
Quercetin Increases Mitochondrial Biogenesis and Reduces Free Radicals in Neuronal SH-SY5Y Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Sirtinol, QE, and H2O2 Treatment
2.3. Cell Viability Assay
2.4. Measurement of Mitochondrial Biogenesis
2.5. Measurement of Cellular Adenosine Triphosphate Production
2.6. Measurement of Intracellular ROS
2.7. Measurement of Cellular Apoptosis
2.8. Western Blotting
2.9. Statistical Analysis
3. Results
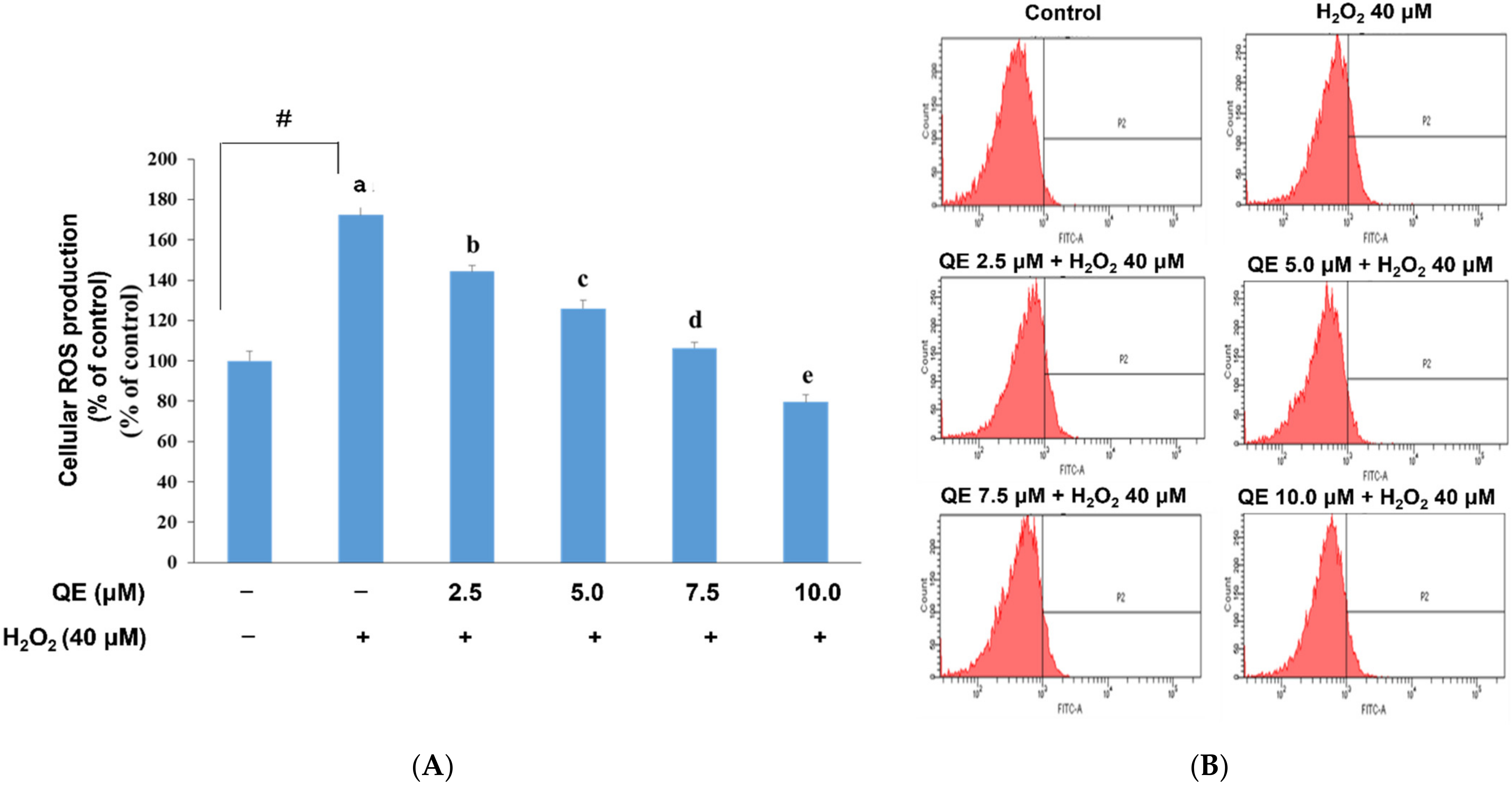
3.1. Effects of QE on Cellular ROS Production, Mitochondrial Biogenesis, and ATP Production in SH-SY5Y Cells with H2O2-Induced Oxidative Stress
3.2. Effects of QE on Mitochondrial Biogenesis-Related Protein Expression in SH-SY5Y Cells with H2O2-Induced Oxidative Stress
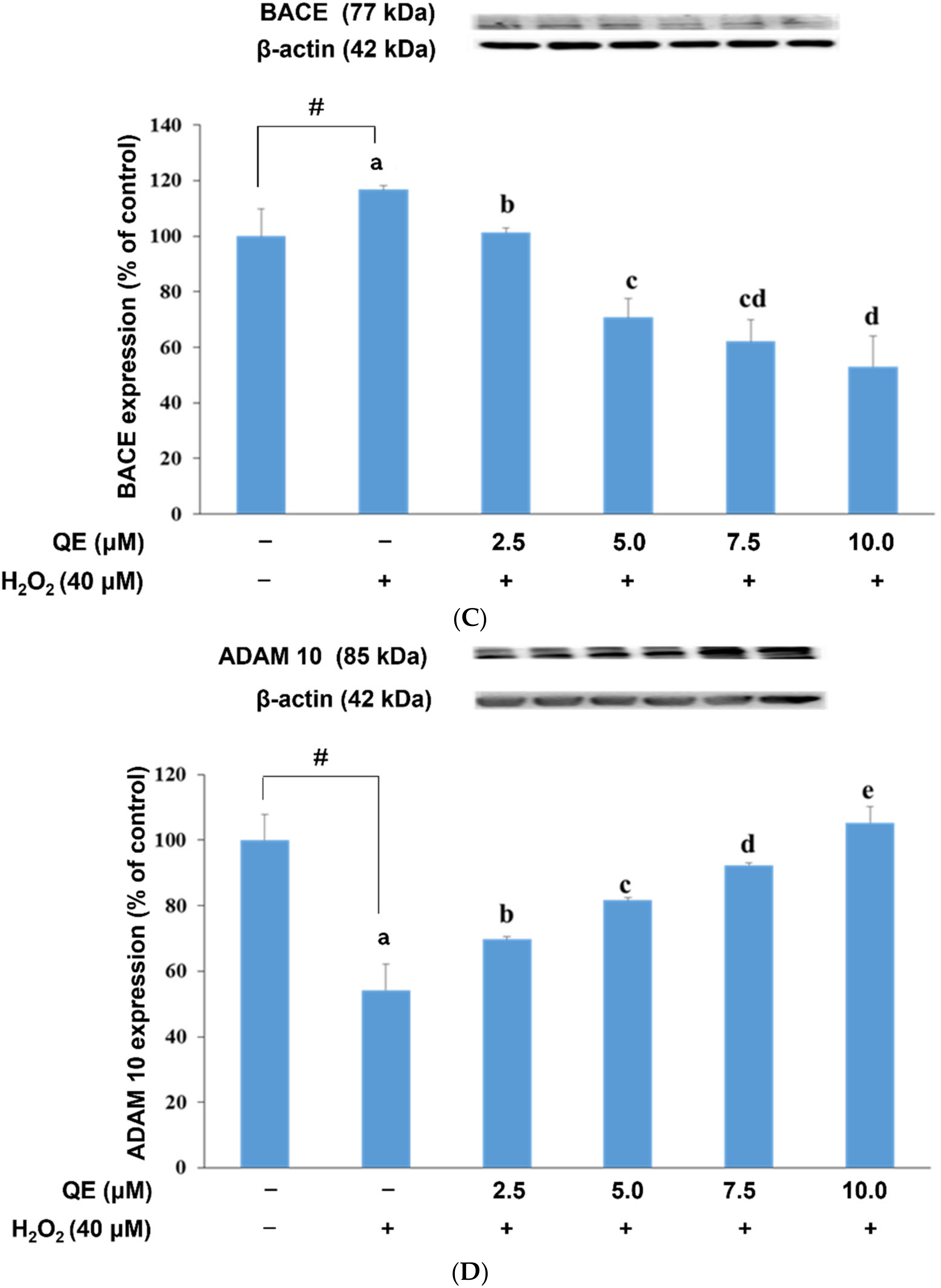
3.3. QE Reduced the Expression of Proteins Involved in the APP Amyloidogenic Pathway in SH-SY5Y Cells with H2O2-Induced Oxidative Stress
3.4. QE Reduced Cellular Apoptosis and Active Caspase-3 Expression in SH-SY5Y Cells with H2O2-Induced Oxidative Stress
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sekler, A.; Jimenez, J.M.; Rojo, L.; Pastene, E.; Fuentes, P.; Slachevsky, A.; Maccioni, R.B. Cognitive impairment and Alzheimer’s disease: Links with oxidative stress and cholesterol metabolism. Neuropsychiatr. Dis. Treat. 2008, 4, 715–722. [Google Scholar]
- Cabiscol, E.; Tamarit, J.; Ros, J. Protein carbonylation: Proteomics, specificity and relevance to aging. Mass Spectrom. Rev. 2014, 33, 21–48. [Google Scholar] [CrossRef]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp. Neurol. 2009, 218, 286–292. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003. [Google Scholar]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F. Mitochondria-ros crosstalk in the control of cell death and aging. J. Signal Transduct. 2012, 2012, 1–17. [Google Scholar] [CrossRef]
- Milton, N.G. Role of hydrogen peroxide in the aetiology of Alzheimer’s disease: Implications for treatment. Drugs Aging 2004, 21, 81–100. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Lindblom, R.; Higgins, G.; Coughlan, M.; de Haan, J.B. Targeting mitochondria and reactive oxygen species-driven pathogenesis in diabetic nephropathy. Rev. Diabet. Stud. RDS 2015, 12, 134. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant mechanisms in renal injury and disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef]
