
Glycochenodeoxycholate Affects Iron Homeostasis via Up-Regulating Hepcidin Expression


Abstract
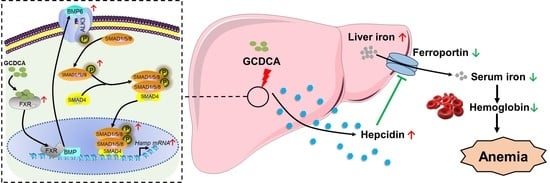
:
1. Introduction
2. Materials and Methods
2.1. Determination of Population Experimental Indexes
2.2. Chemicals
2.3. Cell Culture
2.4. Cell Viability Determination
2.5. Quantitative Real-Time Polymerase Chain Reaction Analysis
2.6. Western Blotting Analysis
2.7. Animal Treatment
2.8. Analysis of GCDCA Concentration in Mice
2.9. Analyses of Iron Parameters and Serum Hepcidin Level
2.10. Statistical Analyses
3. Results
3.1. Imbalance of Iron Homeostasis Occurred Simultaneously with Increased Serum GCDCA Level in Patients with CKD
3.2. GCDCA Up-Regulated Hepcidin in a Dose-Related Manner
3.3. BMP6/ALK3-SMAD Signaling Mainly Mediated GCDCA-Induced Hepcidin Expression
3.4. FXR Was Triggered by GCDCA for Hepcidin Expression
3.5. GCDCA Disturbed Iron Homeostasis in Mice
3.6. GCDCA Activated FXR-SMAD Signaling for Hepcidin Up-Regulation in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roy, C.N.; Andrews, N.C. Anemia of inflammation: The hepcidin link. Curr. Opin. Hematol. 2005, 12, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a putative mediator of anemia of in-flammation, is a type II acute-phase protein. Blood 2003, 101, 2461–2463. [Google Scholar] [CrossRef]
- Malyszko, J.; Mysliwiec, M. Hepcidin in anemia and inflammation in chronic kidney disease. Kidney Blood Press. Res. 2007, 30, 15–30. [Google Scholar] [CrossRef]
- Auerbach, M. Anemia management and mortality risk in incident hemodialysis patients. J. Am. Med. Assoc. 2010, 304, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, S.; Li, S.; Zhao, L.; Hao, Y.; Qin, J.; Zhang, L.; Zhang, C.; Bian, W.; Zuo, L.; et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 2020, 69, 2131–2142. [Google Scholar] [CrossRef]
- Chen, H.; Cao, G.; Chen, D.-Q.; Wang, M.; Vaziri, N.D.; Zhang, Z.-H.; Mao, J.-R.; Bai, X.; Zhao, Y.-Y. Metabolomics insights into activated redox signaling and lipid metabolism dysfunction in chronic kidney disease progression. Redox Biol. 2016, 10, 168–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-H.; Chuang, J.-H.; Yang, Y.-L.; Huang, C.-C.; Wu, C.-L.; Chen, C.-L. Cholestasis downregulate hepcidin expression through inhibiting IL-6-induced phosphorylation of signal transducer and activator of transcription 3 signaling. Lab. Investig. 2009, 89, 1128–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.-R.; Feng, L.; Jiang, W.-D.; Wu, P.; Liu, Y.; Jiang, J.; Kuang, S.-Y.; Tang, L.; Zhou, X.-Q. Supplementation exogenous bile acid improved growth and intestinal immune function associated with NF-kappa B and TOR signalling pathways in on-growing grass carp (Ctenopharyngodon idella): Enhancement the effect of protein-sparing by dietary lipid. Fish Shellfish Immunol. 2019, 92, 552–569. [Google Scholar] [CrossRef] [PubMed]
- Morehouse, Z.P.; Easparro, B.; Proctor, C.; Nash, R.J.; Atwood, J. Evaluation of preparation techniques for optimal hepcidin detection in human hepatocyte culture. FASEB J. 2018, 32, 873.11. [Google Scholar] [CrossRef]
