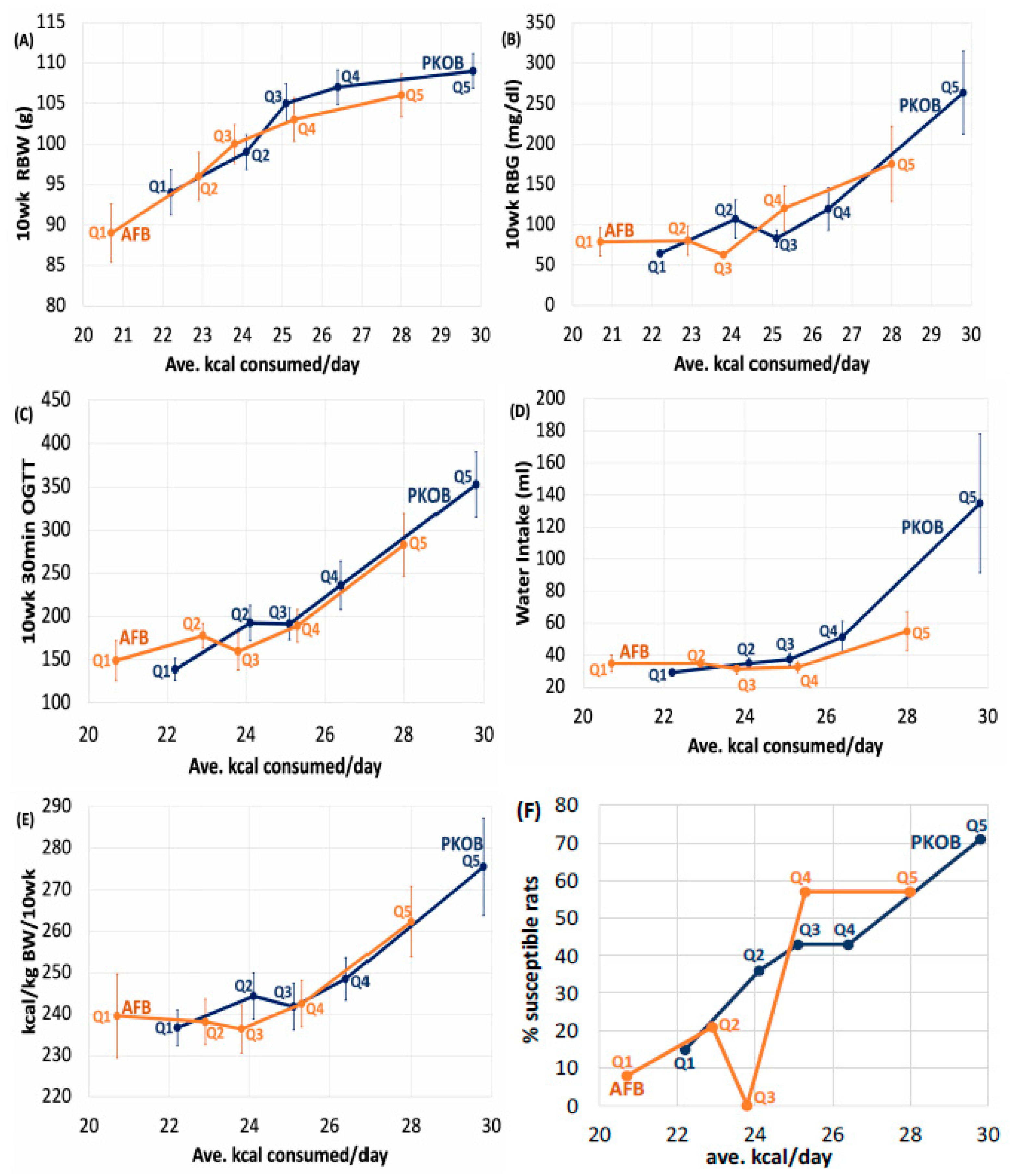
3.2. Analysis by Dietary Quintiles of Caloric Intake (Qkcal)
3.2.1. CHO:Fat Score and Diabetes
3.2.2. Diabetes Highlighted as RBG
3.2.3. Caloric Quintiles (Qkcal) Sorted as Resistant/Susceptible Rats
3.2.4. Kcal Sort on BW and Food Efficiency
3.2.5. Kcal/Day Relates to Dietary cumGLoad
3.2.6. Kcal Effect on Water Intake
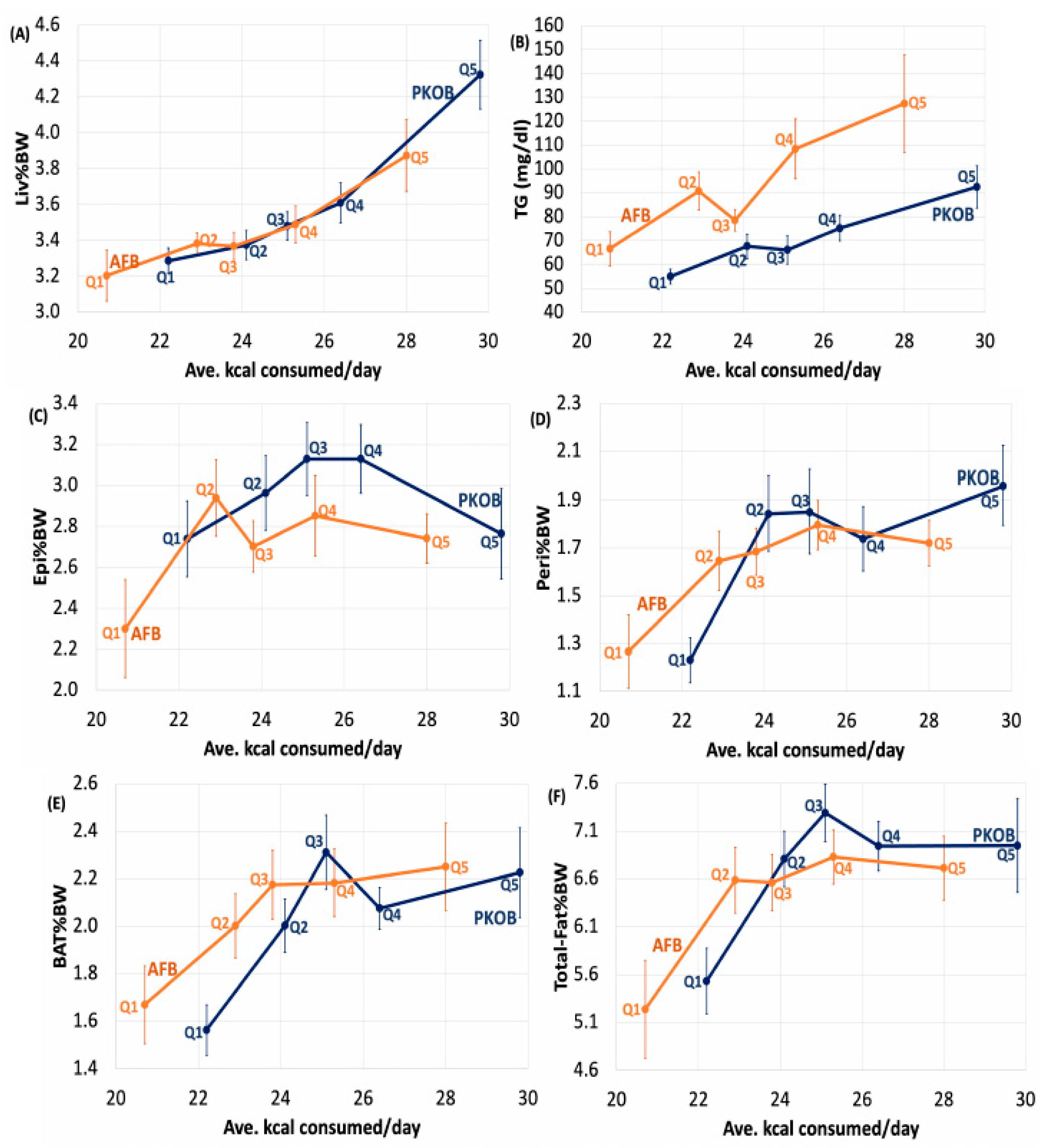
3.2.7. Kcal Effect on Organ Weights
3.2.8. Kcal Effect on Plasma Lipids
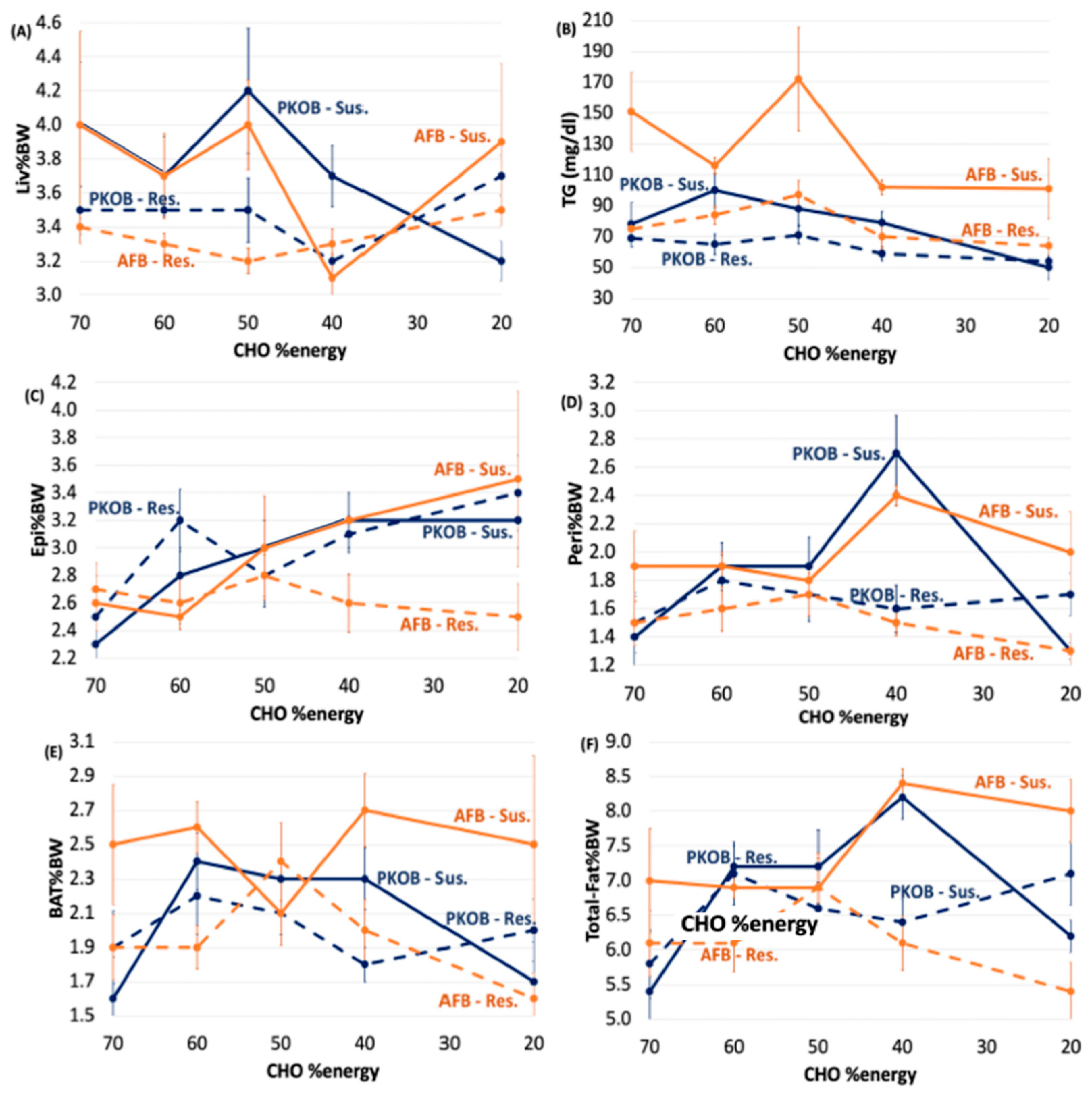
3.3. Analysis by Quintiles of RBG <75 mg/dL> across Diets
3.3.1. RBG (Q1–Q5RBG)