- Boengler, K.; Bulic, M.; Schreckenberg, R.; Schlüter, K.D.; Schulz, R. The gap junction modifier ZP1609 decreases cardiomyocyte hypercontracture following ischaemia/reperfusion independent from mitochondrial connexin 43. Br. J. Pharmacol. 2017, 174, 2060–2073. [Google Scholar] [CrossRef]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662–3670. [Google Scholar] [CrossRef]
- Burns, E.; Kruckeberg, T.; Comerford, L.; Buschmann, M.T. Thinning of capillary walls and declining numbers of endothelial mitochondria in the cerebral cortex of the aging primate, Macaca nemestrina. J. Gerontol. 1979, 34, 642–650. [Google Scholar] [CrossRef]
- Genova, M.L.; Ventura, B.; Giuliano, G.; Bovina, C.; Formiggini, G.; Castelli, G.P.; Lenaz, G. The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron–sulfur cluster N2. FEBS Lett. 2001, 505, 364–368. [Google Scholar] [CrossRef]
- Lenaz, G.; D’Aurelio, M.; Pich, M.M.; Genova, M.; Ventura, B.; Bovina, C.; Formiggini, G.; Castelli, G.P. Mitochondrial bioenergetics in aging. Biochim. Biophys. Acta (BBA)-Bioenerg. 2000, 1459, 397–404. [Google Scholar] [CrossRef]
- Dencher, N.A.; Frenzel, M.; Reifschneider, N.H.; Sugawa, M.; Krause, F. Proteome alterations in rat mitochondria caused by aging. Ann. N. Y. Acad. Sci. 2007, 1100, 291–298. [Google Scholar] [CrossRef]
- Ojaimi, J.; Masters, C.L.; Opeskin, K.; McKelvie, P.; Byrne, E. Mitochondrial respiratory chain activity in the human brain as a function of age. Mech. Ageing Dev. 1999, 111, 39–47. [Google Scholar] [CrossRef]
- Gureev, A.P.; Syromyatnikov, M.Y.; Gorbacheva, T.M.; Starkov, A.A.; Popov, V.N. Methylene blue improves sensorimotor phenotype and decreases anxiety in parallel with activating brain mitochondria biogenesis in mid-age mice. Neurosci. Res. 2016, 113, 19–27. [Google Scholar] [CrossRef]
- Conley, K.E.; Jubrias, S.A.; Esselman, P.C. Oxidative capacity and ageing in human muscle. J. Physiol. 2000, 526, 203–210. [Google Scholar] [CrossRef]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef]
- Jeong, S.H.; Kim, H.K.; Song, I.-S.; Noh, S.J.; Marquez, J.; Ko, K.S.; Rhee, B.D.; Kim, N.; Mishchenko, N.P.; Fedoreyev, S.A. Echinochrome A Increases Mitochondrial Mass and Function by Modulating Mitochondrial Biogenesis Regulatory Genes. Mar. Drugs 2014, 12, 4602–4615. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim. Biophys. Acta BBA Gene Struct. Expr. 2002, 1576, 1–14. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta BBA Mol. Cell Res. 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Velliquette, R.A.; O’Connor, T.; Vassar, R. Energy inhibition elevates β-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: Possible early events in Alzheimer’s disease pathogenesis. J. Neurosci. 2005, 25, 10874–10883. [Google Scholar] [CrossRef]