- Zaritsky, J.; Young, B.; Wang, H.; Nemeth, E.; Ganz, T.; Rivera, S.; Nissenson, A.; Salusky, I. Hepcidin- A potential novel biomarker for iron status and erythropoietin resistance in pediatric chronic kidney disease. J. Investig. Med. 2009, 57, 180. [Google Scholar] [CrossRef]
- Young, B.; Zaritsky, J. Hepcidin for Clinicians. Clin. J. Am. Soc. Nephrol. 2009, 4, 1384–1387. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, M.; Flores, S.R.L.; Woloshun, R.R.; Yang, C.; Yin, L.; Xiang, P.; Xu, X.; Garrick, M.D.; Vidyasagar, S.; et al. Oral gavage of ginger nanoparticle-derived lipid vectors carrying Dmt1 siRNA blunts iron loading in murine hereditary hemo-chromatosis. Mol. Ther. 2019, 27, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Rubab, Z.; Amin, H.; Abbas, K.; Hussain, S.; Ullah, M.I.; Mohsin, S. Serum hepcidin levels in patients with end-stage renal disease on hemodialysis. Saudi J. Kidney Dis. Transplant. 2015, 26, 19–25. [Google Scholar] [CrossRef]
- El Sewefy, D.A.; Farweez, B.A.; Behairy, M.A.; Yassin, N.R. Impact of serum hepcidin and inflammatory markers on resistance to erythropoiesis-stimulating therapy in haemodialysis patients. Int. Urol. Nephrol. 2019, 51, 325–334. [Google Scholar] [CrossRef]
- Lim, J.H.; Park, Y.W.; Lee, S.H.; Do, J.Y.; Kim, S.H.; Han, S.; Jung, H.Y.; Choi, J.Y.; Cho, J.H.; Kim, C.D.; et al. Association of hepcidin with anemia parameters in incident dialysis patients: Differences between dialysis modalities. Ther. Apher. Dial. 2020, 24, 4–16. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Rhee, D.K.; Malhotra, R.; Mayeur, C.; Hurst, L.A.; Ager, E.; Shelton, G.; Kramer, Y.; McCulloh, D.; Keefe, D.; et al. Progesterone receptor membrane component-1 regulates hepcidin biosynthesis. J. Clin. Investig. 2016, 126, 389–401. [Google Scholar] [CrossRef]
- Malyszko, J.; Malyszko, J.S.; Pawlak, K.; Mysliwiec, M. Hepcidin, iron status, and renal function in chronic renal failure, kidney transplantation, and hemodialysis. Am. J. Hematol. 2006, 81, 832–837. [Google Scholar] [CrossRef]
- Xue, D.; He, X.; Zhou, C. Serum hepcidin level correlates with hyperlipidemia status in patients following allograft renal transplantation. Transplant. Proc. 2014, 46, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.R.; An, W.; Feola, M.; Glassberg, J.A.; Ginzburg, Y. Hepcidin predicts decrease in erythroid expansion in sickle cell disease patients treated with inhaled steroids. Blood 2018, 132, 2339. [Google Scholar] [CrossRef]
- Ikeda, Y.; Tajima, S.; Izawa-Ishizawa, Y.; Kihira, Y.; Ishizawa, K.; Tomita, S.; Tsuchiya, K.; Tamaki, T. Estrogen regulates hepcidin expression via GPR30-BMP6-dependent signaling in hepatocytes. PLoS ONE 2012, 7, e0040465. [Google Scholar] [CrossRef]
