

The Evolution of Ketosis: Potential Impact on Clinical Conditions

 , , , , ,
, , , , , 
Abstract
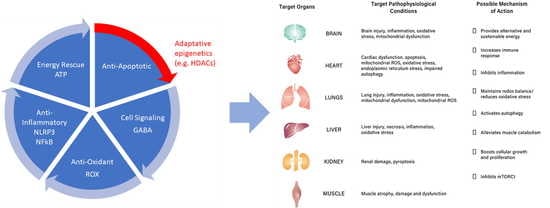
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
3. Metabolism of Ketones: Ketogenesis and Ketolysis
4. Endogenous Sources of Ketone Bodies
5. Exogenous Sources of Ketone Bodies
6. Metabolic Functions of Ketone Bodies in Vital Organs
7. Biological Properties of Ketone Bodies
8. Role of Ketone Bodies in Pathophysiological Ailments
8.1. Heart Failure
8.2. Kidneys and Liver Diseases
8.3. Brain Injury and Neuronal Diseases
8.4. Muscle Weakness
9. Neuroprotective Actions of Ketone Bodies
Ketone Bodies Influence Neurotransmitters
10. Discussion
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: A physiological basis for exogenous supplementation. J. Physiol. 2016, 595, 2857–2871. [Google Scholar] [CrossRef]
- Rui, L. Energy Metabolism in the Liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arbelaez, D.; Crujeiras, A.B.; Castro, A.I.; Goday, A.; Mas-Lorenzo, A.; Bellon, A.; Tejera, C.; Bellido, D.; Galban, C.; Sajoux, I.; et al. Acid–base safety during the course of a very low-calorie-ketogenic diet. Endocrine 2017, 58, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.L.; Holcomb, L.E.; Kolwicz, S.C., Jr. Ketogenic Diets and Exercise Performance. Nutrients 2019, 11, 2296. [Google Scholar] [CrossRef] [PubMed]
- Rowe, P.; O’Neill, C.; DeWitt, E.; Kolwicz, S.C. Endurance Exercise Capacity and Substrate Metabolism in Male and Female Mice. FASEB J. 2019, 33, 698.1. [Google Scholar] [CrossRef]
- Wentz, A.; D’Avignon, D.A.; Weber, M.L.; Cotter, D.G.; Doherty, J.M.; Kerns, R.; Nagarajan, R.; Reddy, N.; Sambandam, N.; Crawford, P.A. Adaptation of Myocardial Substrate Metabolism to a Ketogenic Nutrient Environment. J. Biol. Chem. 2010, 285, 24447–24456. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, A.; Crepaldi, C.; Miola, M.; Maran, A.; Pengo, V.; Tiengo, A.; Del Prato, S. High blood ketone body concentration in Type 2 non-insulin dependent diabetic patients. J. Endocrinol. Investig. 1996, 19, 99–105. [Google Scholar] [CrossRef]
- Guerci, B.; Benichou, M.; Floriot, M.; Bohme, P.; Fougnot, S.; Franck, P.; Drouin, P. Accuracy of an Electrochemical Sensor for Measuring Capillary Blood Ketones by Fingerstick Samples During Metabolic Deterioration After Continuous Subcutaneous Insulin Infusion Interruption in Type 1 Diabetic Patients. Diabetes Care 2003, 26, 1137–1141. [Google Scholar] [CrossRef]
- Aksu, N.; Akcora, Z.; Ilhan, B.; Bayar, O.; Akkas, M. Ketacidosis due to starvation. Eur. J. Emerg. Med. 2018, 17, 39–40. [Google Scholar]
- Cabrera-Mulero, A.; Tinahones, A.; Bandera, B.; Moreno-Indias, I.; Macías-González, M.; Tinahones, F.J. Keto microbiota: A powerful contributor to host disease recovery. Rev. Endocr. Metab. Disord. 2019, 20, 415–425. [Google Scholar] [CrossRef]
- Alsharairi, N.A. The Role of Short-Chain Fatty Acids in the Interplay between a Very Low-Calorie Ketogenic Diet and the Infant Gut Microbiota and Its Therapeutic Implications for Reducing Asthma. Int. J. Mol. Sci. 2020, 21, 9580. [Google Scholar] [CrossRef] [PubMed]