- Bischoff, S.C. Quercetin: Potentials in the prevention and therapy of disease. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 733–740. [Google Scholar] [CrossRef]
- Boots, A.W.; Li, H.; Schins, R.P.; Duffin, R.; Heemskerk, J.W.; Bast, A.; Haenen, G.R. The quercetin paradox. Toxicol. Appl. Pharmacol. 2007, 222, 89–96. [Google Scholar] [CrossRef]
- Davis, J.M.; Murphy, E.A.; Carmichael, M.D.; Davis, B. Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2009, 296, R1071–R1077. [Google Scholar] [CrossRef] [PubMed]
- Suematsu, N.; Hosoda, M.; Fujimori, K. Protective effects of quercetin against hydrogen peroxide-induced apoptosis in human neuronal SH-SY5Y cells. Neurosci. Lett. 2011, 504, 223–227. [Google Scholar] [CrossRef]
- Adiwidjaja, J.; McLachlan, A.J.; Boddy, A.V. Curcumin as a clinically-promising anti-cancer agent: Pharmacokinetics and drug interactions. Expert Opin. Drug Metab. Toxicol. 2017, 13, 953–972. [Google Scholar] [CrossRef] [PubMed]
- Mailliot, C.; Bussière, T.; Caillet-Boudin, M.L.; Delacourte, A.; Buée, L. Alzheimer-specific epitope of AT100 in transfected cell lines with tau: Toward an efficient cell model of tau abnormal phosphorylation. Neurosci. Lett. 1998, 255, 13–16. [Google Scholar] [CrossRef]
- Zhong, J.; Iqbal, K.; Grundke-Iqbal, I. Hyperphosphorylated tau in SY5Y cells: Similarities and dissimilarities to abnormally hyperphosphorylated tau from Alzheimer disease brain. FEBS Lett. 1999, 453, 224–228. [Google Scholar] [CrossRef]
- Misonou, H.; Morishima-Kawashima, M.; Ihara, Y. Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry 2000, 39, 6951–6959. [Google Scholar] [CrossRef]
- Wang, J.-z.; Tung, Y.C.; Wang, Y.; Li, X.T.; Iqbal, K.; Grundke-Iqbal, I. Hyperphosphorylation and accumulation of neurofilament proteins in Alzheimer disease brain and in okadaic acid-treated SY5Y cells. FEBS Lett. 2001, 507, 81–87. [Google Scholar] [CrossRef]
- Olivieri, G.; Novakovic, M.; Savaskan, E.; Meier, F.; Baysang, G.; Brockhaus, M.; Müller-Spahn, F. The effects of beta-estradiol on SHSY5Y neuroblastoma cells during heavy metal induced oxidative stress, neurotoxicity and beta-amyloid secretion. Neuroscience 2002, 113, 849–855. [Google Scholar] [CrossRef]