- Silvestri, L.; Nai, A.; Dulja, A.; Pagani, A. Hepcidin and the BMP-SMAD pathway: An unexpected liaison. In Iron Metabolism: Hepcidin; Litwack, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 110, pp. 71–99. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A red carpet for iron metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchi, C.; Montosi, G.; Zhang, K.; Lamberti, I.; Duncan, S.A.; Kaufman, R.J.; Pietrangelo, A. ER stress controls iron metabolism through induction of hepcidin. Science 2009, 325, 877–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, Y.; Nakagawa, K.; Imai, Y.; Katagiri, T.; Koike, T.; Takaoka, K. Cyclic AMP enhances smad-mediated BMP signaling through PKA-CREB pathway. J. Bone Miner. Metab. 2008, 26, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Li, C.L.; Xu, X.L.; Zheng, Y.; Xiao, C.Y.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A role of SMAD4 in iron metabolism through the positive regulation of hepicidin expression. Cell Metab. 2005, 2, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Yung, L.-M.; Cheng, W.-H.; Yu, P.B.; Babitt, J.L.; Lin, H.Y.; Xia, Y. Hepcidin regulation by BMP signaling in macrophages is lipopolysaccharide dependent. PLoS ONE 2012, 7, e0044622. [Google Scholar] [CrossRef]
- Andriopoulos, B., Jr.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Herman-Edelstein, M.; Weinstein, T.; Levi, M. Bile acid receptors and the kidney. Curr. Opin. Nephrol. Hyperten. 2018, 27, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Meng, X.; Li, Y.; Sun, L.; Jiang, L.; Xuan, H.; Chen, X. Bile acids elevated in chronic periaortitis could activate far-nesoid-X-receptor to suppress IL-6 production by macrophages. Front. Immunol. 2021, 12, 632864. [Google Scholar] [CrossRef]
- Xiong, H.; Zhang, C.; Han, L.; Xu, T.; Saeed, K.; Han, J.; Liu, J.; Klaassen, C.D.; Gonzalez, F.J.; Lu, Y.; et al. Suppressed farnesoid X receptor by iron overload in mice and humans potentiates iron-induced hepatotoxicity. Hepatology 2022, 76, 387–403. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.X.; Jiang, T.; Takahashi, H.; Li, J.; Iwahashi, M.; Sutherland, E.; Levi, M. FXR modulates renal lipid metabolism and fibrosis in diabetic nephropathy. Faseb J. 2007, 21, A665. [Google Scholar] [CrossRef]
- Babitt, J.L.; Lin, H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Male | Female | |||||
|---|---|---|---|---|---|---|
| Indicators | Healthy (n = 37) | CKD (n = 147) | p-Value | Healthy (n = 32) | CKD (n = 76) | p-Value |
| RBC (1012/L) | 4.98 ± 0.43 | 3.55 ± 0.49 *** | <0.001 | 4.53 ± 0.21 | 3.48 ± 0.4 *** | <0.001 |
| HGB (g/L) | 151.00 ± 11.03 | 108.64 ± 15.81 *** | <0.001 | 133.91 ± 7.92 | 105.03 ± 12.97 *** | <0.001 |
| HCT (%) | 45.90 ± 3.74 | 33.85 ± 4.52 *** | <0.001 | 40.87 ± 1.95 | 32.88 ± 4.04 *** | <0.001 |
| MCH (g/L) | 30.93 ± 1.31 | 30.21 ± 2.12 * | 0.0137 | 29.59 ± 1.49 | 30.19 ± 1.49 | 0.0701 |
| MCHC (pg) | 336.05 ± 9.33 | 325.98 ± 13.32 *** | <0.001 | 327.56 ± 6.4 | 319.92 ± 11.37 *** | <0.001 |
| MCV (fL) | 92.14 ± 5.04 | 93 ± 6.28 | 0.3810 | 90.3 ± 3.48 | 94.71 ± 5.01 *** | <0.001 |
| Indicators | Fe (μmol/L) | FERR (ng/mL) | TIBC (μmol/L) | UIBC (μmol/L) | TSAT (%) | TRF (g/L) |
|---|---|---|---|---|---|---|
| Male | 12.89 ± 5.61 | 349.91 ± 418.82 | 45.75 ± 10.78 | 31.38 ± 10.49 | 29.5 ± 14.14 | 1.89 ± 0.52 |
| Female | 10.89 ± 4.39 | 403.44 ± 291.04 | 44.55 ± 12.3 | 29.81 ± 9.29 | 26.25 ± 11.31 | 1.84 ± 0.51 |
| Male | Female | |||||
|---|---|---|---|---|---|---|