- Alsharairi, N.A. The Role of Short-Chain Fatty Acids in Mediating Very Low-Calorie Ketogenic Diet-Infant Gut Microbiota Relationships and Its Therapeutic Potential in Obesity. Nutrients 2021, 13, 3702. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. β-hydroxybutyrate: Much more than a metabolite. Diabetes Res. Clin. Pract. 2014, 106, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Sasaki, D.; Hannya, A. In vitro human colonic microbiota utilises D--hydroxybutyrate to increase butyro-genesis. Sci. Rep. 2020, 10, 8516. [Google Scholar] [CrossRef]
- Tzika, E.; Dreker, T.; Imhof, A. Epigenetics and Metabolism in Health and Disease. Front. Genet. 2018, 9, 361. [Google Scholar] [CrossRef]
- Prescott, S.; Saffery, R. The role of epigenetic dysregulation in the epidemic of allergic disease. Clin. Epigenet. 2011, 2, 223–232. [Google Scholar] [CrossRef]
- Moher, D.; Shamseer, L.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; Group, P.-P. Preferred reporting items for systematic review and meta-analysis protocols (prisma-p) 2015 statement. Syst. Rev. 2015, 4, 1. [Google Scholar] [CrossRef]
- Van Enst, W.A.; Ochodo, E.; Scholten, R.J.P.M.; Hooft, L.; Leeflang, M.M. Investigation of publication bias in meta-analyses of diagnostic test accuracy: A meta-epidemiological study. BMC Med. Res. Methodol. 2014, 14, 70. [Google Scholar] [CrossRef]
- Desrochers, S.; David, F.; Garneau, M.; Jetté, M.; Brunengraber, H. Metabolism of R- and S-1,3-butanediol in perfused livers from meal-fed and starved rats. Biochem. J. 1992, 285, 647–653. [Google Scholar] [CrossRef]
- Najac, C.; Radoul, M.; Le Page, L.M.; Batsios, G.; Subramani, E.; Viswanath, P.; Gillespie, A.M.; Ronen, S.M. In vivo investigation of hyperpolarized [1,3-13C2]acetoacetate as a metabolic probe in normal brain and in glioma. Sci. Rep. 2019, 9, 3402. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed]
- Fukao, T.; Song, X.-Q.; A Mitchell, G.; Yamaguchi, S.; Sukegawa, K.; Or, T.; Kondo, N. Enzymes of Ketone Body Utilization in Human Tissues: Protein and Messenger RNA Levels of Succinyl-Coenzyme A (CoA):3-Ketoacid CoA Transferase and Mitochondrial and Cytosolic Acetoacetyl-CoA Thiolases. Pediatr. Res. 1997, 42, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Covián, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; De Los Reyes-Gavilán, C.G.; Salazar, N. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Gerhart, D.Z.; Enerson, B.E.; Zhdankina, O.Y.; Leino, R.L.; Drewes, L.R. Expression of monocarboxylate transporter MCT1 by brain endothelium and glia in adult and suckling rats. Am. J. Physiol. Metab. 1997, 273, E207–E213. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef]
- Chowdhury, G.M.; Jiang, L.; Rothman, D.L.; Behar, K.L. The Contribution of Ketone Bodies to Basal and Activity-Dependent Neuronal Oxidation in Vivo. J. Cereb. Blood Flow Metab. 2014, 34, 1233–1242. [Google Scholar] [CrossRef]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef]
- Indrio, F.; Martini, S.; Francavilla, R.; Corvaglia, L.; Cristofori, F.; Mastrolia, S.A.; Neu, J.; Rautava, S.; Spena, G.R.; Raimondi, F.; et al. Epigenetic Matters: The Link between Early Nutrition, Microbiome, and Long-term Health Development. Front. Pediatr. 2017, 5, 178. [Google Scholar] [CrossRef]