- Jämsä, A.; Hasslund, K.; Cowburn, R.F.; Bäckström, A.; Vasänge, M. The retinoic acid and brain-derived neurotrophic factor differentiated SH-SY5Y cell line as a model for Alzheimer’s disease-like tau phosphorylation. Biochem. Biophys. Res. Commun. 2004, 319, 993–1000. [Google Scholar] [CrossRef]
- Ferrari, A.; Hoerndli, F.; Baechi, T.; Nitsch, R.M.; Götz, J. β-Amyloid induces paired helical filament-like tau filaments in tissue culture. J. Biol. Chem. 2003, 278, 40162–40168. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Kleemann, R.; Verschuren, L.; Morrison, M.; Zadelaar, S.; van Erk, M.J.; Wielinga, P.Y.; Kooistra, T. Anti-inflammatory, anti-proliferative and anti-atherosclerotic effects of quercetin in human in vitro and in vivo models. Atherosclerosis 2011, 218, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Hollensworth, S.B.; Shen, C.-C.; Sim, J.E.; Spitz, D.R.; Wilson, G.L.; LeDoux, S.P. Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic. Biol. Med. 2000, 28, 1161–1174. [Google Scholar] [CrossRef]
- Bao, D.; Wang, J.; Pang, X.; Liu, H. Protective effect of quercetin against oxidative stress-induced cytotoxicity in rat pheochromocytoma (PC-12) cells. Molecules 2017, 22, 1122. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, Z.; Shen, R.; Zhong, W.; Zheng, H.; Chen, Z.; Tang, J.; Zhu, J. Resveratrol Improves Mitochondrial Biogenesis Function and Activates PGC-1α Pathway in a Preclinical Model of Early Brain Injury Following Subarachnoid Hemorrhage. Front. Mol. Biosci. 2021, 8, 223. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Viswakarma, N.; Jia, Y.; Bai, L.; Vluggens, A.; Borensztajn, J.; Xu, J.; Reddy, J.K. Coactivators in PPAR-regulated gene expression. PPAR Res. 2010, 2010, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator–activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1α expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.J.; Dong, S.Y.; Cui, X.X.; Feng, Y.; Liu, T.; Yin, M.; Kuo, S.H.; Tan, E.K.; Zhao, W.J.; Wu, Y.C. Resveratrol alleviates MPTP-induced motor impairments and pathological changes by autophagic degradation of α-synuclein via SIRT1-deacetylated LC3. Mol. Nutr. Food Res. 2016, 60, 2161–2175. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Yan, S.S. Role of mitochondrial amyloid-β in Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, S569–S578. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Feng, L.; Dong, G.; Tao, Y.; Mei, L.; Xie, Z.-J.; Dong, Z. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc. Natl. Acad. Sci. USA 2007, 104, 11649–11654. [Google Scholar] [CrossRef] [PubMed]