3.3.2. RBG Quintiles (Q1–Q5RBG) Separates Resistant/Susceptible Rats
3.3.3. CHO:Fat Score, Gload, BW and 18:2(n-6)
3.3.4. Organ Weights and Plasma Lipids
4. Discussion
4.1. CHO and GLoad
- (1)
- The extra calories had to reflect an increased GLoad within an overall increase in caloric intake, and
- (2)
- Individual host genetics/epigenetics had to be vulnerable to processing the increased glucose burden.
4.2. Dietary Fat
4.3. Minor Components
4.4. Caloric Intake Issue
4.5. Human Data Support
Epidemiological Data
4.6. Highly-Processed Food, Caloric Intake, and T2DM
4.7. Potential Genes Involved
4.8. Diurnal Rhythm Compromised
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Subramaniam, A.; Landstrom, M.; Luu, A.; Hayes, K.C. The Nile Rat (Arvicanthis niloticus) as a Superior Carbohydrate-Sensitive Model for Type 2 Diabetes Mellitus (T2DM). Nutrients 2018, 10, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, K.G.M.M.; Zimmet, P.; Shaw, J.; IDF Epidemiology Task Force Consensus Group. The Metabolic Syndrome—A New Worldwide Definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Global Report on Diabetes. Available online: https://www.who.int/publications-detail-redirect/9789241565257 (accessed on 16 July 2021).