- Bridgman, S.L.; Azad, M.B.; Field, C.; Haqq, A.M.; Becker, A.B.; Mandhane, P.J.; Subbarao, P.; Turvey, S.; Sears, M.R.; Scott, J.A.; et al. Fecal Short-Chain Fatty Acid Variations by Breastfeeding Status in Infants at 4 Months: Differences in Relative versus Absolute Concentrations. Front. Nutr. 2017, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zouhal, H.; Saeidi, A.; Salhi, A.; Li, H.; Essop, M.F.; Laher, I.; Rhibi, F.; Amani-Shalamzari, S.; Ben Abderrahman, A. Exercise Training and Fasting: Current Insights. Open Access J. Sports Med. 2020, 11, 224919. [Google Scholar] [CrossRef] [PubMed]
- Kesl, S.L.; Poff, A.M.; Ward, N.P.; Fiorelli, T.N.; Ari, C.; Van Putten, A.J.; Sherwood, J.W.; Arnold, P.; D’Agostino, D.P. Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague–Dawley rats. Nutr. Metab. 2016, 13, 9. [Google Scholar] [CrossRef]
- White, H.; Heffernan, A.J.; Worrall, S.; Grunsfeld, A.; Thomas, M. A Systematic Review of Intravenous β-Hydroxybutyrate Use in Humans–A Promising Future Therapy? Front. Med. 2021, 8, 740374. [Google Scholar] [CrossRef] [PubMed]
- Cox, P.J.; Kirk, T.; Ashmore, T.; Willerton, K.; Evans, R.; Smith, A.; Murray, A.J.; Stubbs, B.; West, J.; McLure, S.W.; et al. Nutritional Ketosis Alters Fuel Preference and Thereby Endurance Performance in Athletes. Cell Metab. 2016, 24, 256–268. [Google Scholar] [CrossRef]
- Murray, A.J.; Knight, N.S.; Cole, M.A.; Cochlin, L.E.; Carter, E.; Tchabanenko, K.; Pichulik, T.; Gulston, M.K.; Atherton, H.J.; Schroeder, M.A.; et al. Novel ketone diet enhances physical and cognitive performance. FASEB J. 2016, 30, 4021–4032. [Google Scholar] [CrossRef]
- Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 309–319. [Google Scholar] [CrossRef]
- Paoli, A. Ketogenic Diet for Obesity: Friend or Foe? Int. J. Environ. Res. Public Health 2014, 11, 2092–2107. [Google Scholar] [CrossRef]
- Wheless, J.W. History of the ketogenic diet. Epilepsia 2008, 49 (Suppl. 8), 3–5. [Google Scholar] [CrossRef]
- Browning, J.D.; A Baker, J.; Rogers, T.; Davis, J.; Satapati, S.; Burgess, S.C. Short-term weight loss and hepatic triglyceride reduction: Evidence of a metabolic advantage with dietary carbohydrate restriction. Am. J. Clin. Nutr. 2011, 93, 1048–1052. [Google Scholar] [CrossRef]
- Garbow, J.R.; Doherty, J.M.; Schugar, R.C.; Travers, S.; Weber, M.L.; Wentz, A.; Ezenwajiaku, N.; Cotter, D.G.; Brunt, E.M.; Crawford, P.A. Hepatic steatosis, inflammation, and ER stress in mice maintained long term on a very low-carbohydrate ketogenic diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G956–G967. [Google Scholar] [CrossRef] [PubMed]
- De Koning, L.; Fung, T.T.; Liao, X.; Chiuve, S.E.; Rimm, E.B.; Willett, W.C.; Spiegelman, D.; Hu, F.B. Low-carbohydrate diet scores and risk of type 2 diabetes in men. Am. J. Clin. Nutr. 2011, 93, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Dashti, H.M.; Al-Zaid, N.S.; Mathew, T.C.; Al-Mousawi, M.; Talib, H.; Asfar, S.K.; Behbahani, A.I. Long Term Effects of Ketogenic Diet in Obese Subjects with High Cholesterol Level. Mol. Cell. Biochem. 2006, 286, 19641727. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1060–H1076. [Google Scholar] [CrossRef]