| Bile Acids | Healthy (n = 37) (µg/L) | CKD (n = 147) (µg/L) | p-Value | Healthy (n = 32) (µg/L) | CKD (n = 76) (µg/L) | p-Value |
| Glycochenodeoxycholate | 690.48 ± 358.83 | 1238.75 ± 1005.47 *** | <0.001 | 501.15 ± 269.94 | 983.74 ± 623.56 *** | <0.001 |
| Chenodeoxycholic acid | 508.63 ± 355.17 | 186.53 ± 185.29 *** | <0.001 | 298.42 ± 125.93 | 143.43 ± 126.36 *** | <0.001 |
| Deoxycholate | 437.25 ± 294.28 | 129.36 ± 103.56 *** | <0.001 | 233.16 ± 91.1 | 125.92 ± 117.79 *** | <0.001 |
| Glycodeoxycholate | 160.23 ± 118.07 | 364.68 ± 387.49 *** | <0.001 | 77.97 ± 62.73 | 214.48 ± 236.24 *** | <0.001 |
| Glycoursodeoxycholic acid | 164.60 ± 112.96 | 189.18 ± 247.74 | 0.3949 | 100.66 ± 133.89 | 241.88 ± 308.93 ** | 0.0018 |
| Cholic acid | 108.62 ± 139.56 | 41.57 ± 61.98 * | 0.020 | 45.10 ± 28.06 | 24.63 ± 17.86 ** | 0.0022 |
| Glycocholic acid | 104.68 ± 86.29 | 239.67 ± 263.58 *** | <0.001 | 77.97 ± 62.73 | 214.48 ± 236.24 *** | <0.001 |
| Ursodeoxycholic acid | 70.96 ± 55.96 | 34.25 ± 39.02 ** | 0.0028 | 34.31 ± 23.71 | 36.13 ± 46.22 | 0.8037 |
| Taurochenodeoxycholate | 52.38 ± 66.25 | 224.17 ± 297.08 *** | <0.001 | 46.55 ± 24.59 | 129.01 ± 118.69 *** | <0.001 |
| Glycochenodeoxycholate 3-glucuronide | 39.46 ± 22.75 | 60.77 ± 64.43 ** | 0.0017 | 22.41 ± 15.31 | 37.25 ± 48.82 * | 0.0263 |
| Taurodeoxycholate | 14.86 ± 21.46 | 87.21 ± 140.56 *** | <0.001 | 29.70 ± 20.77 | 68.40 ± 82.71 *** | <0.001 |
| Taurocholic acid | 9.58 ± 9.97 | 67.15 ± 92.9 *** | <0.001 | 10.98 ± 5.27 | 39.34 ± 46.04 *** | <0.001 |
| Tauroursodeoxycholate | 6.38 ± 5.38 | 15.33 ± 18.95 *** | <0.001 | 2.88 ± 1.73 | 14.63 ± 16.29 *** | <0.001 |
| Conjugated bile acids | 1053.84 ± 550.55 | 2290.00 ± 1720.81 *** | <0.001 | 840.59 ± 412.21 | 1801.83 ± 1183.49 *** | <0.001 |
| Unconjugated bile acids | 794.52 ± 316.46 | 364.97 ± 257.36 *** | <0.001 | 553.15 ± 219.42 | 289.46 ± 169.32 *** | <0.001 |
| Conjugated bile acids/Unconjugated bile acids | 1.35 ± 0.83 | 7.99 ± 6.57 *** | <0.001 | 1.85 ± 1.4 | 7.34 ± 5.76 *** | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.-j.; Zhao, G.-p.; Wang, X.-f.; Liu, X.-x.; Li, Y.-x.; Qiu, L.-l.; Wang, X.-y.; Ren, F.-z. Glycochenodeoxycholate Affects Iron Homeostasis via Up-Regulating Hepcidin Expression. Nutrients 2022, 14, 3176. https://doi.org/10.3390/nu14153176
Wang L-j, Zhao G-p, Wang X-f, Liu X-x, Li Y-x, Qiu L-l, Wang X-y, Ren F-z. Glycochenodeoxycholate Affects Iron Homeostasis via Up-Regulating Hepcidin Expression. Nutrients. 2022; 14(15):3176. https://doi.org/10.3390/nu14153176
Chicago/Turabian StyleWang, Long-jiao, Guo-ping Zhao, Xi-fan Wang, Xiao-xue Liu, Yi-xuan Li, Li-li Qiu, Xiao-yu Wang, and Fa-zheng Ren. 2022. "Glycochenodeoxycholate Affects Iron Homeostasis via Up-Regulating Hepcidin Expression" Nutrients 14, no. 15: 3176. https://doi.org/10.3390/nu14153176
APA StyleWang, L. -j., Zhao, G. -p., Wang, X. -f., Liu, X. -x., Li, Y. -x., Qiu, L. -l., Wang, X. -y., & Ren, F. -z. (2022). Glycochenodeoxycholate Affects Iron Homeostasis via Up-Regulating Hepcidin Expression. Nutrients, 14(15), 3176. https://doi.org/10.3390/nu14153176