- Risérus, U.; Willett, W.C.; Hu, F.B. Dietary fats and prevention of type 2 diabetes. Prog. Lipid Res. 2009, 48, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Halton, T.L.; Liu, S.; Manson, J.E.; Hu, F.B. Low-Carbohydrate-Diet Score and Risk of Type 2 Diabetes in Women. Am. J. Clin. Nutr. 2008, 87, 339–346. [Google Scholar] [CrossRef] [PubMed]
- de Koning, L.; Fung, T.T.; Liao, X.; Chiuve, S.E.; Rimm, E.B.; Willett, W.C.; Spiegelman, D.; Hu, F.B. Low-Carbohydrate Diet Scores and Risk of Type 2 Diabetes in Men. Am. J. Clin. Nutr. 2011, 93, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Seidelmann, S.B.; Claggett, B.; Cheng, S.; Henglin, M.; Shah, A.; Steffen, L.M.; Folsom, A.R.; Rimm, E.B.; Willett, W.C.; Solomon, S.D. Dietary Carbohydrate Intake and Mortality: A Prospective Cohort Study and Meta-Analysis. Lancet Public Health 2018, 3, e419–e428. [Google Scholar] [CrossRef] [Green Version]
- Willett, W.C. Dietary Fat Plays a Major Role in Obesity: No. Obes. Rev. 2002, 3, 59–68. [Google Scholar] [CrossRef]
- Willett, W.; Manson, J.; Liu, S. Glycemic Index, Glycemic Load, and Risk of Type 2 Diabetes. Am. J. Clin. Nutr. 2002, 76, 274S–280S. [Google Scholar] [CrossRef] [Green Version]
- Schulze, M.B.; Liu, S.; Rimm, E.B.; Manson, J.E.; Willett, W.C.; Hu, F.B. Glycemic Index, Glycemic Load, and Dietary Fiber Intake and Incidence of Type 2 Diabetes in Younger and Middle-Aged Women. Am. J. Clin. Nutr. 2004, 80, 348–356. [Google Scholar] [CrossRef]
- Greenwood, D.C.; Threapleton, D.E.; Evans, C.E.L.; Cleghorn, C.L.; Nykjaer, C.; Woodhead, C.; Burley, V.J. Glycemic Index, Glycemic Load, Carbohydrates, and Type 2 Diabetes: Systematic Review and Dose-Response Meta-Analysis of Prospective Studies. Diabetes Care 2013, 36, 4166–4171. [Google Scholar] [CrossRef] [Green Version]
- Livesey, G.; Taylor, R.; Livesey, H.F.; Buyken, A.E.; Jenkins, D.J.A.; Augustin, L.S.A.; Sievenpiper, J.L.; Barclay, A.W.; Liu, S.; Wolever, T.M.S.; et al. Dietary Glycemic Index and Load and the Risk of Type 2 Diabetes: A Systematic Review and Updated Meta-Analyses of Prospective Cohort Studies. Nutrients 2019, 11, 1280. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, D.S.; Hu, F.B.; Tappy, L.; Brand-Miller, J. Dietary Carbohydrates: Role of Quality and Quantity in Chronic Disease. BMJ 2018, 361, k2340. [Google Scholar] [CrossRef] [Green Version]
- Hussain, T.A.; Mathew, T.C.; Dashti, A.A.; Asfar, S.; Al-Zaid, N.; Dashti, H.M. Effect of Low-Calorie versus Low-Carbohydrate Ketogenic Diet in Type 2 Diabetes. Nutrition 2012, 28, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.D.; Chen, K.Y.; Guo, J.; Lam, Y.Y.; Leibel, R.L.; Mayer, L.E.; Reitman, M.L.; Rosenbaum, M.; Smith, S.R.; Walsh, B.T.; et al. Energy Expenditure and Body Composition Changes after an Isocaloric Ketogenic Diet in Overweight and Obese Men. Am. J. Clin. Nutr. 2016, 104, 324–333. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, L.; Imamura, F.; Brage, S.; Griffin, S.J.; Wareham, N.J.; Forouhi, N.G. Intakes and Sources of Dietary Sugars and Their Association with Metabolic and Inflammatory Markers. Clin. Nutr. 2018, 37, 1313–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinman, R.D.; Pogozelski, W.K.; Astrup, A.; Bernstein, R.K.; Fine, E.J.; Westman, E.C.; Accurso, A.; Frassetto, L.; Gower, B.A.; McFarlane, S.I.; et al. Dietary Carbohydrate Restriction as the First Approach in Diabetes Management: Critical Review and Evidence Base. Nutrition 2015, 31, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.-J.; Lee, H.-S.; Lee, J.-W. Association of Carbohydrate and Fat Intake with Metabolic Syndrome. Clin. Nutr. 2018, 37, 746–751. [Google Scholar] [CrossRef]
- Ludwig, D.S. The Ketogenic Diet: Evidence for Optimism but High-Quality Research Needed. J. Nutr. 2020, 150, 1354–1359. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, D.S.; Ebbeling, C.B.; Heymsfield, S.B. Improving the Quality of Dietary Research. JAMA 2019, 322, 1549–1550. [Google Scholar] [CrossRef]
- Ludwig, D.S.; Astrup, A.; Bazzano, L.A.; Ebbeling, C.B.; Heymsfield, S.B.; King, J.C.; Willett, W.C. Ultra-Processed Food and Obesity: The Pitfalls of Extrapolation from Short Studies. Cell Metab. 2019, 30, 3–4. [Google Scholar] [CrossRef]