- Kruszynska, Y.T.; McCormack, J.G.; McIntyre, N. Effects of glycogen stores and non-esterified fatty acid availability on insulin-stimulated glucose metabolism and tissue pyruvate dehydrogenase activity in the rat. Diabetologia 1991, 34, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Waagepetersen, H.S. Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 2018, 293, 7108–7116. [Google Scholar] [CrossRef]
- Pearson, T.S.; Akman, C.; Hinton, V.J.; Engelstad, K.; De Vivo, D.C. Phenotypic Spectrum of Glucose Transporter Type 1 Deficiency Syndrome (Glut1 DS). Curr. Neurol. Neurosci. Rep. 2013, 13, 342. [Google Scholar] [CrossRef]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 15953344. [Google Scholar] [CrossRef]
- Koutnik, A.P.; D’Agostino, D.P.; Egan, B. Anticatabolic Effects of Ketone Bodies in Skeletal Muscle. Trends Endocrinol. Metab. 2019, 30, 227–229. [Google Scholar] [CrossRef]
- Roediger, W.E.W. The starved colon—Diminished mucosal nutrition, diminished absorption, and colitis. Dis. Colon Rectum 1990, 33, 858–862. [Google Scholar] [CrossRef]
- Cheng, C.-W.; Biton, M.; Haber, A.L.; Gunduz, N.; Eng, G.; Gaynor, L.T.; Tripathi, S.; Calibasi-Kocal, G.; Rickelt, S.; Butty, V.L.; et al. Ketone Body Signaling Mediates Intestinal Stem Cell Homeostasis and Adaptation to Diet. Cell 2019, 178, 1115–1131.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kann, O.; Kovács, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; Brustovetsky, T.; Jemmerson, R.; Dubinsky, J.M. Calcium-induced Cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J. Neurochem. 2002, 80, 207–218. [Google Scholar] [CrossRef]
- Hansson, M.J.; Mansson, R.; Mattiasson, G.; Ohlsson, J.; Karlsson, J.; Keep, M.F.; Elmer, E. Brain-derived respiring mitochondria exhibit homogeneous, complete and cyclosporin-sensitive permeability transition. J. Neurochem. 2004, 89, 715–729. [Google Scholar] [CrossRef]
- Calabrese, V.; Scapagnini, G.; Stella, A.M.G.; Bates, T.; Clark, J.B. Mitochondrial Involvement in Brain Function and Dysfunction: Relevance to Aging, Neurodegenerative Disorders and Longevity. Neurochem. Res. 2001, 26, 739–764. [Google Scholar] [CrossRef]
- Stafstrom, C.E.; Rho, J.M. The Ketogenic Diet as a Treatment Paradigm for Diverse Neurological Disorders. Front. Pharmacol. 2012, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.M.; Johannsen, D.L.; Ravussin, E. Skeletal Muscle Mitochondria and Aging: A Review. J. Aging Res. 2012, 2012, 194821. [Google Scholar] [CrossRef]
- Olmos, Y.; Sanchez-Gomez, F.J.; Wild, B.; Garcia-Quintans, N.; Cabezudo, S.; Lamas, S.; Monsalve, M. SirT1 Regulation of Antioxidant Genes Is Dependent on the Formation of a FoxO3a/PGC-1α Complex. Antioxidants Redox Signal. 2013, 19, 1507–1521. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Ciriolo, M.R. Extranuclear localization of SIRT1 and PGC-1α: An insight into possible roles in diseases associated with mitochondrial dysfunction. Curr. Mol. Med. 2013, 13, 140–154. [Google Scholar] [CrossRef]
- Milder, J.; Patel, M. Modulation of oxidative stress and mitochondrial function by the ketogenic diet. Epilepsy Res. 2011, 100, 295–303. [Google Scholar] [CrossRef]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2016, 42, 35–49. [Google Scholar] [CrossRef]