- Fossati, S.; Giannoni, P.; Solesio, M.E.; Cocklin, S.L.; Cabrera, E.; Ghiso, J.; Rostagno, A. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol. Dis. 2016, 86, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Role of free radicals in the neurodegenerative diseases: Therapeutic implications for antioxidant treatment. Drugs Aging 2001, 18, 685–716. [Google Scholar]
- Halliwell, B.; Clement, M.V.; Long, L.H. Hydrogen peroxide in the human body. FEBS Lett. 2000, 486, 10–13. [Google Scholar] [CrossRef]
- Chen, L.; Liu, L.; Yin, J.; Luo, Y.; Huang, S. Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int. J. Biochem. Cell Biol. 2009, 41, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, H.; Sun, Y.; Lin, X.; Chen, B.; Tan, C.; Cao, G.; Wang, Z. Protective effects of salidroside on hydrogen peroxide-induced apoptosis in SH-SY5Y human neuroblastoma cells. Eur. J. Pharmacol. 2007, 564, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Xu, S.; Zhong, M.; Zhang, L.; Wang, Y.; Zhou, Z.; Hao, Y.; Zhang, W.; Yang, X.; Wei, A.; Pei, L. Overexpression of Tfam protects mitochondria against β-amyloid-induced oxidative damage in SH-SY5Y cells. FEBS J. 2009, 276, 3800–3809. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Amić, D.; Davidović-Amić, D.; Bešlo, D.; Trinajstić, N. Structure-radical scavenging activity relationships of flavonoids. Croat. Chem. Acta 2003, 76, 55–61. [Google Scholar]
- Alam, T.I.; Kanki, T.; Muta, T.; Ukaji, K.; Abe, Y.; Nakayama, H.; Takio, K.; Hamasaki, N.; Kang, D. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 2003, 31, 1640–1645. [Google Scholar] [CrossRef]
- Kang, D.; Kim, S.H.; Hamasaki, N. Mitochondrial transcription factor A (TFAM): Roles in maintenance of mtDNA and cellular functions. Mitochondrion 2007, 7, 39–44. [Google Scholar] [CrossRef]
- Fujita, Y.; Izawa, Y.; Ali, N.; Kanematsu, Y.; Tsuchiya, K.; Hamano, S.; Tamaki, T.; Yoshizumi, M. Pramipexole protects against H2O2-induced PC12 cell death. Naunyn Schmiedeberg’s Arch. Pharmacol. 2006, 372, 257–266. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of mitochondrial biogenesis as a way for active longevity: Interaction between the Nrf2 and PGC-1α signaling pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Labinskyy, N.; Pinto, J.T.; Ballabh, P.; Zhang, H.; Losonczy, G.; Pearson, K.; de Cabo, R.; Pacher, P.; Zhang, C. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H13–H20. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.-W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in cellular functions: Role of polyphenols. Arch. Biochem. Biophys. 2010, 501, 79–90. [Google Scholar] [CrossRef]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta (BBA)-Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Tamagno, E.; Parola, M.; Bardini, P.; Piccini, A.; Borghi, R.; Guglielmotto, M.; Santoro, G.; Davit, A.; Danni, O.; Smith, M. β-Site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J. Neurochem. 2005, 92, 628–636. [Google Scholar] [CrossRef]
- Prinzen, C.; Muller, U.; Endres, K.; Fahrenholz, F.; Postina, R. Genomic structure and functional characterization of the human ADAM10 promoter. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1522–1524. [Google Scholar] [CrossRef]
- Tippmann, F.; Hundt, J.; Schneider, A.; Endres, K.; Fahrenholz, F. Up-regulation of the alpha-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 1643–1654. [Google Scholar] [CrossRef]
- Wang, R.; Li, J.J.; Diao, S.; Kwak, Y.D.; Liu, L.; Zhi, L.; Bueler, H.; Bhat, N.R.; Williams, R.W.; Park, E.A.; et al. Metabolic stress modulates Alzheimer’s beta-secretase gene transcription via SIRT1-PPARgamma-PGC-1 in neurons. Cell Metab. 2013, 17, 685–694. [Google Scholar] [CrossRef]
- Li, Y.-P.; Bushnell, A.F.; Lee, C.-M.; Perlmutter, L.S.; Wong, S.K.-F. β-Amyloid induces apoptosis in human-derived neurotypic SH-SY5Y cells. Brain Res. 1996, 738, 196–204. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, C.-L.; Kao, N.-J.; Lin, C.-I.; Cross, T.-W.L.; Lin, S.-H. Quercetin Increases Mitochondrial Biogenesis and Reduces Free Radicals in Neuronal SH-SY5Y Cells. Nutrients 2022, 14, 3310. https://doi.org/10.3390/nu14163310
Ho C-L, Kao N-J, Lin C-I, Cross T-WL, Lin S-H. Quercetin Increases Mitochondrial Biogenesis and Reduces Free Radicals in Neuronal SH-SY5Y Cells. Nutrients. 2022; 14(16):3310. https://doi.org/10.3390/nu14163310
Chicago/Turabian StyleHo, Chia-Ling, Ning-Jo Kao, Ching-I Lin, Tzu-Wen L. Cross, and Shyh-Hsiang Lin. 2022. "Quercetin Increases Mitochondrial Biogenesis and Reduces Free Radicals in Neuronal SH-SY5Y Cells" Nutrients 14, no. 16: 3310. https://doi.org/10.3390/nu14163310
APA StyleHo, C. -L., Kao, N. -J., Lin, C. -I., Cross, T. -W. L., & Lin, S. -H. (2022). Quercetin Increases Mitochondrial Biogenesis and Reduces Free Radicals in Neuronal SH-SY5Y Cells. Nutrients, 14(16), 3310. https://doi.org/10.3390/nu14163310