- Chaabo, F.; Pronczuk, A.; Maslova, E.; Hayes, K. Nutritional Correlates and Dynamics of Diabetes in the Nile Rat (Arvicanthis niloticus): A Novel Model for Diet-Induced Type 2 Diabetes and the Metabolic Syndrome. Nutr. Metab. 2010, 7, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilu, C.; Zimmet, P.; Vishnevskia-Dai, V.; Einat, H.; Agam, G.; Grossman, E.; Kronfeld-Schor, N. Diurnality, Type 2 Diabetes, and Depressive-Like Behavior. J. Biol. Rhythm. 2019, 34, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Bilu, C.; Einat, H.; Barak, O.; Zimmet, P.; Vishnevskia-Dai, V.; Govrin, A.; Agam, G.; Kronfeld-Schor, N. Linking Type 2 Diabetes Mellitus, Cardiac Hypertrophy and Depression in a Diurnal Animal Model. Sci. Rep. 2019, 9, 11865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulman, G.I. Cellular Mechanisms of Insulin Resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci. 2017, 38, 649–665. [Google Scholar] [CrossRef]
- Reali, F.; Morine, M.J.; Kahramanoğulları, O.; Raichur, S.; Schneider, H.-C.; Crowther, D.; Priami, C. Mechanistic Interplay between Ceramide and Insulin Resistance. Sci. Rep. 2017, 7, 41231. [Google Scholar] [CrossRef]
- Hla, T.; Kolesnick, R. C16: 0-Ceramide Signals Insulin Resistance. Cell Metab. 2014, 20, 703–705. [Google Scholar] [CrossRef] [Green Version]
- Hayes, K.C. Synthetic and Modified Glycerides: Effects on Plasma Lipids. Curr. Opin. Lipidol. 2001, 12, 55–60. [Google Scholar] [CrossRef]
- Chorell, E.; Otten, J.; Stomby, A.; Ryberg, M.; Waling, M.; Hauksson, J.; Svensson, M.; Olsson, T. Improved Peripheral and Hepatic Insulin Sensitivity after Lifestyle Interventions in Type 2 Diabetes Is Associated with Specific Metabolomic and Lipidomic Signatures in Skeletal Muscle and Plasma. Metabolites 2021, 11, 834. [Google Scholar] [CrossRef]
- Salmerón, J.; Ascherio, A.; Rimm, E.B.; Colditz, G.A.; Spiegelman, D.; Jenkins, D.J.; Stampfer, M.J.; Wing, A.L.; Willett, W.C. Dietary Fiber, Glycemic Load, and Risk of NIDDM in Men. Diabetes Care 1997, 20, 545–550. [Google Scholar] [CrossRef]
- Rueda, R.; Ramírez, M.; García-Salmerón, J.L.; Maldonado, J.; Gil, A. Gestational Age and Origin of Human Milk Influence Total Lipid and Fatty Acid Contents. Ann. Nutr. Metab. 1998, 42, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Hayes, K.C. The Omega-6 versus Omega-3 Fatty Acid Modulation of Lipoprotein Metabolism. In Omega-3 Fatty Acids; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2001; Volume 788, pp. 37–53. [Google Scholar] [CrossRef]
- Guasch-Ferré, M.; Becerra-Tomás, N.; Ruiz-Canela, M.; Corella, D.; Schröder, H.; Estruch, R.; Ros, E.; Arós, F.; Gómez-Gracia, E.; Fiol, M.; et al. Total and Subtypes of Dietary Fat Intake and Risk of Type 2 Diabetes Mellitus in the Prevención Con Dieta Mediterránea (PREDIMED) Study. Am. J. Clin. Nutr. 2017, 105, 723–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, A.; Landstrom, M.; Hayes, K.C. Genetic Permissiveness and Dietary Glycemic Load Interact to Predict Type-II Diabetes in the Nile Rat (Arvicanthis niloticus). Nutrients 2019, 11, 1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livesey, G.; Taylor, R.; Livesey, H.F.; Buyken, A.E.; Jenkins, D.J.A.; Augustin, L.S.A.; Sievenpiper, J.L.; Barclay, A.W.; Liu, S.; Wolever, T.M.S.; et al. Dietary Glycemic Index and Load and the Risk of Type 2 Diabetes: Assessment of Causal Relations. Nutrients 2019, 11, 1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolsinger, J.; Pronczuk, A.; Sambanthamurthi, R.; Hayes, K.C. Anti-Diabetic Effects of Palm Fruit Juice in the Nile Rat (Arvicanthis niloticus). J. Nutr. Sci. 2014, 3, e5. [Google Scholar] [CrossRef] [Green Version]
- Bolsinger, J.; Pronczuk, A.; Hayes, K.C. Dietary Carbohydrate Dictates Development of Type 2 Diabetes in the Nile Rat. J. Nutr. Biochem. 2013, 24, 1945–1952. [Google Scholar] [CrossRef]
- Bolsinger, J.; Landstrom, M.; Pronczuk, A.; Auerbach, A.; Hayes, K.C. Low Glycemic Load Diets Protect against Metabolic Syndrome and Type 2 Diabetes Mellitus in the Male Nile Rat. J. Nutr. Biochem. 2017, 42, 134–148. [Google Scholar] [CrossRef]