- Haces, M.L.; Hernández-Fonseca, K.; Medina-Campos, O.N.; Montiel, T.; Pedraza-Chaverri, J.; Massieu, L. Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp. Neurol. 2008, 211, 85–96. [Google Scholar] [CrossRef]
- Vergati, M.; Krasniqi, E.; Monte, G.D.; Riondino, S.; Vallone, D.; Guadagni, F.; Ferroni, P.; Roselli, M. Ketogenic Diet and Other Dietary Intervention Strategies in the Treatment of Cancer. Curr. Med. Chem. 2017, 24, 1170–1185. [Google Scholar] [CrossRef]
- Veech, R.L. Ketone ester effects on metabolism and transcription. J. Lipid Res. 2014, 55, 2004–2006. [Google Scholar] [CrossRef]
- Gough, S.M.; Casella, A.; Ortega, K.J.; Hackam, A.S. Neuroprotection by the Ketogenic Diet: Evidence and Controversies. Front. Nutr. 2021, 8, 782657. [Google Scholar] [CrossRef]
- Greco, T.; Glenn, T.; A Hovda, D.; Prins, M.L. Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J. Cereb. Blood Flow Metab. 2016, 36, 1603–1613. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.-D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Guo, M.; Wang, X.; Zhao, Y.; Yang, Q.; Ding, H.; Dong, Q.; Chen, X.; Cui, M. Ketogenic Diet Improves Brain Ischemic Tolerance and Inhibits NLRP3 Inflammasome Activation by Preventing Drp1-Mediated Mitochondrial Fission and Endoplasmic Reticulum Stress. Front. Mol. Neurosci. 2018, 11, 86. [Google Scholar] [CrossRef]
- Forsythe, C.E.; Phinney, S.D.; Fernandez, M.L.; Quann, E.E.; Wood, R.J.; Bibus, D.M.; Kraemer, W.J.; Feinman, R.D.; Volek, J.S. Comparison of Low Fat and Low Carbohydrate Diets on Circulating Fatty Acid Composition and Markers of Inflammation. Lipids 2007, 43, 65–77. [Google Scholar] [CrossRef]
- Offermanns, S. Hydroxy-Carboxylic Acid Receptor Actions in Metabolism. Trends Endocrinol. Metab. 2017, 28, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Rho, J.M. The New Ketone Alphabet Soup: BHB, HCA, and HDAC. Epilepsy Curr. 2014, 14, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Ono, J.G.; Worgall, T.S.; Worgall, S. 17q21 locus and ORMDL3: An increased risk for childhood asthma. Pediatr. Res. 2013, 75, 165–170. [Google Scholar] [CrossRef]
- Bae, H.R.; Kim, D.H.; Park, M.H.; Lee, B.; Kim, M.J.; Lee, E.K.; Chung, K.W.; Kim, S.M.; Im, D.S.; Chung, H.Y. β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget 2016, 7, 66444–66454. [Google Scholar] [CrossRef]
- Sada, N.; Fujita, Y.; Mizuta, N.; Ueno, M.; Furukawa, T.; Yamashita, T. Inhibition of HDAC increases BDNF expression and promotes neuronal rewiring and functional recovery after brain injury. Cell Death Dis. 2020, 11, 655. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Meroni, E.; Papini, N.; Criscuoli, F.; Casiraghi, M.C.; Massaccesi, L.; Basilico, N.; Erba, D. Metabolic Responses in Endothelial Cells Following Exposure to Ketone Bodies. Nutrients 2018, 10, 250. [Google Scholar] [CrossRef]
- Gunst, J. Recovery from critical illness-induced organ failure: The role of autophagy. Crit. Care 2017, 21, 209. [Google Scholar] [CrossRef]
- Flower, L.; Page, A.; Puthucheary, Z. Should nutritional therapy be modified to account for mitochondrial dysfunction in critical illness? J. Parenter. Enter. Nutr. 2021, 45, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Morales, P.; Pedraza-Chaverri, J.; Tapia, E. Ketone bodies, stress response, and redox homeostasis. Redox Biol. 2019, 29, 101395. [Google Scholar] [CrossRef] [PubMed]
- McClave, S.A.; Taylor, B.E.; Martindale, R.G.; Warren, M.M.; Johnson, D.R.; Braunschweig, C.; McCarthy, M.S.; Davanos, E.; Rice, T.W.; Cresci, G.A.; et al. Guidelines for the Provision and Assessment of Nutrition Support Therapy in the Adult Critically Ill Patient: Society of Critical Care Medicine (SCCM) and American Society for Parenteral and Enteral Nutrition (ASPEN). JPEN J. Parenter. Enter. Nutr. 2016, 40, 159–211. [Google Scholar] [CrossRef] [PubMed]
- Yao, A.; Li, Z.; Lyu, J.; Yu, L.; Wei, S.; Xue, L.; Wang, H.; Chen, G.-Q. On the nutritional and therapeutic effects of ketone body d-β-hydroxybutyrate. Appl. Microbiol. Biotechnol. 2021, 105, 6229–6243. [Google Scholar] [CrossRef]
- Amjad, S.; Nisar, S.; Bhat, A.A.; Shah, A.R.; Frenneaux, M.P.; Fakhro, K.; Haris, M.; Reddy, R.; Patay, Z.; Baur, J.; et al. Role of NAD+ in regulating cellular and metabolic signaling pathways. Mol. Metab. 2021, 49, 101195. [Google Scholar] [CrossRef]
- Waldman, H.S.; McAllister, M.J. Exogenous Ketones as Therapeutic Signaling Molecules in High-Stress Occupations: Implications for Mitigating Oxidative Stress and Mitochondrial Dysfunction in Future Research. Nutr. Metab. Insights 2020, 13, 1178638820979029. [Google Scholar] [CrossRef]
- Bradshaw, P.C.; Seeds, W.A.; Miller, A.C.; Mahajan, V.R.; Curtis, W.M. COVID-19: Proposing a Ketone-Based Metabolic Therapy as a Treatment to Blunt the Cytokine Storm. Oxidative Med. Cell. Longev. 2020, 2020, 6401341. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Ho, K.L.; Pherwani, S.; Ketema, E.B. Ketone metabolism in the failing heart. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2020, 1865, 158813. [Google Scholar] [CrossRef]
- Stubbs, B.J.; Koutnik, A.P.; Goldberg, E.L.; Upadhyay, V.; Turnbaugh, P.J.; Verdin, E.; Newman, J.C. Investigating Ketone Bodies as Immunometabolic Countermeasures against Respiratory Viral Infections. Med 2020, 1, 43–65. [Google Scholar] [CrossRef]
- Wood, T.R.; Stubbs, B.J.; Juul, S.E. Exogenous Ketone Bodies as Promising Neuroprotective Agents for Developmental Brain Injury. Dev. Neurosci. 2018, 40, 451–462. [Google Scholar] [CrossRef]
- Simeone, T.A.; Simeone, K.A.; Rho, J.M. Ketone Bodies as Anti-Seizure Agents. Neurochem. Res. 2017, 42, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Berg, J.; Yellen, G. Ketogenic Diet Metabolites Reduce Firing in Central Neurons by Opening KATP Channels. J. Neurosci. 2007, 27, 3618–3625. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2013, 25, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.L.; Matsumoto, J.H. The collective therapeutic potential of cerebral ketone metabolism in traumatic brain injury. J. Lipid Res. 2014, 55, 2450–2457. [Google Scholar] [CrossRef]
- White, H.; Venkatesh, B. Clinical review: Ketones and brain injury. Crit. Care 2011, 15, 219. [Google Scholar] [CrossRef] [PubMed]