- Leow, S.-S.; Bolsinger, J.; Pronczuk, A.; Hayes, K.C.; Sambanthamurthi, R. Hepatic Transcriptome Implications for Palm Fruit Juice Deterrence of Type 2 Diabetes Mellitus in Young Male Nile Rats. Genes. Nutr. 2016, 11, 29. [Google Scholar] [CrossRef] [Green Version]
- Qian, F.; Korat, A.A.; Malik, V.; Hu, F.B. Metabolic Effects of Monounsaturated Fatty Acid-Enriched Diets Compared with Carbohydrate or Polyunsaturated Fatty Acid-Enriched Diets in Patients with Type 2 Diabetes: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Diabetes Care 2016, 39, 1448–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, F.; Liu, G.; Hu, F.B.; Bhupathiraju, S.N.; Sun, Q. Association between Plant-Based Dietary Patterns and Risk of Type 2 Diabetes. JAMA Intern. Med. 2019, 179, 1335–1344. [Google Scholar] [CrossRef]
- Storlien, L.H.; Pan, D.A.; Kriketos, A.D.; O’Connor, J.; Caterson, I.D.; Cooney, G.J.; Jenkins, A.B.; Baur, L.A. Skeletal Muscle Membrane Lipids and Insulin Resistance. Lipids 1996, 31, S261–S265. [Google Scholar] [CrossRef] [PubMed]
- Khosla, P.; Hayes, K.C. Cholesterolaemic Effects of the Saturated Fatty Acids of Palm Oil. Food Nutr. Bull. 1994, 15, 1–7. [Google Scholar] [CrossRef]
- Hayes, K.C. Saturated Fats and Blood Lipids: New Slant on an Old Story. Can. J. Cardiol. 1995, 11 (Suppl. G), 39G–46G. [Google Scholar] [PubMed]
- Hayes, K.C. Dietary Fat and Heart Health: In Search of the Ideal Fat. Asia Pac. J. Clin. Nutr. 2002, 11, S394–S400. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S.; Dietz, W.H. Trends in Energy Intake among Adults in the United States: Findings from NHANES. Am. J. Clin. Nutr. 2013, 97, 848–853. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, C.E.; Keast, D.R.; Fulgoni, V.L.; Nicklas, T.A. Food Sources of Energy and Nutrients among Adults in the US: NHANES 2003–2006. Nutrients 2012, 4, 2097–2120. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.R.; Wagner, J.D. Type 2 Diabetes-an Introduction to the Development and Use of Animal Models. ILAR J. 2006, 47, 181–185. [Google Scholar] [CrossRef] [Green Version]
- Field, A.E.; Willett, W.C.; Lissner, L.; Colditz, G.A. Dietary Fat and Weight Gain among Women in the Nurses’ Health Study. Obesity 2007, 15, 967–976. [Google Scholar] [CrossRef]
- Wolever, T.M. Effect of Macronutrients on the Glycemic Index. Am. J. Clin. Nutr. 2017, 106, 704–705. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, D.S. The Glycemic Index: Physiological Mechanisms Relating to Obesity, Diabetes, and Cardiovascular Disease. JAMA 2002, 287, 2414–2423. [Google Scholar] [CrossRef]
- Leibowitz, G.; Yuli, M.; Donath, M.Y.; Nesher, R.; Melloul, D.; Cerasi, E.; Gross, D.J.; Kaiser, N. Beta-Cell Glucotoxicity in the Psammomys Obesus Model of Type 2 Diabetes. Diabetes 2001, 50 (Suppl. 1), S113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, B.P.; Digitale, E.K.; Stanhope, K.L.; Graham, J.L.; Baskin, D.G.; Reed, B.J.; Sweet, I.R.; Griffen, S.C.; Havel, P.J. Development and Characterization of a Novel Rat Model of Type 2 Diabetes Mellitus: The UC Davis Type 2 Diabetes Mellitus UCD-T2DM Rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1782–R1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajardo, R.J.; Karim, L.; Calley, V.I.; Bouxsein, M.L. A Review of Rodent Models of Type 2 Diabetic Skeletal Fragility. J. Bone Miner. Res. 2014, 29, 1025–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, J.; Brown, F.; Remillard, D.E.; Bolsinger, J.; Hayes, K. Pathology of the Nile rat developing type 2 diabetes. FASEB J. 2013, 27, 874.13. [Google Scholar] [CrossRef]
- Libby, P. The Changing Landscape of Atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Noakes, T.; Volek, J.S.; Phinney, S.D. Low-Carbohydrate Diets for Athletes: What Evidence? Br. J. Sports Med. 2014, 48, 1077–1078. [Google Scholar] [CrossRef] [Green Version]
- Phinney, S.D.; Bistrian, B.R.; Wolfe, R.R.; Blackburn, G.L. The Human Metabolic Response to Chronic Ketosis without Caloric Restriction: Physical and Biochemical Adaptation. Metabolism 1983, 32, 757–768. [Google Scholar] [CrossRef]
- Phinney, S.D. Ketogenic Diets and Physical Performance. Nutr. Metab. 2004, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Gershuni, V.M.; Yan, S.L.; Medici, V. Nutritional Ketosis for Weight Management and Reversal of Metabolic Syndrome. Curr. Nutr. Rep. 2018, 7, 97–106. [Google Scholar] [CrossRef]
- Shan, R.; Duan, W.; Liu, L.; Qi, J.; Gao, J.; Zhang, Y.; Du, S.; Han, T.; Pang, X.; Sun, C.; et al. Low-Carbohydrate, High-Protein, High-Fat Diets Rich in Livestock, Poultry and Their Products Predict Impending Risk of Type 2 Diabetes in Chinese Individuals That Exceed Their Calculated Caloric Requirement. Nutrients 2018, 10, 77. [Google Scholar] [CrossRef] [Green Version]
- Bach, A.C.; Babayan, V.K. Medium-Chain Triglycerides: An Update. Am. J. Clin. Nutr. 1982, 36, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Babayan, V.K. Medium Chain Triglycerides and Structured Lipids. Lipids 1987, 22, 417–420. [Google Scholar] [CrossRef]
- Ludwig, D.S.; Willett, W.C.; Volek, J.S.; Neuhouser, M.L. Dietary Fat: From Foe to Friend? Science 2018, 362, 764–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsythe, C.E.; Phinney, S.D.; Fernandez, M.L.; Quann, E.E.; Wood, R.J.; Bibus, D.M.; Kraemer, W.J.; Feinman, R.D.; Volek, J.S. Comparison of Low Fat and Low Carbohydrate Diets on Circulating Fatty Acid Composition and Markers of Inflammation. Lipids 2008, 43, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Leite, J.O.; DeOgburn, R.; Ratliff, J.C.; Su, R.; Volek, J.S.; McGrane, M.M.; Dardik, A.; Fernandez, M.L. Low-Carbohydrate Diet Disrupts the Association between Insulin Resistance and Weight Gain. Metabolism 2009, 58, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Volek, J.S.; Sharman, M.J.; Love, D.M.; Avery, N.G.; Gómez, A.L.; Scheett, T.P.; Kraemer, W.J. Body Composition and Hormonal Responses to a Carbohydrate-Restricted Diet. Metabolism 2002, 51, 864–870. [Google Scholar] [CrossRef]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv. Nutr. 2020, 11, 77–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerich, J.E. Hypoglycaemia and Counterregulation in Type 2 Diabetes. Lancet 2000, 356, 1946–1947. [Google Scholar] [CrossRef]
- Gerich, J.E. Insulin Resistance Is Not Necessarily an Essential Component of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2000, 85, 2113–2115. [Google Scholar] [CrossRef]
- Brown, J.M.; Scarlett, J.M.; Schwartz, M.W. Rethinking the Role of the Brain in Glucose Homeostasis and Diabetes Pathogenesis. J. Clin. Investig. 2019, 129, 3035–3037. [Google Scholar] [CrossRef] [Green Version]
- Krajmalnik-Brown, R.; Ilhan, Z.-E.; Kang, D.-W.; DiBaise, J.K. Effects of Gut Microbes on Nutrient Absorption and Energy Regulation. Nutr. Clin. Pract. 2012, 27, 201–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, M.H.; Wang, H.; Pan, J.N.; Clark, W.C.; Cui, S.; Eckwahl, M.J.; Pan, D.W.; Parisien, M.; Owens, S.M.; Cheng, B.L.; et al. Microbiome Characterization by High-Throughput Transfer RNA Sequencing and Modification Analysis. Nat. Commun. 2018, 9, 5353. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.D.; Farooqi, I.S.; Friedman, J.M.; Klein, S.; Loos, R.J.F.; Mangelsdorf, D.J.; O’Rahilly, S.; Ravussin, E.; Redman, L.M.; Ryan, D.H.; et al. The Energy Balance Model of Obesity: Beyond Calories in, Calories Out. Am. J. Clin. Nutr. 2022, 115, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Augustin, L.S.A.; Aas, A.-M.; Astrup, A.; Atkinson, F.S.; Baer-Sinnott, S.; Barclay, A.W.; Brand-Miller, J.C.; Brighenti, F.; Bullo, M.; Buyken, A.E.; et al. Dietary Fibre Consensus from the International Carbohydrate Quality Consortium (ICQC). Nutrients 2020, 12, 2553. [Google Scholar] [CrossRef]
- Cardona, F.; Andrés-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of Polyphenols on Gut Microbiota and Implications in Human Health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Sonnenburg, J.L.; Bäckhed, F. Diet-Microbiota Interactions as Moderators of Human Metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef]
- Daïen, C.I.; Pinget, G.V.; Tan, J.K.; Macia, L. Detrimental Impact of Microbiota-Accessible Carbohydrate-Deprived Diet on Gut and Immune Homeostasis: An Overview. Front. Immunol. 2017, 8, 548. [Google Scholar] [CrossRef] [Green Version]