- Altayyar, M.; Nasser, J.A.; Thomopoulos, D.; Bruneau, M. The Implication of Physiological Ketosis on The Cognitive Brain: A Narrative Review. Nutrients 2022, 14, 513. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. d -β-Hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef]
- Koutnik, A.P.; Poff, A.M.; Ward, N.; DeBlasi, J.; Soliven, M.A.; Romero, M.A.; Roberson, P.A.; Fox, C.; Roberts, M.D.; D’Agostino, D.P. Ketone Bodies Attenuate Wasting in Models of Atrophy. J. Cachex Sarcopenia Muscle 2020, 11, 973–996. [Google Scholar] [CrossRef]
- Goossens, C.; Weckx, R.; Derde, S.; Perre, S.V.; Derese, I.; Van Veldhoven, P.P.; Ghesquière, B.; Berghe, G.V.D.; Langouche, L. Altered cholesterol homeostasis in critical illness-induced muscle weakness: Effect of exogenous 3-hydroxybutyrate. Crit. Care 2021, 25, 252. [Google Scholar] [CrossRef]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8767. [Google Scholar] [CrossRef] [PubMed]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal Lipid Metabolism: Multiple Pathways Driving Functional Outcomes in Health and Disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Crawford, M.A. Survival of the fattest: Fat babies were the key to evolution of the large human brain. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2003, 136, 17–26. [Google Scholar] [CrossRef]
- Ahmed, T.B.; Eggesbø, M.; Criswell, R.; Uhl, O.; Demmelmair, H.; Koletzko, B. Total Fatty Acid and Polar Lipid Species Composition of Human Milk. Nutrients 2021, 14, 158. [Google Scholar] [CrossRef]
- Békési, A.; Williamson, D.H. An Explanation for Ketogenesis by the Intestine of the Suckling Rat: The Presence of an Active Hydroxymethylglutaryl-Coenzyme A Pathway. Neonatology 1990, 58, 160–165. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Crawford, M.A. Energetic and nutritional constraints on infant brain development: Implications for brain expansion during human evolution. J. Hum. Evol. 2014, 77, 88–98. [Google Scholar] [CrossRef]
- Vannucci, S.J.; Simpson, I.A. Developmental switch in brain nutrient transporter expression in the rat. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1127–E1134. [Google Scholar] [CrossRef]
- Larqué, E.; Zamora, S.; Gil, A. Dietary trans Fatty Acids Affect the Essential Fatty-Acid Concentration of Rat Milk. J. Nutr. 2000, 130, 847–851. [Google Scholar] [CrossRef]
- Steiner, P. Brain Fuel Utilization in the Developing Brain. Ann. Nutr. Metab. 2019, 75 (Suppl. 1), 8–18. [Google Scholar] [CrossRef]
- Tildon, J.T.; McKenna, M.C.; Stevenson, J.H. Transport of 3-hydroxybutyrate by cultured rat brain astrocytes. Neurochem. Res. 1994, 19, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S. Glutamate as a Neurotransmitter in the Brain: Review of Physiology and Pathology. J. Nutr. 2000, 130, 1007S–1015S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yudkoff, M.; Daikhin, Y.; Nissim, I.; Grunstein, R.; Nissim, I. Effects of Ketone Bodies on Astrocyte Amino Acid Metabolism. J. Neurochem. 2002, 69, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Yudkoff, M.; Nelson, D.; Daikhin, Y.; Erecińska, M. Tricarboxylic acid cycle in rat brain synaptosomes. Fluxes and interactions with aspartate aminotransferase and malate/aspartate shuttle. J. Biol. Chem. 1994, 269, 27414–27420. [Google Scholar] [CrossRef]