- Machado, P.P.; Steele, E.M.; Levy, R.B.; Sui, Z.; Rangan, A.; Woods, J.; Gill, T.; Scrinis, G.; Monteiro, C.A. Ultra-Processed Foods and Recommended Intake Levels of Nutrients Linked to Non-Communicable Diseases in Australia: Evidence from a Nationally Representative Cross-Sectional Study. BMJ Open 2019, 9, e029544. [Google Scholar] [CrossRef] [Green Version]
- Hall, K.D.; Ayuketah, A.; Brychta, R.; Cai, H.; Cassimatis, T.; Chen, K.Y.; Chung, S.T.; Costa, E.; Courville, A.; Darcey, V.; et al. Ultra-Processed Diets Cause Excess Calorie Intake and Weight Gain: An Inpatient Randomized Controlled Trial of Ad Libitum Food Intake. Cell Metab. 2019, 30, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Slyper, A. Oral Processing, Satiation and Obesity: Overview and Hypotheses. Diabetes Metab. Syndr. Obes. 2021, 14, 3399–3415. [Google Scholar] [CrossRef]
- Qian, F.; Sun, Q. Plant-Based Dietary Patterns and Incidence of Type 2 Diabetes-Reply. JAMA Intern. Med. 2019, 179, 1604–1605. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, C.A.; Cannon, G.; Moubarac, J.-C.; Levy, R.B.; Louzada, M.L.C.; Jaime, P.C. The UN Decade of Nutrition, the NOVA Food Classification and the Trouble with Ultra-Processing. Public Health Nutr. 2018, 21, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez Steele, E.; Baraldi, L.G.; Louzada, M.L.d.C.; Moubarac, J.-C.; Mozaffarian, D.; Monteiro, C.A. Ultra-Processed Foods and Added Sugars in the US Diet: Evidence from a Nationally Representative Cross-Sectional Study. BMJ Open 2016, 6, e009892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, C.A. Nutrition and Health. The Issue Is Not Food, nor Nutrients, so Much as Processing. Public Health Nutr. 2009, 12, 729–731. [Google Scholar] [CrossRef] [Green Version]
- Karra, E.; Chandarana, K.; Batterham, R.L. The Role of Peptide YY in Appetite Regulation and Obesity. J. Physiol. 2009, 587 Pt 1, 19–25. [Google Scholar] [CrossRef]
- Noda, K.; Melhorn, M.I.; Zandi, S.; Frimmel, S.; Tayyari, F.; Hisatomi, T.; Almulki, L.; Pronczuk, A.; Hayes, K.C.; Hafezi-Moghadam, A. An Animal Model of Spontaneous Metabolic Syndrome: Nile Grass Rat. FASEB J. 2010, 24, 2443–2453. [Google Scholar] [CrossRef] [Green Version]
- The Truth about Fat. KPBS. Available online: https://www.kpbs.org/news/2020/apr/07/nova-truth-about-fat/ (accessed on 19 June 2020).
- Kalman, R.; Adler, J.H.; Lazarovici, G.; Bar-On, H.; Ziv, E. The Efficiency of Sand Rat Metabolism Is Responsible for Development of Obesity and Diabetes. J. Basic Clin. Physiol. Pharmacol. 1993, 4, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.M.; Sun, E.W.; Keating, D.J. Mechanisms Controlling Hormone Secretion in Human Gut and Its Relevance to Metabolism. J. Endocrinol. 2019, 244, R1–R15. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramaniam, A.; Park, B.; Raphael, D.; Landstrom, M.; Hayes, K.C. Dietary Carbohydrate as Glycemic Load, Not Fat, Coupled with Genetic Permissiveness Favoring Rapid Growth and Extra Calories, Dictate Metabolic Syndrome and Diabetes Induction in Nile Rats (Arvicanthis niloticus). Nutrients 2022, 14, 3064. https://doi.org/10.3390/nu14153064
Subramaniam A, Park B, Raphael D, Landstrom M, Hayes KC. Dietary Carbohydrate as Glycemic Load, Not Fat, Coupled with Genetic Permissiveness Favoring Rapid Growth and Extra Calories, Dictate Metabolic Syndrome and Diabetes Induction in Nile Rats (Arvicanthis niloticus). Nutrients. 2022; 14(15):3064. https://doi.org/10.3390/nu14153064
Chicago/Turabian StyleSubramaniam, Avinaash, Bumjoon Park, Domenick Raphael, Michelle Landstrom, and K. C. Hayes. 2022. "Dietary Carbohydrate as Glycemic Load, Not Fat, Coupled with Genetic Permissiveness Favoring Rapid Growth and Extra Calories, Dictate Metabolic Syndrome and Diabetes Induction in Nile Rats (Arvicanthis niloticus)" Nutrients 14, no. 15: 3064. https://doi.org/10.3390/nu14153064
APA StyleSubramaniam, A., Park, B., Raphael, D., Landstrom, M., & Hayes, K. C. (2022). Dietary Carbohydrate as Glycemic Load, Not Fat, Coupled with Genetic Permissiveness Favoring Rapid Growth and Extra Calories, Dictate Metabolic Syndrome and Diabetes Induction in Nile Rats (Arvicanthis niloticus). Nutrients, 14(15), 3064. https://doi.org/10.3390/nu14153064