- Hasselbalch, S.G.; Knudsen, G.M.; Jakobsen, J.; Hageman, L.P.; Holm, S.; Paulson, O.B. Blood-brain barrier permeability of glucose and ketone bodies during short-term starvation in humans. Am. J. Physiol. Metab. 1995, 268, E1161–E1166. [Google Scholar] [CrossRef] [PubMed]
- Kuzawa, C.W.; Chugani, H.T.; Grossman, L.I.; Lipovich, L.; Muzik, O.; Hof, P.R.; Wildman, D.E.; Sherwood, C.C.; Leonard, W.R.; Lange, N. Metabolic costs and evolutionary implications of human brain development. Proc. Natl. Acad. Sci. USA 2014, 111, 13010–13015. [Google Scholar] [CrossRef]
- Kirmse, K.; Witte, O.W.; Holthoff, K. GABA Depolarizes Immature Neocortical Neurons in the Presence of the Ketone Body -Hydroxybutyrate. J. Neurosci. 2010, 30, 16002–16007. [Google Scholar] [CrossRef]
- Cotter, D.G.; D’Avignon, D.A.; Wentz, A.E.; Weber, M.L.; Crawford, P.A. Obligate Role for Ketone Body Oxidation in Neonatal Metabolic Homeostasis. J. Biol. Chem. 2011, 286, 6902–6910. [Google Scholar] [CrossRef]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar] [CrossRef]
- Stubbs, B.J.; Cox, P.J.; Evans, R.D.; Santer, P.; Miller, J.J.; Faull, O.K.; Magor-Elliott, S.; Hiyama, S.; Stirling, M.; Clarke, K. On the Metabolism of Exogenous Ketones in Humans. Front. Physiol. 2017, 8, 848. [Google Scholar] [CrossRef]
- Suissa, L.; Kotchetkov, P.; Guigonis, J.-M.; Doche, E.; Osman, O.; Pourcher, T.; Lindenthal, S. Ingested Ketone Ester Leads to a Rapid Rise of Acetyl-CoA and Competes with Glucose Metabolism in the Brain of Non-Fasted Mice. Int. J. Mol. Sci. 2021, 22, 524. [Google Scholar] [CrossRef]
- Bhatt, D.P.; Houdek, H.M.; Watt, J.A.; Rosenberger, T.A. Acetate supplementation increases brain phosphocreatine and reduces AMP levels with no effect on mitochondrial biogenesis. Neurochem. Int. 2013, 62, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Rodríguez, D.; Giménez-Cassina, A. Ketone Bodies in the Brain Beyond Fuel Metabolism: From Excitability to Gene Expression and Cell Signaling. Front. Mol. Neurosci. 2021, 14, 732120. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dilliraj, L.N.; Schiuma, G.; Lara, D.; Strazzabosco, G.; Clement, J.; Giovannini, P.; Trapella, C.; Narducci, M.; Rizzo, R. The Evolution of Ketosis: Potential Impact on Clinical Conditions. Nutrients 2022, 14, 3613. https://doi.org/10.3390/nu14173613
Dilliraj LN, Schiuma G, Lara D, Strazzabosco G, Clement J, Giovannini P, Trapella C, Narducci M, Rizzo R. The Evolution of Ketosis: Potential Impact on Clinical Conditions. Nutrients. 2022; 14(17):3613. https://doi.org/10.3390/nu14173613
Chicago/Turabian StyleDilliraj, Latha Nagamani, Giovanna Schiuma, Djidjell Lara, Giovanni Strazzabosco, James Clement, PierPaolo Giovannini, Claudio Trapella, Marco Narducci, and Roberta Rizzo. 2022. "The Evolution of Ketosis: Potential Impact on Clinical Conditions" Nutrients 14, no. 17: 3613. https://doi.org/10.3390/nu14173613
APA StyleDilliraj, L. N., Schiuma, G., Lara, D., Strazzabosco, G., Clement, J., Giovannini, P., Trapella, C., Narducci, M., & Rizzo, R. (2022). The Evolution of Ketosis: Potential Impact on Clinical Conditions. Nutrients, 14(17), 3613. https://doi.org/10.3390/nu14173613